
Current and Novel Antiplatelet Therapies for the Treatment of Cardiovascular Diseases

 ,
,  , and
, and
Abstract
1. Introduction
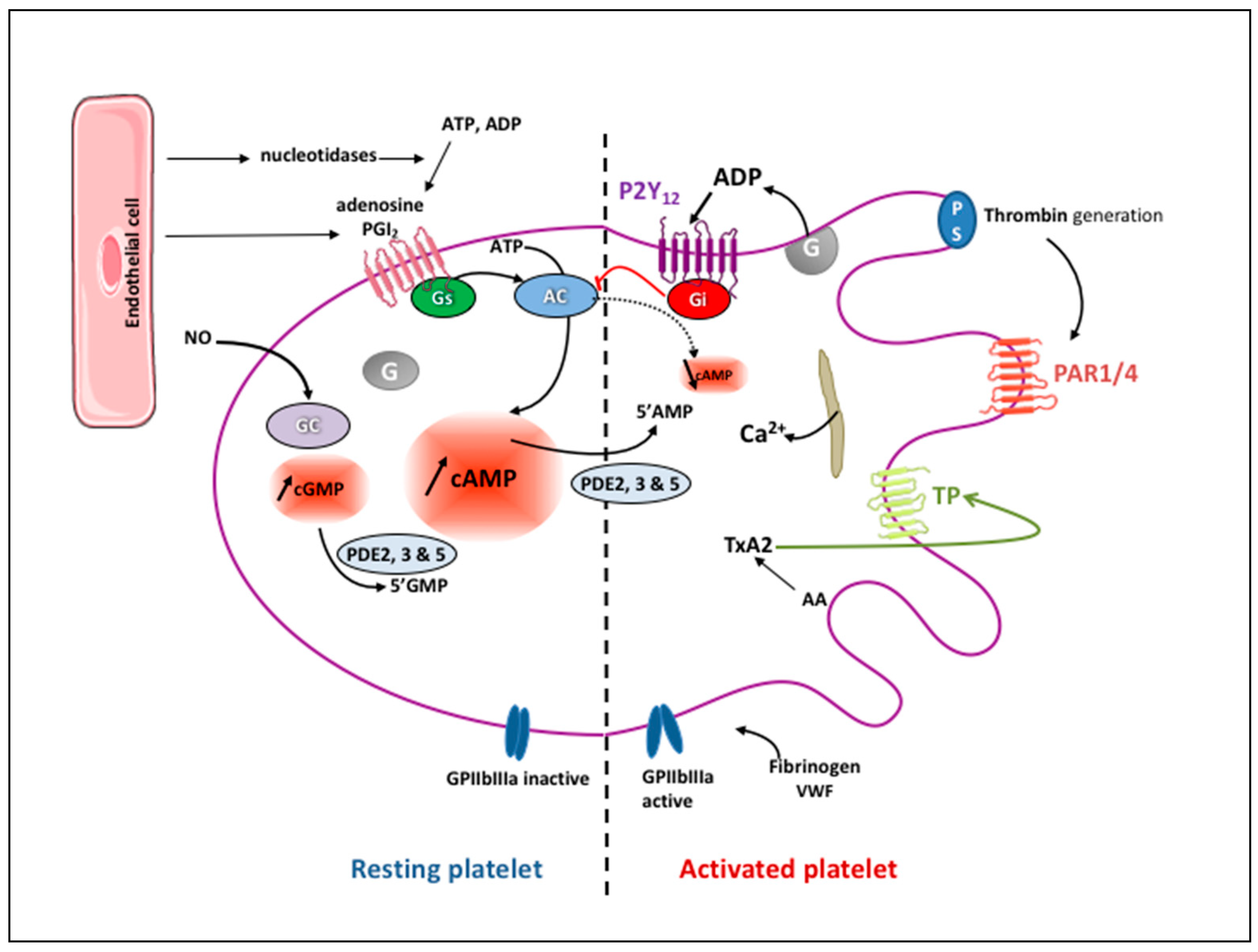
2. Platelet Physiology
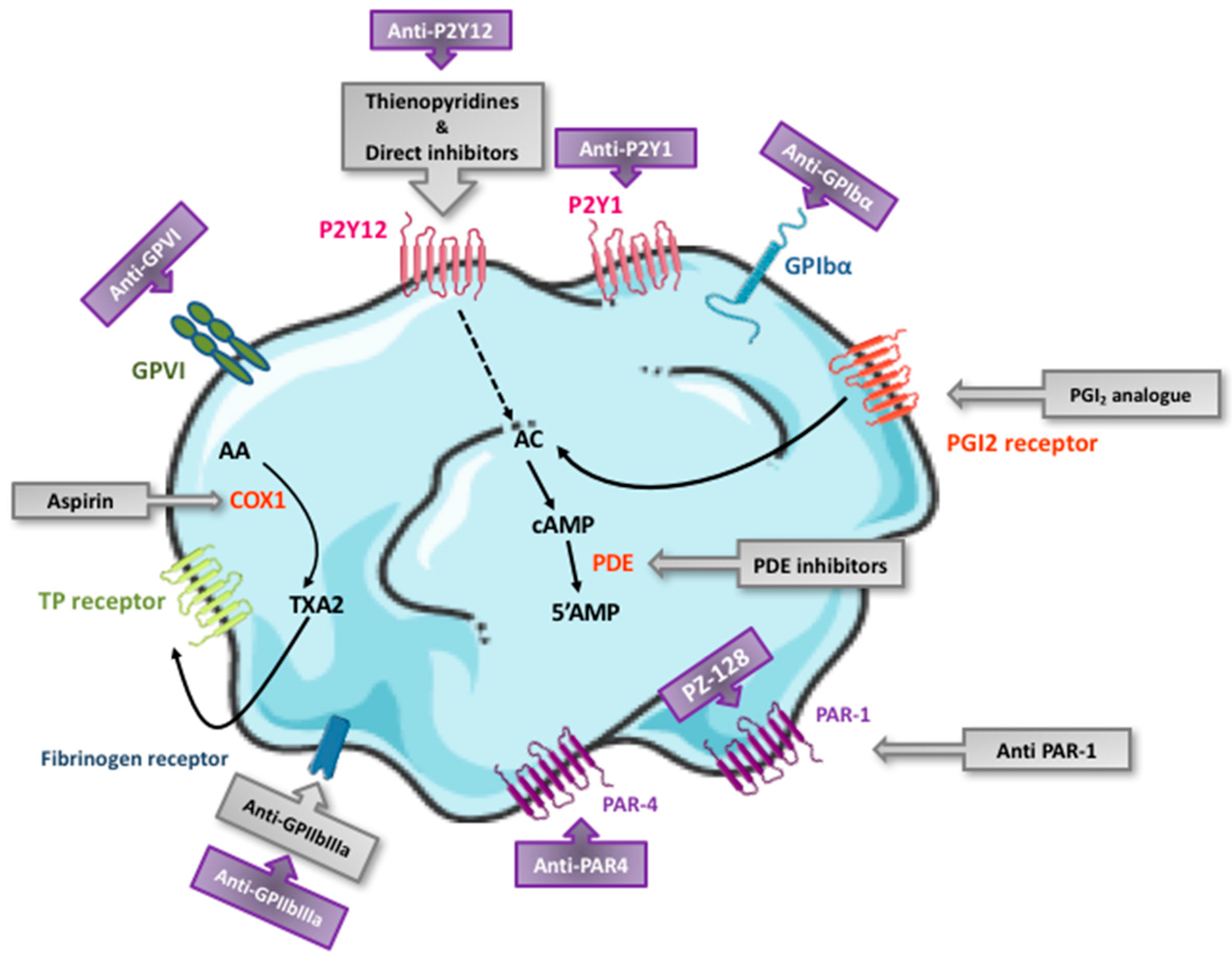
3. Currently Available Antiplatelet Agents: Targets and Pharmacological Characteristics
3.1. Aspirin
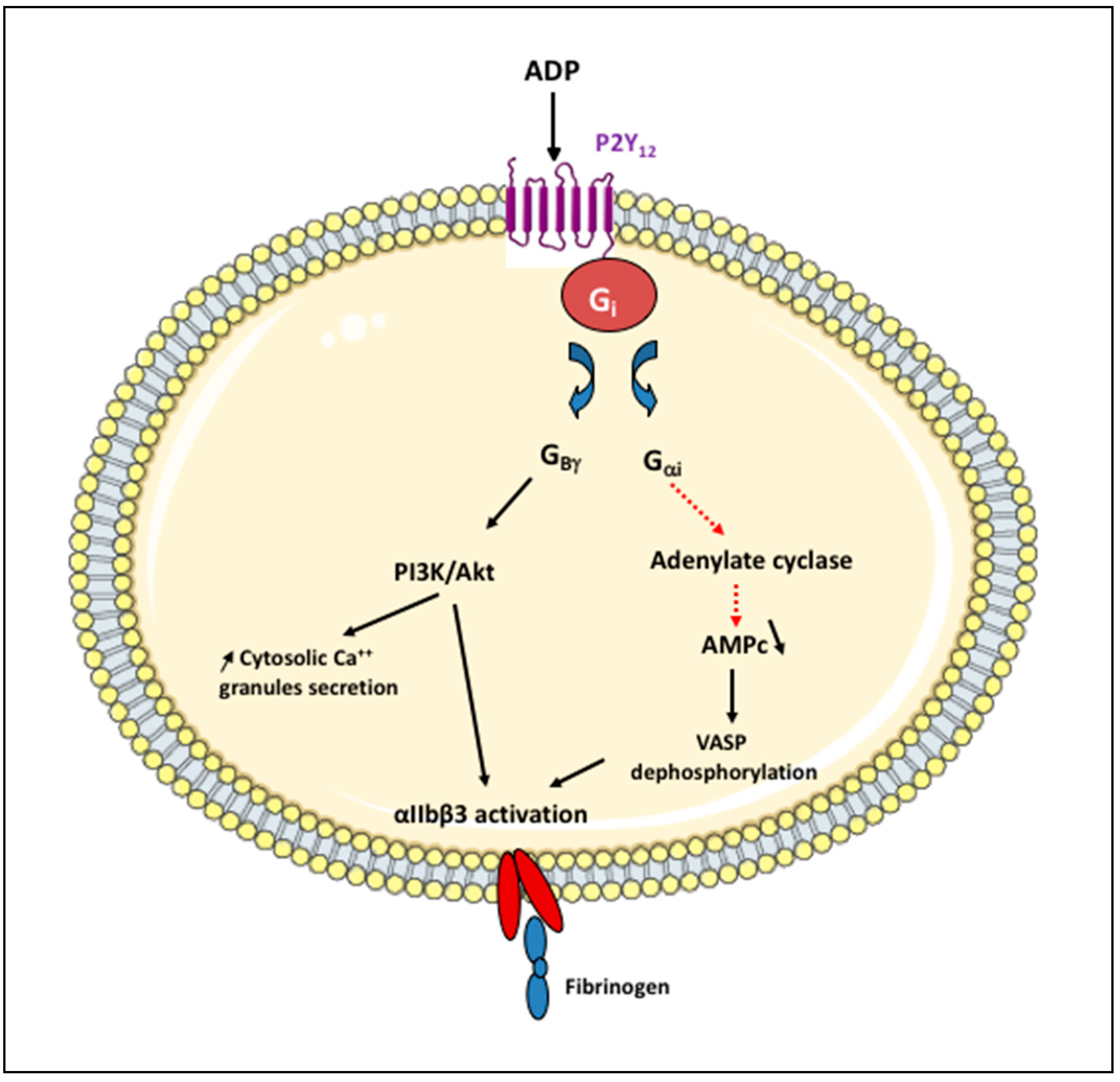
3.2. P2Y12 Receptor Antagonists
3.2.1. Thienopyridines
3.2.2. ATP Analogues
3.3. GPIIbIIIa Inhibitors
3.4. Phosphodiesterase Inhibitors
3.5. Prostacyclin Analogue
3.6. PAR1 Antagonist
4. Indications of the Currently Available Antiplatelet Agents
4.1. Primary Prevention of CVD
4.2. Acute Coronary Syndrome
4.3. Stable Coronary Artery Disease
4.4. Peripheral Artery Disease
4.5. Stroke and Transient Ischemic Attack
5. Antiplatelet Agents under Preclinical/Clinical Development
5.1. Novel PAR1 Antagonists
5.2. PAR4 Antagonists
5.3. GPVI Antagonists
5.4. C-Type Lectin-Like Receptor Inhibitors
5.5. Bruton Tyrosine Kinase Inhibitors
5.6. Inhibitors of the von Willebrand-GPIbα Axis
5.7. Phosphoinositide 3-Kinase-β Inhibitors
5.8. Protein Disulfide Isomerase Inhibitors
5.9. Novel GPIIbIIIa Inhibitors
5.10. Novel P2Y12 and P2Y1 Receptors Antagonists
5.11. 12-Lipoxygenase Inhibitor
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Capodanno, D.; Ingala, S.; Calderone, D.; Angiolillo, M.J. Aspirin for the primary prevention of cardiovascular disease: Latest evidence. Expert Rev. Cardiovasc. Ther. 2019, 17, 633–643. [Google Scholar] [CrossRef]
- Lefrancais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.; David, T.; Coughlin, T.D.S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Bergmeier, W. Negative regulators of platelet activation and adhesion. J. Thromb. Haemost. JTH 2018, 16, 220–230. [Google Scholar] [CrossRef]
- Nagy, Z.; Smolenski, A. Cyclic nucleotide-dependent inhibitory signaling interweaves with activating pathways to determine platelet responses. Res. Pract. Thromb. Haemost. 2018, 2, 558–571. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Kulkarni, S.; Ulsemer, P.; Cranmer, S.L.; Yap, C.L.; Nesbitt, W.; Harper, I.; Mistry, N.; Dopheide, S.M.; Hughan, S.C.; et al. The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells. J. Biol. Chem. 1999, 274, 36241–36251. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.P.; Auger, J.M.; McCarty, O.J.T.; Pearce, A.C. GPVI and integrin alphaIIb beta3 signaling in platelets. J. Thromb. Haemost. JTH 2005, 3, 1752–1762. [Google Scholar] [CrossRef]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling during platelet adhesion and activation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef]
- Ruiz, F.A.; Lea, C.R.; Oldfield, E.; Decampo, R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J. Biol. Chem. 2004, 279, 44250–44257. [Google Scholar] [CrossRef] [PubMed]
- Israels, S.J.; Gerrard, J.M.; Jacques, Y.V.; McNicol, A.; Cham, B.; Nishibori, M.; Bainton, D.F. Platelet dense granule membranes contain both granulophysin and P-selectin (GMP-140). Blood 1992, 80, 143–152. [Google Scholar] [CrossRef]
- Angiolillo, D.J.; Ueno, M.; Goto, S. Basic principles of platelet biology and clinical implications. Circ. J. 2010, 74, 597–607. [Google Scholar] [CrossRef]
- Shattil, S.J.; Newman, P.J. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood 2004, 104, 1606–1615. [Google Scholar] [CrossRef]
- Huang, Y.; Joshi, S.; Xiang, B.; Kanaho, Y.; Li, Z.; Bouchard, B.A.; Moncman, C.L.; Whiteheart, S. Arf6 controls platelet spreading and clot retraction via integrin αIIbβ3 trafficking. Blood 2016, 127, 1459–1467. [Google Scholar] [CrossRef]
- Agbani, E.O.; Bosch, M.T.V.D.; Brown, E.; Williams, C.M.; Mattheij, N.J.; Cosemans, J.M.; Collins, P.W.; Heemskerk, J.W.; Hers, I.; Poole, A.W. Coordinated Membrane Ballooning and Procoagulant Spreading in Human Platelets. Circulation 2015, 132, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Nieman, M.T. Protease-activated receptors in hemostasis. Blood 2016, 128, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.; Hoffhines, A. The discovery of aspirin’s antithrombotic effects. Tex. Hear. Inst. J. 2007, 34, 179–186. [Google Scholar]
- Roth, G.J.; Majerus, P.W. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J. Clin. Investig. 1975, 56, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Awtry, E.H.; Loscalzo, J. Aspirin. Circulation 2000, 101, 1206–1218. [Google Scholar] [CrossRef]
- Patrono, C.; Baigent, C.; Hirsh, J.; Roth, G. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008, 133, 199S–233S. [Google Scholar] [CrossRef]
- Wolberg, A.S.; Rosendaal, F.R.; Weitz, J.I.; Jaffer, I.H.; Agnelli, G.; Baglin, T.; Mackman, N. Venous thrombosis. Nat. Rev. Dis. Primers 2015, 1, 15006. [Google Scholar] [CrossRef]
- Undas, A.; Brummel-Ziedins, K.E.; Mann, K.G. Antithrombotic properties of aspirin and resistance to aspirin: Beyond strictly antiplatelet actions. Blood 2007, 109, 2285–2292. [Google Scholar] [CrossRef]
- Cipollone, F.; Rocca, B.; Patrono, C. Cyclooxygenase-2 expression and inhibition in atherothrombosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Coller, B.; FitzGerald, G.A.; Hirsh, J.; Roth, G. Platelet-active drugs: The relationships among dose, effectiveness, and side effects: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004, 126, 234S–264S. [Google Scholar] [CrossRef]
- Patrono, C. Aspirin as an antiplatelet drug. N. Engl. J. Med. 1994, 330, 1287–1294. [Google Scholar] [CrossRef]
- Patrono, C.; García Rodríguez, L.A.; Landolfi, R.; Baigent, C. Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med. 2005, 353, 2373–2383. [Google Scholar] [CrossRef]
- Nagelschmitz, J.; Blunck, M.; Kraetzcshmar, J.; Ludwig, M.; Wensing, G.; Hohelfeld, T. Pharmacokinetics and pharmacodynamics of acetylsalicylic acid after intravenous and oral administration to healthy volunteers. Clin. Pharmacol. 2014, 6, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Roffi, M.; Patrono, C.; Collet, J.-P.; Mueller, C.; Valgimigli, M.; Andreotti, F.; Bax, J.J.; Borger, M.; Brotons, C.; Chew, D.P.; et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 267–315. [Google Scholar] [CrossRef]
- Aboyans, V.; Ricco, J.-B.; Bartelink, M.-L.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.-P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS): Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteriesEndorsed by: The European Stroke Organization (ESO)The Task Force for the Diagnosis and Treatment of Peripheral Arterial Diseases of the European Society of Cardiology (ESC) and of the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177. [Google Scholar] [CrossRef]
- Valgimigli, M.; Bueno, H.; Byrne, R.; Collet, J.-P.; Costa, F.; Jeppsson, A.; Jüni, P.; Kastrati, A.; Kolh, P.; Mauri, L.; et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: The Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2018, 39, 213–260. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.S.; Mulder, H.; Wruck, L.M.; Pencina, M.J.; Kripalani, S.; Muñoz, D.; Crenshaw, D.L.; Effron, M.B.; Re, R.N.; Gupta, K.; et al. Comparative Effectiveness of Aspirin Dosing in Cardiovascular Disease. N. Engl. J. Med. 2021, 384, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Rocca, B.; Santilli, F.; Pitocco, D.; Mucci, L.; Petrucci, G.; Vitacolonna, E.; Lattanzio, S.; Mattoscio, D.; Zaccardi, F.; Liani, R.; et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J. Thromb. Haemost. JTH 2012, 10, 1220–1230. [Google Scholar] [CrossRef]
- Pascale, S.; Petrucci, G.; Dragani, A.; Habib, A.; Zaccardi, F.; Pagliaccia, F.; Pocaterra, D.; Ragazzoni, E.; Rolandi, G.; Rocca, B.; et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 2012, 119, 3595–3603. [Google Scholar] [CrossRef]
- Godier, A.; Garrigue, D.; Lasne, D.; Fontana, P.; Bonhomme, F.; Collet, J.-P.; De Maistre, E.; Ickx, B.; Gruel, Y.; Mazighi, M.; et al. Management of antiplatelet therapy for non-elective invasive procedures or bleeding complications: Proposals from the French Working Group on Perioperative Haemostasis (GIHP) and the French Study Group on Thrombosis and Haemostasis (GFHT), in collaboration with the French Society for Anaesthesia and Intensive Care (SFAR). Arch. Cardiovasc. Dis. 2019, 112, 199–216. [Google Scholar] [CrossRef]
- Savonitto, S.; Caracciolo, M.; Cattaneo, M.; DEServi, S. Management of patients with recently implanted coronary stents on dual antiplatelet therapy who need to undergo major surgery. J. Thromb. Haemost. JTH 2011, 9, 2133–2142. [Google Scholar] [CrossRef]
- Hankey, G.J.; Eikelboom, J.W. Aspirin resistance. Lancet 2006, 367, 606–617. [Google Scholar] [CrossRef]
- Würtz, M.; Grove, E.L.; Wulff, L.N.; Kaltoft, A.K.; Tilsted, H.H.; Jensen, L.O.; Hvas, A.-M.; Kristensen, S.D. Patients with previous definite stent thrombosis have a reduced antiplatelet effect of aspirin and a larger fraction of immature platelets. JACC Cardiovasc. Interv. 2010, 3, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Vermillet, A.; Boval, B.; Guyetand, C.; Petroni, T.; Dillinger, J.-G.; Sideris, G.; Sollier, C.B.D.; Drouet, L.; Henry, P. 24-hour time-dependent aspirin efficacy in patients with stable coronary artery disease. Thromb. Haemost. 2011, 105, 336–344. [Google Scholar] [CrossRef]
- Perneby, C.; Wallén, N.H.; Rooney, C.; Fitzgerald, D.; Hjemdahl, P. Dose- and time-dependent antiplatelet effects of aspirin. Thromb. Haemost. 2006, 95, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Dillinger, J.-G.; Drissa, A.; Sideris, G.; Sollier, C.B.D.; Voicu, S.; Silberman, S.M.; Logeart, D.; Drouet, L.; Henry, P. Biological efficacy of twice daily aspirin in type 2 diabetic patients with coronary artery disease. Am. Heart J. 2012, 164, 600–606.e1. [Google Scholar] [CrossRef] [PubMed]
- Lordkipanidzé, M.; Pharand, C.; Schampaert, E.; Palisaitis, D.A.; Diodati, J.G. Heterogeneity in platelet cyclooxygenase inhibition by aspirin in coronary artery disease. Int. J. Cardiol. 2011, 150, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Rocca, B.; Petrucci, G. Variability in the responsiveness to low-dose aspirin: Pharmacological and disease-related mechanisms. Thrombosis 2012, 2012, 376721. [Google Scholar] [CrossRef]
- Bagoly, Z.; Homoródi, N.; Kovács, E.G.; Sarkady, F.; Csiba, L.; Édes, I.; Muszbek, L. How to test the effect of aspirin and clopidogrel in patients on dual antiplatelet therapy? Platelets 2016, 27, 59–65. [Google Scholar] [CrossRef]
- Catella-Lawson, F.; Reilly, M.; Kapoor, S.C.; Cucchiara, A.J.; Demarco, S.; Tournier, B.; Vyas, S.N.; Fitzgerald, G.A. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N. Engl. J. Med. 2001, 345, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Massimi, I.; Lotti, L.V.; Temperilli, F.; Mancone, M.; Sardella, G.; Calcagno, S.; Turriziani, O.; Frati, L.; Pulcinelli, F.M. Enhanced platelet MRP4 expression and correlation with platelet function in patients under chronic aspirin treatment. Thromb. Haemost. 2016, 116, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Mattiello, T.; Guerriero, R.; Lotti, L.V.; Trifirò, E.; Felli, M.P.; Barbarulo, A.; Pucci, B.; Gazzaniga, P.; Gaudio, C.; Frati, L.; et al. Aspirin extrusion from human platelets through multidrug resistance protein-4-mediated transport: Evidence of a reduced drug action in patients after coronary artery bypass grafting. J. Am. Coll. Cardiol. 2011, 58, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Massimi, I.; Guerriero, R.; Lotti, L.V.; Lulli, V.; Borgognone, A.; Romani, F.; Barilla’, F.; Gaudio, C.; Gabbianelli, M.; Frati, L.; et al. Aspirin influences megakaryocytic gene expression leading to up-regulation of multidrug resistance protein-4 in human platelets. Br. J. Clin. Pharmacol. 2014, 78, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, B.; Rubboli, A.; Castelvetri, C.; Milandri, M. Ticlopidine versus oral anticoagulation for coronary stenting. Cochrane Database Syst. Rev. 2001, CD002133. [Google Scholar] [CrossRef]
- Hollopeter, G.; Jantzen, H.-M.; Vincent, D.; Li, G.; England, L.; Ramakrishnan, V.; Yang, R.-B.; Nurden, P.; Nurden, A.; Julius, D.; et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [Google Scholar] [CrossRef]
- Kazui, M.; Nishiya, Y.; Ishizuka, T.; Hagihara, K.; Farid, N.A.; Okazaki, O.; Ikeda, T.; Kurihara, A. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 2010, 38, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Sangkuhl, K.; Klein, T.E.; Altman, R.B. Clopidogrel pathway. Pharm. Genom. 2010, 20, 463–465. [Google Scholar] [CrossRef]
- Yusuf, S.; Zhao, F.; Mehta, S.R.; Chrolavicius, S.; Tognoni, G.; Fox, K.K. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N. Engl. J. Med. 2001, 345, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Erlinge, D.; Varenhorst, C.; Braun, O.Ö.; James, S.; Winters, K.J.; Jakubowski, J.A.; Brandt, J.T.; Sugidachi, A.; Siegbahn, A.; Wallentin, L. Patients with poor responsiveness to thienopyridine treatment or with diabetes have lower levels of circulating active metabolite, but their platelets respond normally to active metabolite added ex vivo. J. Am. Coll. Cardiol. 2008, 52, 1968–1977. [Google Scholar] [CrossRef]
- O’Donoghue, M.; Wiviott, S.D. Clopidogrel response variability and future therapies: Clopidogrel: Does one size fit all? Circulation 2006, 114, e600–e606. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, T.; Payne, C.D.; Winters, K.J.; Darstein, C.; Brandt, J.T.; Jakubowski, J.A.; Naganuma, H.; Siegbahn, A.; Wallentin, L. Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur. Heart J. 2006, 27, 1166–1173. [Google Scholar] [CrossRef]
- Brandt, J.T.; Payne, C.D.; Wiviott, S.D.; Weerakkody, G.; Farid, N.A.; Small, D.S.; Jakubowski, J.A.; Naganuma, H.; Winters, K.J. A comparison of prasugrel and clopidogrel loading doses on platelet function: Magnitude of platelet inhibition is related to active metabolite formation. Am. Heart J. 2007, 153, e9–e16. [Google Scholar] [CrossRef]
- Nakamura, M.; Iizuka, T.; Sagawa, K.; Abe, K.; Chikada, S.; Arai, M. Prasugrel for Japanese patients with acute coronary syndrome in short-term clinical practice (PRASFIT-Practice I): A postmarketing observational study. Cardiovasc. Interv. Ther. 2018, 33, 135–145. [Google Scholar] [CrossRef]
- Farid, N.A.; Smith, R.L.; Gillespie, T.A.; Rash, T.J.; Blair, P.E.; Kurihara, A.; Goldberg, M.J. The disposition of prasugrel, a novel thienopyridine, in humans. Drug Metab. Dispos. 2007, 35, 1096–1104. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Antman, E.M.; Braunwald, E. Prasugrel. Circulation 2010, 122, 394–403. [Google Scholar] [CrossRef]
- Rehmel, J.L.F.; Eckstein, J.A.; Farid, N.A.; Heim, J.B.; Kasper, S.C.; Kurihara, A.; Wrighton, S.A.; Ring, B.J. Interactions of two major metabolites of prasugrel, a thienopyridine antiplatelet agent, with the cytochromes P450. Drug Metab. Dispos. 2006, 34, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Farid, N.A.; Kurihara, A.; Wrighton, S.A. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans. J. Clin. Pharmacol. 2010, 50, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, J.A.; Matsushima, N.; Asai, F.; Naganuma, H.; Brandt, J.T.; Hirota, T.; Freestone, S.; Winters, K.J. A multiple dose study of prasugrel (CS-747), a novel thienopyridine P2Y12 inhibitor, compared with clopidogrel in healthy humans. Br. J. Clin. Pharmacol. 2007, 63, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L. P2Y(12) inhibitors: Differences in properties and mechanisms of action and potential consequences for clinical use. Eur. Heart J. 2009, 30, 1964–1977. [Google Scholar] [CrossRef]
- Nylander, S.; Femia, E.A.; Scavone, M.; Berntsson, P.; Asztély, A.-K.; Nelander, K.; Löfgren, L.; Nilsson, R.G.; Cattaneo, M. Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y12 antagonism. J. Thromb. Haemost. JTH 2013, 11, 1867–1876. [Google Scholar] [CrossRef]
- Butler, K.; Teng, R. Pharmacokinetics, pharmacodynamics, safety and tolerability of multiple ascending doses of ticagrelor in healthy volunteers. Br. J. Clin. Pharmacol. 2010, 70, 65–77. [Google Scholar] [CrossRef]
- Teng, R. Pharmacokinetic, pharmacodynamic and pharmacogenetic profile of the oral antiplatelet agent ticagrelor. Clin. Pharmacokinet. 2012, 51, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; James, S.; Storey, R.; Armstrong, M.; Barratt, B.; Horrow, J.; Husted, S.; Katus, H.; Steg, P.G.; Shah, S.H.; et al. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: A genetic substudy of the PLATO trial. Lancet 2010, 376, 1320–1328. [Google Scholar] [CrossRef]
- Capodanno, D.; Milluzzo, R.P.; Angiolillo, D.J. Intravenous antiplatelet therapies (glycoprotein IIb/IIIa receptor inhibitors and cangrelor) in percutaneous coronary intervention: From pharmacology to indications for clinical use. Ther. Adv. Cardiovasc. Dis. 2019, 13, 1753944719893274. [Google Scholar] [CrossRef]
- Baker, D.E.; Ingram, K.T. Cangrelor. Hosp. Pharm. 2015, 50, 922–929. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Stone, G.W.; Mahaffey, K.W.; Gibson, C.M.; Steg, P.G.; Hamm, C.W.; Price, M.J.; Leonardi, S.; Gallup, D.; Bramucci, E.; et al. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N. Engl. J. Med. 2013, 368, 1303–1313. [Google Scholar] [CrossRef]
- Angiolillo, D.J.; Firstenberg, M.S.; Price, M.J.; Tummala, P.E.; Hutyra, M.; Welsby, I.; Voeltz, M.D.; Chandna, H.; Ramaiah, C.; Brtko, M.; et al. Bridging antiplatelet therapy with cangrelor in patients undergoing cardiac surgery: A randomized controlled trial. JAMA 2012, 307, 265–274. [Google Scholar] [CrossRef]
- Salahuddin, H.; Dawod, G.; Zaidi, S.F.; Shawver, J.; Burgess, R.; Jumaa, M.A. Safety of Low Dose Intravenous Cangrelor in Acute Ischemic Stroke: A Case Series. Front. Neurol. 2021, 12, 636682. [Google Scholar] [CrossRef]
- De Luca, G.; Savonitto, S.; van’t Hof, A.W.J.; Suryapranata, H. Platelet GP IIb-IIIa Receptor Antagonists in Primary Angioplasty: Back to the Future. Drugs 2015, 75, 1229–1253. [Google Scholar] [CrossRef] [PubMed]
- Steinhubl, S.R.; Kottke-Marchant, K.; Moliterno, D.J.; Rosenthal, M.L.; Godfrey, N.K.; Coller, B.S.; Topol, E.; Lincoff, A.M. Attainment and maintenance of platelet inhibition through standard dosing of abciximab in diabetic and nondiabetic patients undergoing percutaneous coronary intervention. Circulation 1999, 100, 1977–1982. [Google Scholar] [CrossRef]
- Kereiakes, D.J.; Broderick, T.M.; Roth, E.M.; Whang, D.; Shimshak, T.; Runyon, J.P.; Hattemer, C.; Schneider, J.; Lacock, P.; Mueller, M.; et al. Time course, magnitude, and consistency of platelet inhibition by abciximab, tirofiban, or eptifibatide in patients with unstable angina pectoris undergoing percutaneous coronary intervention. Am. J. Cardiol. 1999, 84, 391–395. [Google Scholar] [CrossRef]
- Ahn, H.S.; Crim, W.; Romano, M.; Sybertz, E.; Pitts, B. Effects of selective inhibitors on cyclic nucleotide phosphodiesterases of rabbit aorta. Biochem. Pharmacol. 1989, 38, 3331–3339. [Google Scholar] [CrossRef]
- Gresele, P.; Momi, S.; Falcinelli, E. Anti-platelet therapy: Phosphodiesterase inhibitors. Br. J. Clin. Pharmacol. 2011, 72, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Kerndt, C.C.; Nagalli, S. Dipyridamole; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Noma, K.; Higashi, Y. Cilostazol for treatment of cerebral infarction. Expert Opin. Pharmacother. 2018, 19, 1719–1726. [Google Scholar] [CrossRef]
- Shichinohe, H.; Tan, C.; Abumiya, T.; Nakayama, N.; Kazumata, K.; Hokari, M.; Houkin, K.; Kuroda, S. Neuroprotective effects of cilostazol are mediated by multiple mechanisms in a mouse model of permanent focal ischemia. Brain Res. 2015, 1602, 53–61. [Google Scholar] [CrossRef]
- Liu, Y.; Shakur, Y.; Yoshitake, M.; Kambayashi Ji, J. Cilostazol (pletal): A dual inhibitor of cyclic nucleotide phosphodiesterase type 3 and adenosine uptake. Cardiovasc. Drug Rev. 2001, 19, 369–386. [Google Scholar] [CrossRef]
- Iwamoto, T.; Kin, K.; Miyazaki, K.; Shin, K.; Takasaki, M. Recovery of platelet function after withdrawal of cilostazol administered orally for a long period. J. Atheroscler. Thromb. 2003, 10, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.M.; Goa, K.L. Iloprost. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in peripheral vascular disease, myocardial ischaemia and extracorporeal circulation procedures. Drugs 1992, 43, 889–924. [Google Scholar] [CrossRef] [PubMed]
- Franchi, F.; Rollini, F.; Park, Y.; Angiolillo, D.J. Platelet thrombin receptor antagonism with vorapaxar: Pharmacology and clinical trial development. Future Cardiol. 2015, 11, 547–564. [Google Scholar] [CrossRef]
- Gremmel, T.; Panzer, S. Oral antiplatelet therapy: Impact for transfusion medicine. Vox Sang. 2017, 112, 511–517. [Google Scholar] [CrossRef]
- Kosoglou, T.; Reyderman, L.; Tiessen, R.G.; Van Vliet, A.A.; Fales, R.R.; Keller, R.; Yang, B.; Cutler, D.L. Pharmacodynamics and pharmacokinetics of the novel PAR-1 antagonist vorapaxar (formerly SCH 530348) in healthy subjects. Eur. J. Clin. Pharmacol. 2012, 68, 249–258. [Google Scholar] [CrossRef]
- McNeil, J.J.; Wolfe, R.; Woods, R.L.; Tonkin, A.M.; Donnan, G.A.; Nelson, M.R.; Reid, C.M.; Lockery, J.E.; Kirpach, B.; Storey, E.; et al. Effect of Aspirin on Cardiovascular Events and Bleeding in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1509–1518. [Google Scholar] [CrossRef]
- ASCEND Study Collaborative Group; Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Brotons, C.; Coppolecchia, R.; Cricelli, C.; Darius, H.; Gorelick, P.B.; Howard, G.; Pearson, T.A.; Rothwell, P.M.; Ruilope, L.M.; et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): A randomised, double-blind, placebo-controlled trial. Lancet 2018, 392, 1036–1046. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Mahase, E. US taskforce advises against low dose aspirin for primary prevention of cardiovascular disease. BMJ 2021, 375, n2521. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, E.D.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Kimura, T.; Ishihara, M.; Nakagawa, Y.; Nakao, K.; Miyauchi, K.; Sakamoto, T.; Tsujita, K.; Hagiwara, N.; Miyazaki, S.; et al. JCS 2018 Guideline on Diagnosis and Treatment of Acute Coronary Syndrome. Circ. J. 2019, 83, 1085–1196. [Google Scholar] [CrossRef]
- Levine, G.N.; Bates, E.R.; Bittl, J.A.; Brindis, R.G.; Fihn, S.D.; Fleisher, L.A.; Granger, C.B.; Lange, R.A.; Mack, M.J.; Mauri, L.; et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2016, 68, 1082–1115. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.-P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, M.; Qaderdan, K.; Willemsen, L.; Hermanides, R.; Bergmeijer, T.; de Vrey, E.; Heestermans, T.; Gin, M.T.J.; Waalewijn, R.; Hofma, S.; et al. Clopidogrel versus ticagrelor or prasugrel in patients aged 70 years or older with non-ST-elevation acute coronary syndrome (POPular AGE): The randomised, open-label, non-inferiority trial. Lancet 2020, 395, 1374–1381. [Google Scholar] [CrossRef]
- Neumann, F.J.; Sousa-Uva, M.; Ahlsson, A.; Alfonso, F.; Banning, A.P.; Benedetto, U.; Byrne, R.A.; Collet, J.P.; Falk, V.; Head, S.J.; et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur. Heart J. 2019, 40, 87–165. [Google Scholar] [CrossRef]
- Mehta, S.R.; Bainey, K.R.; Cantor, W.J.; Lordkipanidze, M.; Marquis-Gravel, G.; Robinson, S.D.; Sibbald, M.; So, D.Y.; Wong, G.C.; Abunassar, J.G.; et al. 2018 Canadian Cardiovascular Society/Canadian Association of Interventional Cardiology Focused Update of the Guidelines for the Use of Antiplatelet Therapy. Can. J. Cardiol. 2018, 34, 214–233. [Google Scholar] [CrossRef]
- January, C.T.; Wann, L.S.; Calkins, H.; Chen, L.Y.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; Furie, K.L.; et al. 2019 AHA/ACC/HRS Focused Update of the 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society in Collaboration With the Society of Thoracic Surgeons. Circulation 2019, 140, e125–e151. [Google Scholar] [CrossRef]
- Saito, Y.; Kobayashi, Y. Update on Antithrombotic Therapy after Percutaneous Coronary Intervention. Intern. Med. 2020, 59, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Goodman, S.G.; Bhatt, D.L.; Eikelboom, J.W.; Price, M.J.; Moliterno, D.J.; Cannon, C.P.; Tanguay, J.-F.; Granger, C.B.; Mauri, L.; et al. Antithrombotic Therapy in Patients With Atrial Fibrillation Treated With Oral Anticoagulation Undergoing Percutaneous Coronary Intervention: A North American Perspective-2018 Update. Circulation 2018, 138, 527–536. [Google Scholar] [CrossRef]
- Lip, G.Y.H.; Collet, J.-P.; Haude, M.; Byrne, R.; Chung, E.H.; Fauchier, L.; Halvorsen, S.; Lau, D.H.; Lopez-Cabanillas, N.; Lettino, M.; et al. 2018 Joint European consensus document on the management of antithrombotic therapy in atrial fibrillation patients presenting with acute coronary syndrome and/or undergoing percutaneous cardiovascular interventions: A joint consensus document of the European Heart Rhythm Association (EHRA), European Society of Cardiology Working Group on Thrombosis, European Association of Percutaneous Cardiovascular Interventions (EAPCI), and European Association of Acute Cardiac Care (ACCA) endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS), Latin America Heart Rhythm Society (LAHRS), and Cardiac Arrhythmia Society of Southern Africa (CASSA). Eur. Soc. Cardiol. 2019, 21, 192–193. [Google Scholar] [CrossRef]
- Baumgartner, H.; Falk, V.; Bax, J.J.; De Bonis, M.; Hamm, C.; Holm, P.J.; Iung, B.; Lancellotti, P.; Lansac, E.; Rodriguez Muñoz, D.; et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 2017, 38, 2739–2791. [Google Scholar] [CrossRef]
- Hamilos, M.; Petousis, S.; Xanthopoulou, I.; Goudevenos, J.; Kanakakis, J.; Sitafidis, G.; Vavouranakis, M.; Skalidis, E.; Kochiadakis, G.; Lekakis, J.; et al. Antiplatelet treatment in diabetic patients with acute coronary syndrome undergoing percutaneous coronary intervention: A GReek AntiPlatElet registry substudy. Coron. Artery Dis. 2018, 29, 53–59. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.-J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.R.; Hamburg, N.; Kinlay, S.; et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2017, 69, 1465–1508. [Google Scholar] [CrossRef] [PubMed]
- Cacoub, P.P.; Bhatt, D.L.; Steg, P.; Topol, E.J.; Creager, M.A. CHARISMA Investigators. Patients with peripheral arterial disease in the CHARISMA trial. Eur. Heart J. 2009, 30, 192–201. [Google Scholar] [CrossRef]
- Morrow, D.A.; Braunwald, E.; Bonaca, M.P.; Ameriso, S.F.; Dalby, A.J.; Fish, M.P.; Fox, K.A.; Lipka, L.J.; Liu, X.; Nicolau, J.C.; et al. Vorapaxar in the secondary prevention of atherothrombotic events. N. Engl. J. Med. 2012, 366, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Tricoci, P.; Huang, Z.; Held, C.; Moliterno, D.J.; Armstrong, P.; Van de Werf, F.; White, H.D.; Aylward, P.; Wallentin, L.; Chen, E.; et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N. Engl. J. Med. 2012, 366, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Bonaca, M.P.; Scirica, B.M.; Creager, M.A.; Olin, J.; Bounameaux, H.; Dellborg, M.; Lamp, J.M.; Murphy, S.A.; Braunwald, E.; Morrow, D.A. Vorapaxar in patients with peripheral artery disease: Results from TRA2{degrees}P-TIMI 50. Circulation 2013, 127, 1522–1529. [Google Scholar] [CrossRef]
- Faxon, D.P.; Creager, M.A.; Smith, S.C., Jr.; Pasternak, R.C.; Olin, J.W.; Bettmann, M.A.; Criqui, M.H.; Milani, R.V.; Loscalzo, J.; Kaufman, J.A.; et al. Atherosclerotic Vascular Disease Conference: Executive summary: Atherosclerotic Vascular Disease Conference proceeding for healthcare professionals from a special writing group of the American Heart Association. Circulation 2004, 109, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Kleindorfer, D.O.; Towfighi, A.; Chaturvedi, S.; Cockroft, K.M.; Gutierrez, J.; Lombardi-Hill, D.; Kamel, H.; Kernan, W.N.; Kittner, S.J.; Leira, E.C.; et al. 2021 Guideline for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack: A Guideline From the American Heart Association/American Stroke Association. Stroke 2021, 52, e364–e467. [Google Scholar] [CrossRef]
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Algra, A.; Chen, Z.; Diener, H.-C.; Norrving, B.; Mehta, Z. Effects of aspirin on risk and severity of early recurrent stroke after transient ischaemic attack and ischaemic stroke: Time-course analysis of randomised trials. Lancet 2016, 388, 365–375. [Google Scholar] [CrossRef]
- Sacco, R.L.; Diener, H.-C.; Yusuf, S.; Cotton, D.; Ôunpuu, S.; Lawton, W.A.; Palesch, Y.; Martin, R.H.; Albers, G.W.; Bath, P.; et al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N. Engl. J. Med. 2008, 359, 1238–1251. [Google Scholar] [CrossRef]
- Sahara, N.; Kuwashiro, T.; Okada, Y. Cerebral infarction and transient ischemic attack. Nihon rinsho. Jpn. J. Clin. Med. 2016, 74, 666–670. [Google Scholar]
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Bliden, K.P.; Turner, S.E.; Tantry, U.S.; Gesheff, M.G.; Barr, T.P.; Covic, L.; Kuliopulos, A. Cell-Penetrating Pepducin Therapy Targeting PAR1 in Subjects With Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 189–197. [Google Scholar] [CrossRef]
- Wilson, S.J.; Ismat, F.A.; Wang, Z.; Cerra, M.; Narayan, H.; Raftis, J.; Gray, T.J.; Connell, S.; Garonzik, S.; Ma, X.; et al. PAR4 (Protease-Activated Receptor 4) Antagonism With BMS-986120 Inhibits Human Ex Vivo Thrombus Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 448–456. [Google Scholar] [CrossRef]
- Ungerer, M.; Rosport, K.; Bültmann, A.; Piechatzek, R.; Uhland, K.; Schlieper, P.; Gawaz, M.P.; Münch, G. Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation 2011, 123, 1891–1899. [Google Scholar] [CrossRef]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.C.; DeFeo-Fraulini, T.; Hutabarat, R.M.; Horvath, C.J.; Merlino, P.G.; Marsh, H.N.; Healy, J.M.; BouFakhreddine, S.; Holohan, T.V.; Schaub, R.G. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation 2007, 116, 2678–2686. [Google Scholar] [CrossRef]
- Markus, H.S.; McCollum, C.; Imray, C.; Goulder, M.A.; Gilbert, J.; King, A. The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy: A randomized trial. Stroke 2011, 42, 2149–2153. [Google Scholar] [CrossRef]
- Nylander, S.; Wågberg, F.; Andersson, M.; Skärby, T.; Gustafsson, D. Exploration of efficacy and bleeding with combined phosphoinositide 3-kinase β inhibition and aspirin in man. J. Thromb. Haemost. JTH 2015, 13, 1494–1502. [Google Scholar] [CrossRef]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellstrom, O.; et al. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. JTH 2012, 10, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Stopa, J.D.; Neuberg, D.; Puligandla, M.; Furie, B.; Flaumenhaft, R.; Zwicker, J.I. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI Insight 2017, 2, e89373. [Google Scholar] [CrossRef]
- Zwicker, J.I.; Schlechter, B.L.; Stopa, J.D.; Liebman, H.A.; Aggarwal, A.; Puligandla, M.; Caughey, T.; Bauer, K.A.; Kuemmerle, N.; Wong, E.; et al. Targeting protein disulfide isomerase with the flavonoid isoquercetin to improve hypercoagulability in advanced cancer. JCI Insight 2019, 4, 125851. [Google Scholar] [CrossRef]
- Covic, L.; Misra, M.; Badar, J.; Singh, C.; Kuliopulos, A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med. 2002, 8, 1161–1165. [Google Scholar] [CrossRef]
- Aisiku, O.; Peters, C.G.; De Ceunynck, K.; Ghosh, C.C.; Dilks, J.R.; Fustolo-Gunnink, S.F.; Huang, M.; Dockendorff, C.; Parikh, S.M.; Flaumenhaft, R. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood 2015, 125, 1976–1985. [Google Scholar] [CrossRef]
- Kuliopulos, A.; Covic, L. Blocking receptors on the inside: Pepducin-based intervention of PAR signaling and thrombosis. Life Sci. 2003, 74, 255–262. [Google Scholar] [CrossRef] [PubMed]
- French, S.L.; Thalmann, C.; Bray, P.F.; Macdonald, L.E.; Murphy, A.; Sleeman, M.W.; Hamilton, J.R. A function-blocking PAR4 antibody is markedly antithrombotic in the face of a hyperreactive PAR4 variant. Blood Adv. 2018, 2, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Gruner, S.; Prostredna, M.; Koch, M.; Miura, Y.; Schulte, V.; Jung, S.M.; Moroi, M.; Nieswandt, B. Relative antithrombotic effect of soluble GPVI dimer compared with anti-GPVI antibodies in mice. Blood 2005, 105, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, K.; Uphaus, T.; Loftus, I.; Poppert, H.; Diener, H.C.; Zobel, J.; Münch, G. Revacept, an Inhibitor of Platelet Adhesion in Symptomatic Carotid Artery Stenosis: Design and Rationale of a Randomized Phase II Clinical Trial. TH Open Companion J. Thromb. Haemost. 2020, 4, e393–e399. [Google Scholar] [CrossRef]
- Schüpke, S.; Hein-Rothweiler, R.; Mayer, K.; Janisch, M.; Sibbing, D.; Ndrepepa, G.; Hilz, R.; Laugwitz, K.-L.; Bernlochner, I.; Gschwendtner, S.; et al. Revacept, a Novel Inhibitor of Platelet Adhesion, in Patients Undergoing Elective PCI-Design and Rationale of the Randomized ISAR-PLASTER Trial. Thromb. Haemost. 2019, 119, 1539–1545. [Google Scholar] [CrossRef]
- Lebozec, K.; Jandrot-Perrus, M.; Avenard, G.; Favre-Bulle, O.; Billiald, P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. MAbs 2017, 9, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Pollitt, A.Y.; Mori, J.; Eble, J.A.; Tomlinson, M.G.; Hartwig, J.H.; O’Callaghan, C.A.; Fütterer, K.; Watson, S.P. CLEC-2 activates Syk through dimerization. Blood 2010, 115, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.L.; Navarro-Nunez, L.; Bénézech, C.; Nayar, S.; Kingston, B.L.; Nieswandt, B.; Barone, F.; Watson, S.P.; Buckley, C.D.; DeSanti, G.E. The expression of mouse CLEC-2 on leucocyte subsets varies according to their anatomical location and inflammatory state. Eur. J. Immunol. 2015, 45, 2484–2493. [Google Scholar] [CrossRef]
- Bourne, J.H.; Colicchia, M.; Di, Y.; Martin, E.; Slater, A.; Roumenina, L.T.; Dimitrov, J.; Watson, S.P.; Rayes, J. Heme induces human and mouse platelet activation through C-type-lectin-like receptor-2. Haematologica 2021, 106, 626–629. [Google Scholar] [CrossRef]
- Suzuki-Inoue, K.; Kato, Y.; Inoue, O.; Kaneko, M.K.; Mishima, K.; Yatomi, Y.; Yamazaki, Y.; Narimatsu, H.; Ozaki, Y. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J. Biol. Chem. 2007, 282, 25993–26001. [Google Scholar] [CrossRef]
- Tsukiji, N.; Inoue, O.; Morimoto, M.; Tatsumi, N.; Nagatomo, H.; Ueta, K.; Shirai, T.; Sasaki, T.; Otake, S.; Tamura, S.; et al. Platelets play an essential role in murine lung development through Clec-2/podoplanin interaction. Blood 2018, 132, 1167–1179. [Google Scholar] [CrossRef]
- Haining, E.J.; Cherpokova, D.; Wolf, K.; Becker, I.C.; Beck, S.; Eble, J.A.; Stegner, D.; Watson, S.P.; Nieswandt, B. CLEC-2 contributes to hemostasis independently of classical hemITAM signaling in mice. Blood 2017, 130, 2224–2228. [Google Scholar] [CrossRef]
- May, F.; Hagedorn, I.; Pleines, I.; Bender, M.; Vögtle, T.; Eble, J.; Elvers, M.; Nieswandt, B. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 2009, 114, 3464–3472. [Google Scholar] [CrossRef]
- Bender, M.; May, F.; Lorenz, V.; Thielmann, I.; Hagedorn, I.; Finney, B.A.; Vögtle, T.; Remer, K.; Braun, A.; Bösl, M.; et al. Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Inoue, K.; Inoue, O.; Ding, G.; Nishimura, S.; Hokamura, K.; Eto, K.; Kashiwagi, H.; Tomiyama, Y.; Yatomi, Y.; Umemura, K.; et al. Essential in vivo roles of the C-type lectin receptor CLEC-2: Embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J. Biol. Chem. 2010, 285, 24494–24507. [Google Scholar] [CrossRef] [PubMed]
- Payne, H.; Ponomaryov, T.; Watson, S.P.; Brill, A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood 2017, 129, 2013–2020. [Google Scholar] [CrossRef]
- Tsukiji, N.; Osada, M.; Sasaki, T.; Shirai, T.; Satoh, K.; Inoue, O.; Umetani, N.; Mochizuki, C.; Saito, T.; Kojima, S.; et al. Cobalt hematoporphyrin inhibits CLEC-2-podoplanin interaction, tumor metastasis, and arterial/venous thrombosis in mice. Blood Adv. 2018, 2, 2214–2225. [Google Scholar] [CrossRef]
- Gitz, E.; Pollitt, A.; Gitz-Francois, J.J.; AlShehri, O.; Mori, J.; Montague, S.; Nash, G.; Douglas, M.R.; Gardiner, E.; Andrews, R.K.; et al. CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood 2014, 124, 2262–2270. [Google Scholar] [CrossRef]
- Newman, D.K. CLEC-2: The inside story. Blood 2015, 125, 3972–3974. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Brown, J.R.; Moslehi, J.; Ewer, M.S.; O’Brien, S.M.; Ghia, P.; Cymbalista, F.; Shanafelt, T.D.; Fraser, G.; Rule, S.; Coutre, S.E.; et al. Incidence of and risk factors for major haemorrhage in patients treated with ibrutinib: An integrated analysis. Br. J. Haematol. 2019, 184, 558–569. [Google Scholar] [CrossRef]
- Busygina, K.; Jamasbi, J.; Seiler, T.; Deckmyn, H.; Weber, C.; Brandl, R.; Lorenz, R.; Siess, W. Oral Bruton tyrosine kinase inhibitors selectively block atherosclerotic plaque-triggered thrombus formation in humans. Blood 2018, 131, 2605–2616. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; De La Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Bartunek, J.; Barbato, E.; Heyndrickx, G.; Vanderheyden, M.; Wijns, W.; Holz, J.-B. Novel antiplatelet agents: ALX-0081, a Nanobody directed towards von Willebrand factor. J. Cardiovasc. Transl. Res. 2013, 6, 355–363. [Google Scholar] [CrossRef]
- Cho, J.; Furie, B.C.; Coughlin, S.R.; Furie, B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J. Clin. Investig. 2008, 118, 1123–1131. [Google Scholar] [CrossRef]
- Hubbard, G.P.; Wolffram, S.; Lovegrove, J.A.; Gibbins, J.M. Ingestion of quercetin inhibits platelet aggregation and essential components of the collagen-stimulated platelet activation pathway in humans. J. Thromb. Haemost. JTH 2004, 2, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Endale, M.; Park, S.-C.; Cho, J.Y.; Rhee, M.H. Dual Roles of Quercetin in Platelets: Phosphoinositide-3-Kinase and MAP Kinases Inhibition, and cAMP-Dependent Vasodilator-Stimulated Phosphoprotein Stimulation. Evid.-Based Complement. Altern. Med. 2012, 2012, 485262. [Google Scholar] [CrossRef]
- Liang, M.-L.; Da, X.-W.; He, A.-D.; Yao, G.-Q.; Xie, W.; Liu, G.; Xiang, J.-Z.; Ming, Z.-Y. Pentamethylquercetin (PMQ) reduces thrombus formation by inhibiting platelet function. Sci. Rep. 2015, 5, 11142. [Google Scholar] [CrossRef]
- Jasuja, R.; Passam, F.H.; Kennedy, D.R.; Kim, S.H.; Van Hessem, L.; Lin, L.; Bowley, S.R.; Joshi, S.S.; Dilks, J.R.; Furie, B.; et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J. Clin. Investig. 2012, 122, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Furie, B.; Zwicker, J.I. Therapeutic implications of protein disulfide isomerase inhibition in thrombotic disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Bassler, N.; Loeffler, C.; Mangin, P.; Yuan, Y.; Schwarz, M.; Hagemeyer, C.E.; Eisenhardt, S.U.; Ahrens, I.; Bode, C.; Jackson, S.; et al. A mechanistic model for paradoxical platelet activation by ligand-mimetic alphaIIb beta3 (GPIIb/IIIa) antagonists. Arterioscler. Thromb. Vasc. Biol. 2007, 27, e9–e15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Vootukuri, S.; Shang, Y.; Negri, A.; Jiang, J.-K.; Nedelman, M.; Diacovo, T.G.; Filizola, M.; Thomas, C.J.; Coller, B.S. RUC-4: A novel αIIbβ3 antagonist for prehospital therapy of myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, J.D.; Wang, X.; Krajewski, S.; Selan, C.; Haller, C.A.; Straub, A.; Chaikof, E.L.; Nandurkar, H.H.; Hagemeyer, C.E.; Peter, K. Delayed targeting of CD39 to activated platelet GPIIb/IIIa via a single-chain antibody: Breaking the link between antithrombotic potency and bleeding? Blood 2013, 121, 3067–3075. [Google Scholar] [CrossRef]
- Stoll, P.; Bassler, N.; Hagemeyer, C.E.; Eisenhardt, S.U.; Chen, Y.C.; Schmidt, R.; Schwarz, M.; Ahrens, I.; Katagiri, Y.; Pannen, B.; et al. Targeting ligand-induced binding sites on GPIIb/IIIa via single-chain antibody allows effective anticoagulation without bleeding time prolongation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Palasubramaniam, J.; Gkanatsas, Y.; Hohmann, J.D.; Westein, E.; Kanojia, R.; Alt, K.; Huang, D.; Jia, F.; Ahrens, I.; et al. Towards effective and safe thrombolysis and thromboprophylaxis: Preclinical testing of a novel antibody-targeted recombinant plasminogen activator directed against activated platelets. Circ. Res. 2014, 114, 1083–1093. [Google Scholar] [CrossRef]
- Shen, B.; Zhao, X.; O’Brien, K.A.; Stojanovic-Terpo, A.; Delaney, M.K.; Kim, K.; Cho, J.; Lam, S.C.-T.; Du, X. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature 2013, 503, 131–135. [Google Scholar] [CrossRef]
- Sinnaeve, P.; Fahrni, G.; Schelfaut, D.; Spirito, A.; Mueller, C.; Frenoux, J.-M.; Hmissi, A.; Bernaud, C.; Ufer, M.; Moccetti, T.; et al. Subcutaneous Selatogrel Inhibits Platelet Aggregation in Patients With Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 75, 2588–2597. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F.; Gurbel, P.A.; Berg, J.T.; Bernaud, C.; Dangas, G.D.; Frenoux, J.-M.; Gorog, D.A.; Hmissi, A.; Kunadian, V.; James, S.K.; et al. Pharmacodynamics, pharmacokinetics, and safety of single-dose subcutaneous administration of selatogrel, a novel P2Y12 receptor antagonist, in patients with chronic coronary syndromes. Eur. Heart J. 2020, 41, 3132–3140. [Google Scholar] [CrossRef]
- Silvain, J.; Zeitouni, M.; Kerneis, M. Selatogrel for Acute Myocardial Infarction: The Promise and Challenges of Self-Medication. J. Am. Coll. Cardiol. 2020, 75, 2598–2601. [Google Scholar] [CrossRef]
- Bach, P.; Antonsson, T.; Bylund, R.; Björkman, J.-A.; Österlund, K.; Giordanetto, F.; van Giezen, J.J.J.; Andersen, S.M.; Zachrisson, H.; Zetterberg, F. Lead optimization of ethyl 6-aminonicotinate acyl sulfonamides as antagonists of the P2Y12 receptor. separation of the antithrombotic effect and bleeding for candidate drug AZD1283. J. Med. Chem. 2013, 56, 7015–7024. [Google Scholar] [CrossRef]
- Delesque-Touchard, N.; Pflieger, A.; Bonnet-Lignon, S.; Millet, L.; Salel, V.; Boldron, C.; Lassalle, G.; Herbert, J.; Savi, P.; Bono, F. SAR216471, an alternative to the use of currently available P2Y12; receptor inhibitors? Thromb. Res. 2014, 134, 693–703. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Lai, A.; Qiao, J.X.; Wang, T.C.; Hua, J.; Price, L.A.; Shen, H.; Chen, X.-Q.; Wong, P.; et al. Discovery of 4-aryl-7-hydroxyindoline-based P2Y1 antagonists as novel antiplatelet agents. J. Med. Chem. 2014, 57, 6150–6164. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Watson, C.; Crain, E.J. The P2Y1 receptor antagonist MRS2500 prevents carotid artery thrombosis in cynomolgus monkeys. J. Thromb. Thrombolysis 2016, 41, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Gremmel, T.; Michelson, A.D.; Frelinger, A.L.; Bhatt, D.L. Novel aspects of antiplatelet therapy in cardiovascular disease. Res. Pract. Thromb. Haemost. 2018, 2, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Gremmel, T.; Yanachkov, I.; Yanachkova, M.I.; Wright, G.E.; Wider, J.; Undyala, V.V.; Michelson, A.D.; FrelingerIII, A.L.; Przyklenk, K. Synergistic Inhibition of Both P2Y1 and P2Y12 Adenosine Diphosphate Receptors As Novel Approach to Rapidly Attenuate Platelet-Mediated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Adili, R.; Tourdot, B.E.; Mast, K.; Yeung, J.; Freedman, J.C.; Green, A.; Luci, D.K.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; et al. First Selective 12-LOX Inhibitor, ML355, Impairs Thrombus Formation and Vessel Occlusion In Vivo With Minimal Effects on Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1828–1839. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Drug Target | Route of Administration | Elimination Half-Life | Onset of Action after Loading Dose | Time to Platelet Function Recovery after Drug Discontinuation | Common Clinical Indication |
|---|---|---|---|---|---|---|
| Aspirin | Cyclooxygenase-1 | oral * | 15–20 min | ~20 min ¶ | 5–7 days | ACS, CAD, PAD, stroke, TIA |
| Clopidogrel | P2Y12 | oral | 30 min # | 2–6 h | 7 days | ACS, CAD, stroke, TIA |
| Prasugrel | P2Y12 | oral | 30–60 min # | 30 min | 7–10 days | ACS |
| Ticagrelor | P2Y12 | oral | 7–9 h | 30 min | 3–5 days | ACS |
| Cangrelor | P2Y12 | IV | 3–6 min | ≤5 min | 30–60 min | ACS |
| Vorapaxar | PAR1 | oral | 5–13 days | - | 4–8 weeks | PAD |
| Dipyridamole | PDE3/5 | oral | 10 h | - | - | Stroke, TIA |
| Cilostazol | PDE3A | oral | 11–13 h | - | 12–16 h | PAD |
| Iloprost | PGI2 analogue | IV | 30 min | - | - | PAD |
| Eptifibatide | GPIIbIIIa | IV | 2.5 h | ≤15 min | 4–8 h | ACS |
| Tirofiban | GPIIbIIIa | IV | 2 h | 20–40 min | 4–8 h | ACS |
| Name | Company | Type | Route of Administration | Target | Completed Clinical Trial | Reference |
|---|---|---|---|---|---|---|
| PZ-128 | Tufts Medical Center | Pepducin | IV | PAR1 | Phase I | [119] |
| BMS-986120 | Bristol-Myers Squibb | Small molecule | oral | PAR4 | Phase I | [120] |
| BMS-986141 | Bristol-Myers Squibb | Small molecule | oral | PAR4 | Phase I | _ |
| Phase II | _ | |||||
| Revacept | Advance Cor | Fusion protein | IV | GPVI ligand | Phase I | [121] |
| Phase II | _ | |||||
| ACT017 | Acticor Biotech | Antibody | IV | GPVI | Phase I | [122] |
| ARC1779 | Archemix | DNA aptamer | IV | VWF | Phase I | [123] |
| Phase II | [124] | |||||
| AZD6482 | AstraZeneca | Small molecule | IV | PI3Kβ | Phase I | [125] |
| Phase I | [126] | |||||
| Isoquercetin | Beth Israel | Small molecule | oral | PDI | Phase I | [127] |
| NHLBI | Small molecule | oral | PDI | Phase II/III | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jourdi, G.; Lordkipanidzé, M.; Philippe, A.; Bachelot-Loza, C.; Gaussem, P. Current and Novel Antiplatelet Therapies for the Treatment of Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 13079. https://doi.org/10.3390/ijms222313079
Jourdi G, Lordkipanidzé M, Philippe A, Bachelot-Loza C, Gaussem P. Current and Novel Antiplatelet Therapies for the Treatment of Cardiovascular Diseases. International Journal of Molecular Sciences. 2021; 22(23):13079. https://doi.org/10.3390/ijms222313079
Chicago/Turabian StyleJourdi, Georges, Marie Lordkipanidzé, Aurélien Philippe, Christilla Bachelot-Loza, and Pascale Gaussem. 2021. "Current and Novel Antiplatelet Therapies for the Treatment of Cardiovascular Diseases" International Journal of Molecular Sciences 22, no. 23: 13079. https://doi.org/10.3390/ijms222313079
APA StyleJourdi, G., Lordkipanidzé, M., Philippe, A., Bachelot-Loza, C., & Gaussem, P. (2021). Current and Novel Antiplatelet Therapies for the Treatment of Cardiovascular Diseases. International Journal of Molecular Sciences, 22(23), 13079. https://doi.org/10.3390/ijms222313079