
Phosphate Metabolism and Pathophysiology in Parathyroid Disorders and Endocrine Tumors

 ,
, 
Abstract
1. Introduction
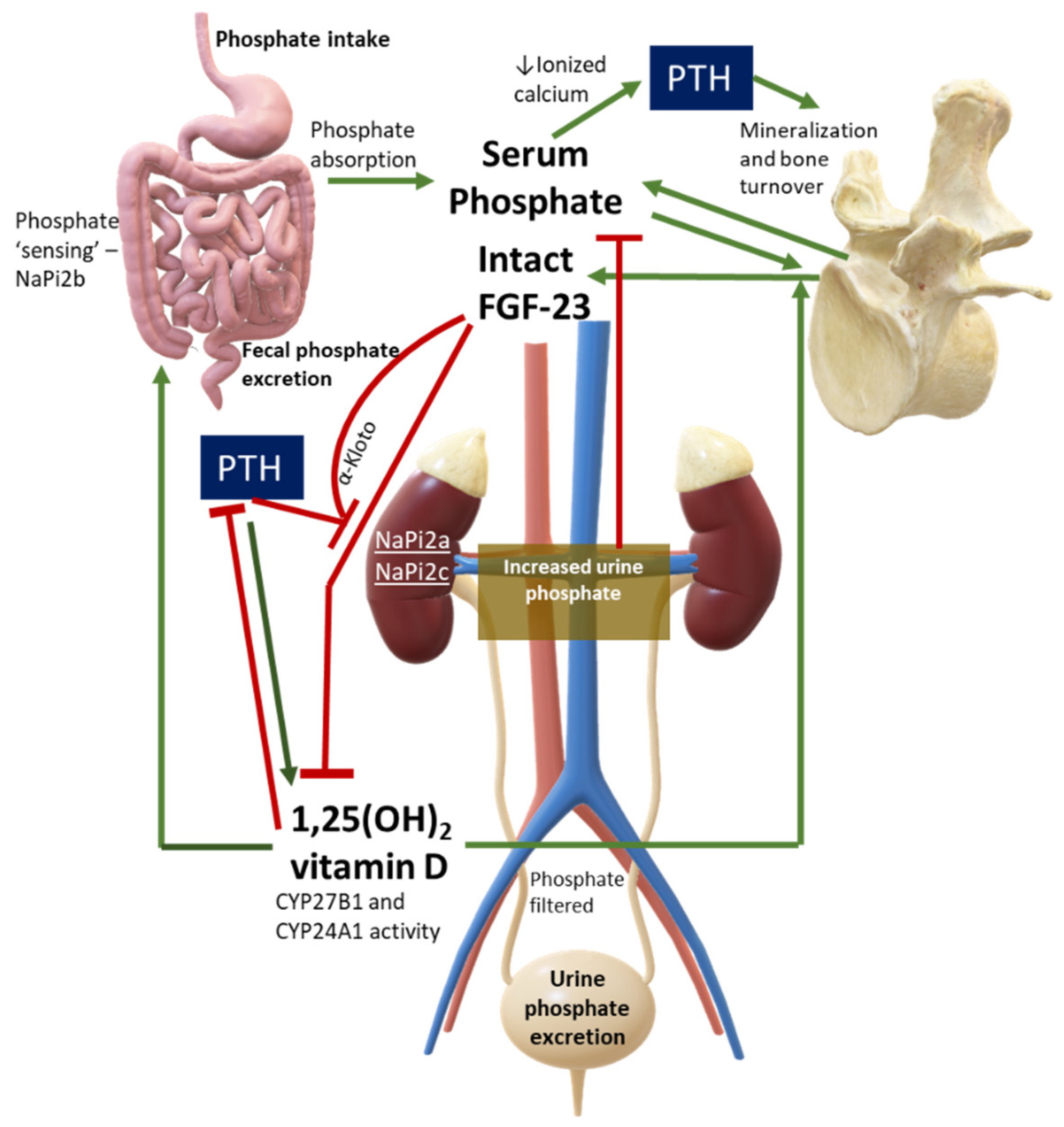
2. Overview of Phosphate Physiology and Pathophysiology
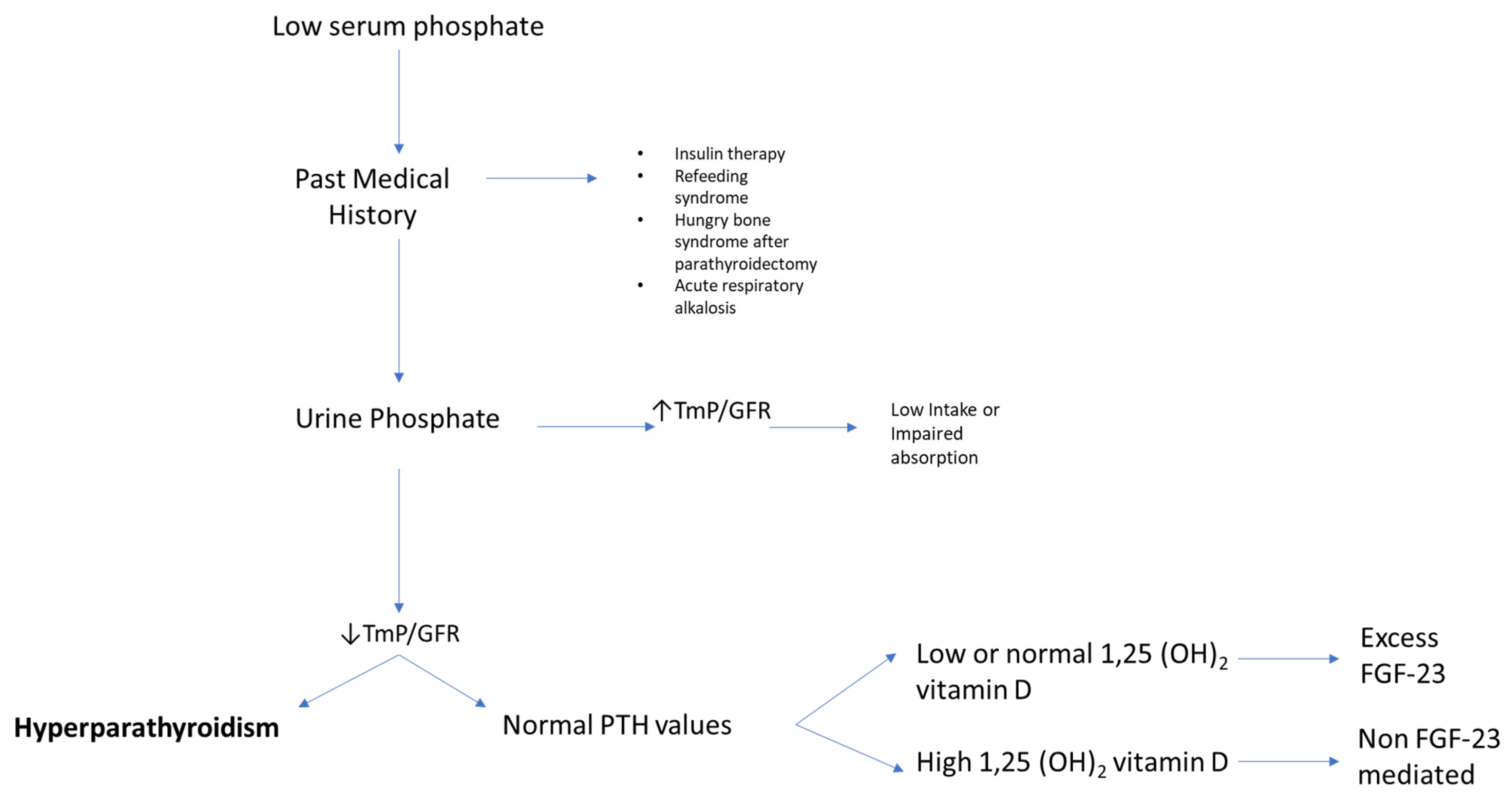
3. Why Measure Serum Phosphate?
4. Phosphate in Primary Hyperparathyroidism
5. Phosphate in Hypoparathyroidism
6. Phosphate in Paraneoplastic Endocrine Tumors
6.1. Tumor-Induced Osteomalacia
6.1.1. Diagnosis
6.1.2. Clinical Presentation
6.2. Other Solid Tumors and Phosphate
7. Conclusions
- In primary hyperparathyroidism, decreased serum phosphate might be associated with a more severe disease. Patients presenting with moderate hypophosphatemia could benefit from a surgical approach, once other underlying causes of hypophosphatemia have been addressed and excluded;
- In patients with hypoparathyroidism, serum phosphate is associated with soft tissue calcification, although evidence of this is still scarce, suggesting that both calcium and phosphate derangements contribute to this complication. Newer treatments based on PTH analogues used as adjunctive therapies are able to quickly restore normal serum phosphate;
- FGF-23 or other phosphatonin-producing tumors cause a rare paraneoplastic syndrome, TIO, which might easily go unrecognized for a long time despite severe, debilitating symptoms. Awareness of this disorder should be enhanced across the medical community to allow prompt endocrinological referral in case of acquired and unexplained hypophosphatemia.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef]
- Levi, M.; Gratton, E.; Forster, I.C.; Hernando, N.; Wagner, C.A.; Biber, J.; Sorribas, V.; Murer, H. Mechanisms of phosphate transport. Nat. Rev. Nephrol. 2019, 15, 482–500. [Google Scholar] [CrossRef]
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706. [Google Scholar] [CrossRef]
- Clarke, B.L. Phosphorus Disorders: Hypophosphatemic Rickets. In Book Metabolic Bone Diseases, A Case-Baesd Approach, 1st ed.; Camacho, P.M., Ed.; Springer Nature Switzerland: Cham, Switzerland, 2019; Volume 3, pp. 83–98. [Google Scholar]
- Brunelli, S.M.; Goldfarb, S. Hypophosphatemia: Clinical consequences and management. J. Am. Soc. Nephrol. 2007, 18, 1999–2003. [Google Scholar] [CrossRef] [PubMed]
- Halevy, J.; Bulvik, S. Severe hypophosphatemia in hospitalized patients. Arch. Intern. Med. 1988, 148, 153–155. [Google Scholar] [CrossRef]
- Larsson, L.; Rebel, K.; Sörbo, B. Severe hypophosphatemia—A hospital survey. Acta Med. Scand. 1983, 214, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Imel, E.A.; Econs, M.J. Approach to the hypophosphatemic patient. J. Clin. Endocrinol. Metab. 2012, 97, 696–706. [Google Scholar] [CrossRef]
- Volk, C.; Schmidt, B.; Brandsch, C.; Kurze, T.; Schlegelmilch, U.; Grosse, I.; Ulrich, C.; Girndt, M.; Stangl, G.I. Acute effects of an inorganic phosphorus additive on mineral metabolism and cardiometabolic risk factors in healthy subjects [published online ahead of print, 27 August 2021]. J. Clin. Endocrinol. Metab. 2021. [Google Scholar] [CrossRef]
- Tenenhouse, H.S. Cellular and molecular mechanisms of renal phosphate transport. J. Bone Miner. Res. 1997, 12, 159–164. [Google Scholar] [CrossRef]
- Silverberg, S.J.; Shane, E.; Clemens, T.L.; Dempster, D.W.; Segre, G.V.; Lindsay, R.; Bilezikian, J.P. The effect of oral phosphate administration on major indices of skeletal metabolism in normal subjects. J. Bone Miner. Res. 1986, 1, 383–388. [Google Scholar] [CrossRef]
- Madeo, B.; Kara, E.; Cioni, K.; Vezzani, S.; Trenti, T.; Santi, D.; Simoni, M.; Rochira, V. Serum Calcium to Phosphorous (Ca/P) Ratio Is a Simple, Inexpensive, and Accurate Tool in the Diagnosis of Primary Hyperparathyroidism. JBMR Plus 2018, 2, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Madeo, B.; De Vincentis, S.; Repaci, A.; Altieri, P.; Vicennati, V.; Kara, E.; Vescini, F.; Amadori, P.; Balestrieri, A.; Pagotto, U.; et al. The calcium-to-phosphorous (Ca/P) ratio in the diagnosis of primary hyperparathyroidism and hypoparathyroidism: A multicentric study. Endocrine 2020, 68, 679–687. [Google Scholar] [CrossRef]
- Silverberg, S.J.; Bilezikian, J.P. Asymptomatic Primary Hyperparathyroidism. In The Parathyroids, Basic and Clinical Concepts, 3rd ed.; Bilezikian, J.P., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Section II; pp. 317–330. [Google Scholar]
- Palermo, A.; Naciu, A.M.; Tabacco, G.; Falcone, S.; Santonati, A.; Maggi, D.; D’Onofrio, L.; Briganti, S.I.; Castellitto, D.; Casini, A.; et al. Clinical, Biochemical, and Radiological Profile of Normocalcemic Primary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2020, 105, dgaa174. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Q.; Lu, C.; Fan, P.; Li, J.; Luo, X.; Chen, D. New parathyroid function index for the differentiation of primary and secondary hyperparathyroidism: A case-control study. BMC Endocr. Disord. 2020, 20, 5. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; King, D.; Small, M.; Gibson, C.; Gardner, R.; Stack, B.C., Jr. The Utility of the Cl:PO4 Ratio in Patients with Variant Versions of Primary Hyperparathyroidism. Otolaryngol. Head Neck Surg. 2021, 164, 308–314. [Google Scholar] [CrossRef]
- Hassani, S.; Afkhamizadeh, M.; Teimouri, A.; Najaf Najafi, M.; Vazifeh Mostaan, L.; Mohebbi, M. Evaluation of Serum Level of FGF23 and 1,25(OH)2D3 in Primary Hyperparathyroidism Patients Before and After Parathyroidectomy. Int. J. Gen. Med. 2020, 13, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Sykała, M.; Szumowski, P.; Mojsak, M.; Abdelrazek, S.; Żukowski, Ł.; Lipińska, D.; Juchnicka, I.; Kozłowska, G.; Szelachowska, M.; Krętowski, A.; et al. Assessment of Clinical Utility of Assaying FGF-23, Klotho Protein, Osteocalcin, NTX, and Sclerostin in Patients with Primary Hyperparathyroidism. J. Clin. Med. 2021, 10, 3089. [Google Scholar] [CrossRef]
- Wermers, R.A.; Khosla, S.; Atkinson, E.J.; Achenbach, S.J.; Oberg, A.L.; Grant, C.S.; Melton, L.J., III. Incidence of primary hyperparathyroidism in Rochester, Minnesota, 1993–2001: An update on the changing epidemiology of the disease. J. Bone Miner. Res. 2006, 21, 171–177. [Google Scholar] [CrossRef]
- Lim, V.; Clarke, B.L. Coexisting primary hyperparathyroidism and sarcoidosis cause increased angiotensin-converting enzyme and decreased parathyroid hormone and phosphate levels. J. Clin. Endocrinol. Metab. 2013, 98, 1939–1945. [Google Scholar] [CrossRef][Green Version]
- Castellano, E.; Attanasio, R.; Boriano, A.; Pellegrino, M.; Borretta, G. Serum Phosphate: A Neglected Test In The Clinical Management Of Primary Hyperparathyroidism [published online ahead of print, 14 September 2021]. J. Clin. Endocrinol. Metab. 2021, dgab676. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Bilezikian, J.; Bone, H.; Gurevich, A.; Lakatos, P.; Misiorowski, W.; Rozhinskaya, L.; Trotman, M.L.; Tóth, M. Cinacalcet normalizes serum calcium in a double-blind randomized, placebo-controlled study in patients with primary hyperparathyroidism with contraindications to surgery. Eur. J. Endocrinol. 2015, 172, 527–535. [Google Scholar] [CrossRef]
- Vestergaard, P. Current pharmacological options for the management of primary hyperparathyroidism. Drugs 2006, 66, 2189–2211. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Cianferotti, L.; Brandi, M.L. Clinical presentation and management of hypoparathyroidism. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 927–939. [Google Scholar] [CrossRef]
- Clarke, B.L.; Vokes, T.J.; Bilezikian, J.P.; Shoback, D.M.; Lagast, H.; Mannstadt, M. Effects of parathyroid hormone rhPTH(1-84) on phosphate homeostasis and vitamin D metabolism in hypoparathyroidism: REPLACE phase 3 study. Endocrine 2017, 55, 273–282. [Google Scholar] [CrossRef]
- Zavatta, G.; Clarke, B.L. Challenges in the management of chronic hypoparathyroidism. Endocr. Connect. 2020, 9, R229–R240. [Google Scholar] [CrossRef]
- Khan, A.A.; Rejnmark, L.; Rubin, M.; Schwarz, P.; Vokes, T.; Clarke, B.; Ahmed, I.; Hofbauer, L.; Marcocci, C.; Pagotto, U.; et al. PaTH Forward: A randomized, double-blind, placebo-controlled phase 2 trial of TransCon PTH in adult hypoparathyroidism [published online ahead of print, 4 August 2021]. J. Clin. Endocrinol. Metab. 2021, dgab577. [Google Scholar] [CrossRef]
- Gupta, A.; Winer, K.; Econs, M.J.; Marx, S.J.; Collins, M.T. FGF-23 is elevated by chronic hyperphosphatemia. J. Clin. Endocrinol. Metab. 2004, 89, 4489–4492. [Google Scholar] [CrossRef] [PubMed]
- Ovejero, D.; Hartley, I.R.; de Castro Diaz, L.F.; Theng, E.; Li, X.; Gafni, R.I.; Collins, M.T. PTH and FGF23 Exert Interdependent Effects on Renal Phosphate Handling: Evidence From Patients With Hypoparathyroidism and Hyperphosphatemic Familial Tumoral Calcinosis Treated with Synthetic Human PTH 1-34. J. Bone Miner. Res. 2021. (ePub ahead of print). [Google Scholar] [CrossRef]
- Bilezikian, J.P. Hypoparathyroidism. J. Clin. Endocrinol. Metab. 2020, 105, 1722–1736. [Google Scholar] [CrossRef]
- Zavatta, G.; Tebben, P.J.; McCollough, C.H.; Yu, L.; Vrieze, T.; Clarke, B.L. Basal Ganglia Calcification Is Associated With Local and Systemic Metabolic Mechanisms in Adult Hypoparathyroidism. J. Clin. Endocrinol. Metab. 2021, 106, 1900–1917. [Google Scholar] [CrossRef]
- Ridder, L.O.; Harsløf, T.; Sikjaer, T.; Underbjerg, L.; Rejnmark, L. Determinants of hypercalciuria and renal calcifications in chronic hypoparathyroidism: A cross-sectional study. Clin. Endocrinol. (Oxf.) 2021, 95, 286–294. [Google Scholar] [CrossRef]
- O’Neill, W.C. The fallacy of the calcium-phosphorus product. Kidney Int. 2007, 72, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Shimada, B.K.; Pomozi, V.; Zoll, J.; Kuo, S.; Martin, L.; Le Saux, O. ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions. Int. J. Mol. Sci. 2021, 22, 4555. [Google Scholar] [CrossRef]
- Arnold, A.; Dennison, E.; Kovacs, C.S.; Mannstadt, M.; Rizzoli, R.; Brandi, M.L.; Clarke, B.; Thakker, R.V. Hormonal regulation of biomineralization. Nat. Rev. Endocrinol. 2021, 17, 261–275. [Google Scholar] [CrossRef]
- McCance, R.A. Osteomalacia with Looser’s nodes (Milkman’s syndrome) due to a raised resistance to vitamin D acquired about the age of 15 years. Q. J. Med. 1947, 16, 33–46. [Google Scholar] [PubMed]
- Brandi, M.L.; Clunie, G.; Houillier, P.; Jan de Beur, S.M.; Minisola, S.; Oheim, R.; Seefried, L. Challenges in the management of tumor-induced osteomalacia (TIO). Bone 2021, 152, 116064. [Google Scholar] [CrossRef]
- Boland, J.M.; Tebben, P.J.; Folpe, A.L. Phosphaturic mesenchymal tumors: What an endocrinologist should know. J. Endocrinol. Investig. 2018, 41, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.B.; Zahradnik, R.; Larsson, T.; White, K.E.; Sugimoto, T.; Imanishi, Y.; Yamamoto, T.; Hampson, G.; Koshiyama, H.; Ljunggren, Ö.; et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N. Engl. J. Med. 2003, 348, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- De Beur, S.M.J. Tumor-induced osteomalacia. JAMA 2005, 294, 1260–1267. [Google Scholar] [CrossRef]
- De Beur, S.M.J.; Streeten, E.A.; Civelek, A.C.; McCarthy, E.F.; Uribe, L.; Marx, S.J.; Onobrakpeya, O.; Raisz, L.G.; Watts, N.B.; Sharon, M.; et al. Localisation of mesenchymal tumours by somatostatin receptor imaging. Lancet 2002, 359, 761–763. [Google Scholar] [CrossRef]
- Salim, M.; Behairy, M.S.; Barengolts, E. TIO Associated with Hyperparathyroidism: A Rarity, a Rule, or a Novel HPT-PMT Syndrome-A Case Study with Literature Review. Case Rep. Endocrinol. 2021, 2021, 5172131. [Google Scholar] [CrossRef]
- Geller, J.L.; Khosravi, A.; Kelly, M.H.; Riminucci, M.; Adams, J.S.; Collins, M.T. Cinacalcet in the management of tumor-induced osteomalacia. J. Bone Miner. Res. 2007, 22, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, A.L.; Chaumet-Riffaud, P.; Blanchard, A.; Dupeux, M.; Rothenbuhler, A.; Lambert, B.; Durand, E.; Boros, E.; Briot, K.; Silve, C.; et al. Hyperparathyroidism in Patients with X-Linked Hypophosphatemia. J. Bone Miner. Res. 2020, 35, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, Y.; Ito, N.; Rhee, Y.; Takeuchi, Y.; Shin, C.S.; Takahashi, Y.; Onuma, H.; Kojima, M.; Kanematsu, M.; Kanda, H.; et al. Interim Analysis of a Phase 2 Open-Label Trial Assessing Burosumab Efficacy and Safety in Patients with Tumor-Induced Osteomalacia. J. Bone Miner. Res. 2021, 36, 262–270. [Google Scholar] [CrossRef] [PubMed]
- De Beur, S.M.J.; Miller, P.D.; Weber, T.J.; Peacock, M.; Insogna, K.; Kumar, R.; Rauch, F.; Luca, D.; Cimms, T.; Roberts, M.S.; et al. Burosumab for the Treatment of Tumor-Induced Osteomalacia. J. Bone Miner. Res. 2021, 36, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Chukir, T.; Liu, Y.; Hoffman, K.; Bilezikian, J.P.; Farooki, A. Calcitriol Elevation Is Associated with a Higher Risk of Refractory Hypercalcemia of Malignancy in Solid Tumors. J. Clin. Endocrinol. Metab. 2020, 105, e1115–e1123. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Etiology | Pathogenesis | |
|---|---|---|---|
| Hormone Excess | |||
| FGF-23 | |||
| Tumor-Induced Osteomalacia, TIO | Mesenchimal tumor | Paraneoplastic | |
| X-linked hypophosphatemia, XLH | PHEX mutation | Inappropriately high production of FGF-23 | |
| Autosomal dominant hypophosphatemic rickets, ADHR | FGF-23 mutation | The mutation makes FGF-23 resistant to cleavage | |
| Autosomal recessive hypophosphatemic rickets type1, ARHR1 | Loss of DMP1 | Impaired osteocyte differentiation | |
| Autosomal recessive hypophosphatemic rickets type2, ARHR2 | ENPP1 mutation | Increased FGF-23 production | |
| Fibrous displasia | GNAS mutation | Dysplastic bone produces excess FGF-23 | |
| Linear nevus sebaceus syndrome | Excess FGF-23 prodution | Dysplastic bone or nevi produce excess FGF-23 | |
| Osteoglophonica dysplasia | FGFR1 mutation | Dysplastic bone produces excess FGF-23 | |
| sFRP4 | TIO | Mesenchimal tumor | Paraneoplastic |
| MEPE | TIO | Mesenchimal tumor | Paraneoplastic |
| FGF-7 | TIO | Mesenchimal tumor | Paraneoplastic |
| Transporter mutation | |||
| Nephrolithiasis/Osteoporosis, Hypophosphatemic, 1 (NPHLOP1) | Heterozygous mutation in the SLC34A1 gene | Renal phosphate wasting, loss-of-function of NaPi-IIa. | |
| Nephrolithiasis/Osteoporosis, Hypophosphatemic, 2 (NPHLOP2) | Heterozygous mutation in the SLC9A3R1 gene | NHERF1 1 mutation and higher responsiveness to PTH, through cAMP production | |
| Fanconi Renotubular Syndrome 2 (FRTS2) | Homozygous mutation in the SLC34A1 gene | Loss-of-function of NaPiIIa. Hypercalciuria due to increased serum 1,25-dihydroxyvitamin D levels and increased intestinal calcium absorption | |
| Hypophosphatemic Rickets and Hyperparathyroidism | Alpha-Klotho translocation | Increased Klotho and FGF-23 | |
| Hypophosphatemic Rickets with Hypercalciuria (HHRH) | Homozygous or compound heterozygous mutation in the sodium-phosphate cotransporter gene SLC34A3 | Loss of function of NaPiIIc Hypercalciuria due to increased serum 1,25-dihydroxyvitamin D levels and increased intestinal calcium absorption |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavatta, G.; Altieri, P.; Vandi, G.; Vicennati, V.; Pagotto, U.; Vescini, F. Phosphate Metabolism and Pathophysiology in Parathyroid Disorders and Endocrine Tumors. Int. J. Mol. Sci. 2021, 22, 12975. https://doi.org/10.3390/ijms222312975
Zavatta G, Altieri P, Vandi G, Vicennati V, Pagotto U, Vescini F. Phosphate Metabolism and Pathophysiology in Parathyroid Disorders and Endocrine Tumors. International Journal of Molecular Sciences. 2021; 22(23):12975. https://doi.org/10.3390/ijms222312975
Chicago/Turabian StyleZavatta, Guido, Paola Altieri, Giulia Vandi, Valentina Vicennati, Uberto Pagotto, and Fabio Vescini. 2021. "Phosphate Metabolism and Pathophysiology in Parathyroid Disorders and Endocrine Tumors" International Journal of Molecular Sciences 22, no. 23: 12975. https://doi.org/10.3390/ijms222312975
APA StyleZavatta, G., Altieri, P., Vandi, G., Vicennati, V., Pagotto, U., & Vescini, F. (2021). Phosphate Metabolism and Pathophysiology in Parathyroid Disorders and Endocrine Tumors. International Journal of Molecular Sciences, 22(23), 12975. https://doi.org/10.3390/ijms222312975