
Integration of Mutational Signature Analysis with 3D Chromatin Data Unveils Differential AID-Related Mutagenesis in Indolent Lymphomas

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Characteristics and Sample Acquisition
2.1.1. Library Preparation and Sequencing
2.1.2. Sequence Alignment and Variant Calling
2.1.3. Mutational Signatures Analysis
- 1.
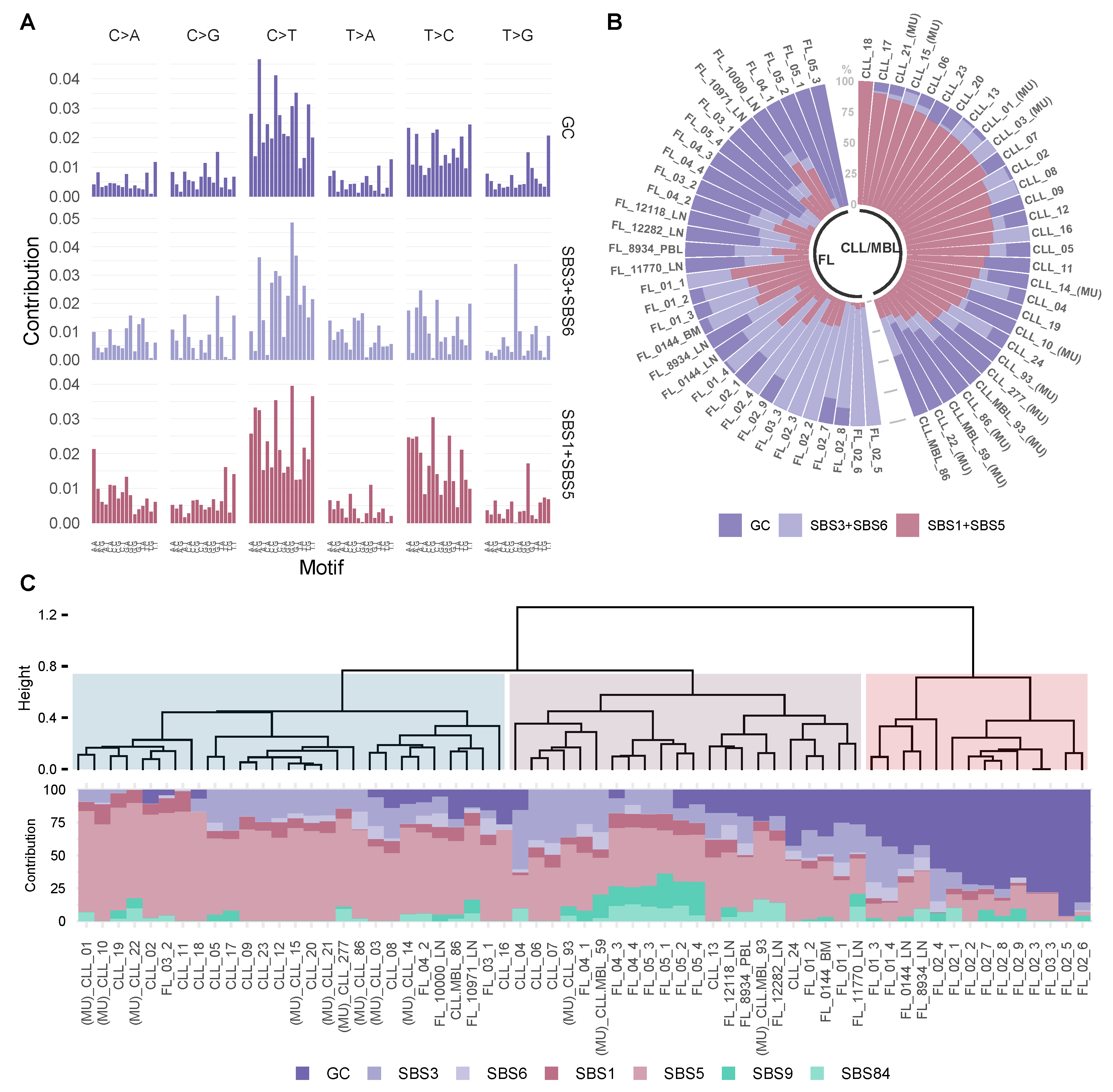
- Variant classification in 96 contexts: Every single nucleotide variant (SNV) was represented for example, as C>A, C>T (by convention beginning with the pyrimidine) within a trinucleotide context (e.g., “GCT>GAT”). This approach yields 96 different possible contexts (six substitution type C>A, C>G, C>T, T>A, T>G, T>C with four possible bases immediately 5’ and 3’ to each substitution). This classification annotation was performed for mutations within localized regions (IG loci) as well as for genome-wide mutations (WES) (Figure 1).
- 2.
- Signature extraction, similarity and fitting: Global and localized mutational signatures were defined by a workflow encompassing a three-step procedure, starting with a de novo signature extraction, followed by an similarity analysis, to allow a final fitting approach. de novo mutational signature extraction was generated by a non-negative matrix factorization (NMF) using R-package SigProfiler [33]. One of the practical drawbacks of the multiplicative NMF algorithm is that the task of selecting the appropriate number of sources is left to the user. Using this tool automatically allows the identification of the optimal number of operative signatures in our dataset (Figure 1, Supplementary Figure S1).
2.1.4. Unsupervised Clustering Analysis
2.1.5. Hi-C Data Analysis and Compartment Identification
3. Results
3.1. Variant Allele Densities and Frequencies in FL and CLL
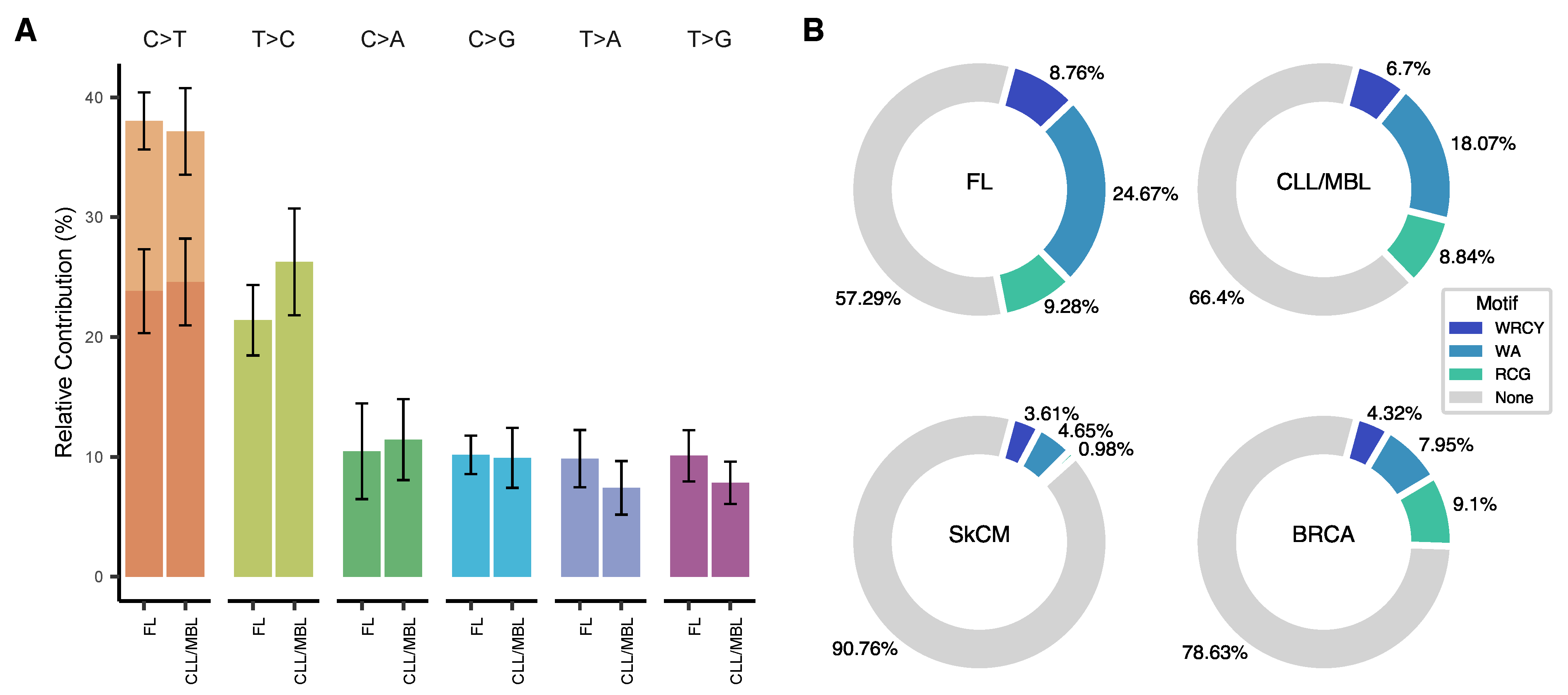
3.2. Somatic Mutations in CLL/MBL and FL Are Frequently Associated with a Deamination Pattern in AID Motifs
3.3. Trinucleotide Context of Somatic Mutations in FL and CLL/MBL
3.4. Inferring the Role of AID as an Underlying Mutagenic Mechanism in FL and CLL/MBL
3.5. FL and CLL Show Differential Distribution of Mutational Signatures across Tridimensional (3D) DNA Compartments
3.6. Analysis of Mutations in Genes Involved in DNA Repair
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Pfeifer, G.P. Patterns of p53 G–>T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis 2001, 22, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Petersen-Mahrt, S.K.; Harris, R.S.; Neuberger, M.S. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 2002, 418, 99–103. [Google Scholar] [CrossRef]
- Di Noia, J.; Neuberger, M.S. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 2002, 419, 43–48. [Google Scholar] [CrossRef]
- Winter, D.B.; Gearhart, P.J. Altered spectra of hypermutation in DNA repair-deficient mice. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2001, 356, 5–11. [Google Scholar] [CrossRef][Green Version]
- Rogozin, I.B.; Kolchanov, N.A. Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighbouring base sequences on mutagenesis. Biochim. Biophys. Acta 1992, 1171, 11–18. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Pavlov, Y.I.; Bebenek, K.; Matsuda, T.; Kunkel, T.A. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat. Immunol. 2001, 2, 530–536. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Lada, A.G.; Goncearenco, A.; Green, M.R.; De, S.; Nudelman, G.; Panchenko, A.R.; Koonin, E.V.; Pavlov, Y.I. Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci. Rep. 2016, 6, 38133. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, Q.; Dose, M.; Pruett, N.; Kieffer-Kwon, K.R.; Resch, W.; Liang, G.; Tang, Z.; Mathe, E.; Benner, C.; et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 2014, 159, 1524–1537. [Google Scholar] [CrossRef]
- Alvarez-Prado, A.F.; Perez-Duran, P.; Perez-Garcia, A.; Benguria, A.; Torroja, C.; de Yebenes, V.G.; Ramiro, A.R. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J. Exp. Med. 2018, 215, 761–771. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Scherer, F.; van der Burgt, M.; Kielbasa, S.M.; Bertinetti-Lapatki, C.; Duhren von Minden, M.; Mikesch, K.; Zirlik, K.; de Wreede, L.; Veelken, H.; Navarrete, M.A. Selection patterns of B-cell receptors and the natural history of follicular lymphoma. Br. J. Haematol. 2016, 175, 972–975. [Google Scholar] [CrossRef] [PubMed]
- Scherer, F.; Navarrete, M.A.; Bertinetti-Lapatki, C.; Boehm, J.; Schmitt-Graeff, A.; Veelken, H. Isotype-switched follicular lymphoma displays dissociation between activation-induced cytidine deaminase expression and somatic hypermutation. Leuk. Lymphoma 2016, 57, 151–160. [Google Scholar] [CrossRef]
- Oppezzo, P.; Vuillier, F.; Vasconcelos, Y.; Dumas, G.; Magnac, C.; Payelle-Brogard, B.; Pritsch, O.; Dighiero, G. Chronic lymphocytic leukemia B cells expressing AID display dissociation between class switch recombination and somatic hypermutation. Blood 2003, 101, 4029–4032. [Google Scholar] [CrossRef][Green Version]
- Burns, A.; Alsolami, R.; Becq, J.; Stamatopoulos, B.; Timbs, A.; Bruce, D.; Robbe, P.; Vavoulis, D.; Clifford, R.; Cabes, M.; et al. Whole-genome sequencing of chronic lymphocytic leukaemia reveals distinct differences in the mutational landscape between IgHV(mut) and IgHV(unmut) subgroups. Leukemia 2018, 32, 332–342. [Google Scholar] [CrossRef]
- Lappalainen, I.; Almeida-King, J.; Kumanduri, V.; Senf, A.; Spalding, J.D.; Ur-Rehman, S.; Saunders, G.; Kandasamy, J.; Caccamo, M.; Leinonen, R.; et al. The European Genome-phenome Archive of human data consented for biomedical research. Nat. Genet. 2015, 47, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H.; et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat. Genet. 2016, 48, 183–188. [Google Scholar] [CrossRef]
- Damm, F.; Mylonas, E.; Cosson, A.; Yoshida, K.; Della Valle, V.; Mouly, E.; Diop, M.; Scourzic, L.; Shiraishi, Y.; Chiba, K.; et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014, 4, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010, 38, e164. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Wingett, S. FastQC. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 November 2021).
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A tool for visualizing and exploring patterns of small mutational events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Lawson, R.G.; Jurs, P.C. New Index for Clustering Tendency and Its Application to Chemical Problems. J. Chem. Inf. Comput. Sci. 1990, 30, 36–41. [Google Scholar] [CrossRef]
- Kaufman, L.; Rousseeuw, P. Finding Groups in Data: An Introduction to Cluster Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: AnRPackage for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Xiong, K.; Ma, J. Revealing Hi-C subcompartments by imputing inter-chromosomal chromatin interactions. Nat. Commun. 2019, 10, 5069. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Layer, R.M.; Quinlan, A.R. Vcfanno: Fast, flexible annotation of genetic variants. Genome Biol. 2016, 17, 1–9. [Google Scholar] [CrossRef]
- Methot, S.P.; Di Noia, J.M. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv. Immunol. 2017, 133, 37–87. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.; Verhoeven, A.J.; Weeks, L.; Kastelein, J.J.; Halley, D.J.; van den Ouweland, A.; Jukema, J.W.; Seidell, J.C.; Birkenhager, J.C. Common C-to-T substitution at position -480 of the hepatic lipase promoter associated with a lowered lipase activity in coronary artery disease patients. Arter. Thromb. Vasc. Biol. 1997, 17, 2837–2842. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef]
- Das, R.; Ghosh, S.K. Genetic variants of the DNA repair genes from Exome Aggregation Consortium (EXAC) database: Significance in cancer. DNA Repair 2017, 52, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Nakano, M.; Sato, R.; Adachi, H.; Kiyota, M.; Kawata, E.; Uoshima, N.; Yasukawa, S.; Chinen, Y.; Mizutani, S.; et al. High-risk follicular lymphomas harbour more somatic mutations including those in the AID-motif. Sci. Rep. 2017, 7, 14039. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Pao, W.; Zhao, Z. Patterns and processes of somatic mutations in nine major cancers. BMC Med. Genom. 2014, 7, 11. [Google Scholar] [CrossRef]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef]
- Kasar, S.; Kim, J.; Improgo, R.; Tiao, G.; Polak, P.; Haradhvala, N.; Lawrence, M.S.; Kiezun, A.; Fernandes, S.M.; Bahl, S.; et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 2015, 6, 8866. [Google Scholar] [CrossRef]
- Meng, F.L.; Du, Z.; Federation, A.; Hu, J.; Wang, Q.; Kieffer-Kwon, K.R.; Meyers, R.M.; Amor, C.; Wasserman, C.R.; Neuberg, D.; et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 2014, 159, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Guglielmino, R.; Houldsworth, J.; Mohr, J.; Aoufouchi, S.; Polakiewicz, R.; Chaganti, R.S.; Dalla-Favera, R. Expression of the AID protein in normal and neoplastic B cells. Blood 2004, 104, 3318–3325. [Google Scholar] [CrossRef]
- Nagai, L.A.E.; Park, S.J.; Nakai, K. Analyzing the 3D chromatin organization coordinating with gene expression regulation in B-cell lymphoma. BMC Med. Genom. 2019, 11, 127. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Bluteau, D.; Lespinasse, J.; Pujol, R.; Vasquez, N.; d’Enghien, C.D.; Stoppa-Lyonnet, D.; Leblanc, T.; Soulier, J.; Surralles, J. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med. 2018, 20, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Bret, C.; Klein, B.; Cartron, G.; Schved, J.F.; Constantinou, A.; Pasero, P.; Moreaux, J. DNA repair in diffuse large B-cell lymphoma: A molecular portrait. Br. J. Haematol. 2015, 169, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Leeksma, O.C.; de Miranda, N.F.; Veelken, H. Germline mutations predisposing to diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e532. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sepulveda-Yanez, J.H.; Alvarez-Saravia, D.; Fernandez-Goycoolea, J.; Aldridge, J.; van Bergen, C.A.M.; Posthuma, W.; Uribe-Paredes, R.; Veelken, H.; Navarrete, M.A. Integration of Mutational Signature Analysis with 3D Chromatin Data Unveils Differential AID-Related Mutagenesis in Indolent Lymphomas. Int. J. Mol. Sci. 2021, 22, 13015. https://doi.org/10.3390/ijms222313015
Sepulveda-Yanez JH, Alvarez-Saravia D, Fernandez-Goycoolea J, Aldridge J, van Bergen CAM, Posthuma W, Uribe-Paredes R, Veelken H, Navarrete MA. Integration of Mutational Signature Analysis with 3D Chromatin Data Unveils Differential AID-Related Mutagenesis in Indolent Lymphomas. International Journal of Molecular Sciences. 2021; 22(23):13015. https://doi.org/10.3390/ijms222313015
Chicago/Turabian StyleSepulveda-Yanez, Julieta H., Diego Alvarez-Saravia, Jose Fernandez-Goycoolea, Jacqueline Aldridge, Cornelis A. M. van Bergen, Ward Posthuma, Roberto Uribe-Paredes, Hendrik Veelken, and Marcelo A. Navarrete. 2021. "Integration of Mutational Signature Analysis with 3D Chromatin Data Unveils Differential AID-Related Mutagenesis in Indolent Lymphomas" International Journal of Molecular Sciences 22, no. 23: 13015. https://doi.org/10.3390/ijms222313015
APA StyleSepulveda-Yanez, J. H., Alvarez-Saravia, D., Fernandez-Goycoolea, J., Aldridge, J., van Bergen, C. A. M., Posthuma, W., Uribe-Paredes, R., Veelken, H., & Navarrete, M. A. (2021). Integration of Mutational Signature Analysis with 3D Chromatin Data Unveils Differential AID-Related Mutagenesis in Indolent Lymphomas. International Journal of Molecular Sciences, 22(23), 13015. https://doi.org/10.3390/ijms222313015