
The Complex Interplay between Autophagy and NLRP3 Inflammasome in Renal Diseases


Abstract
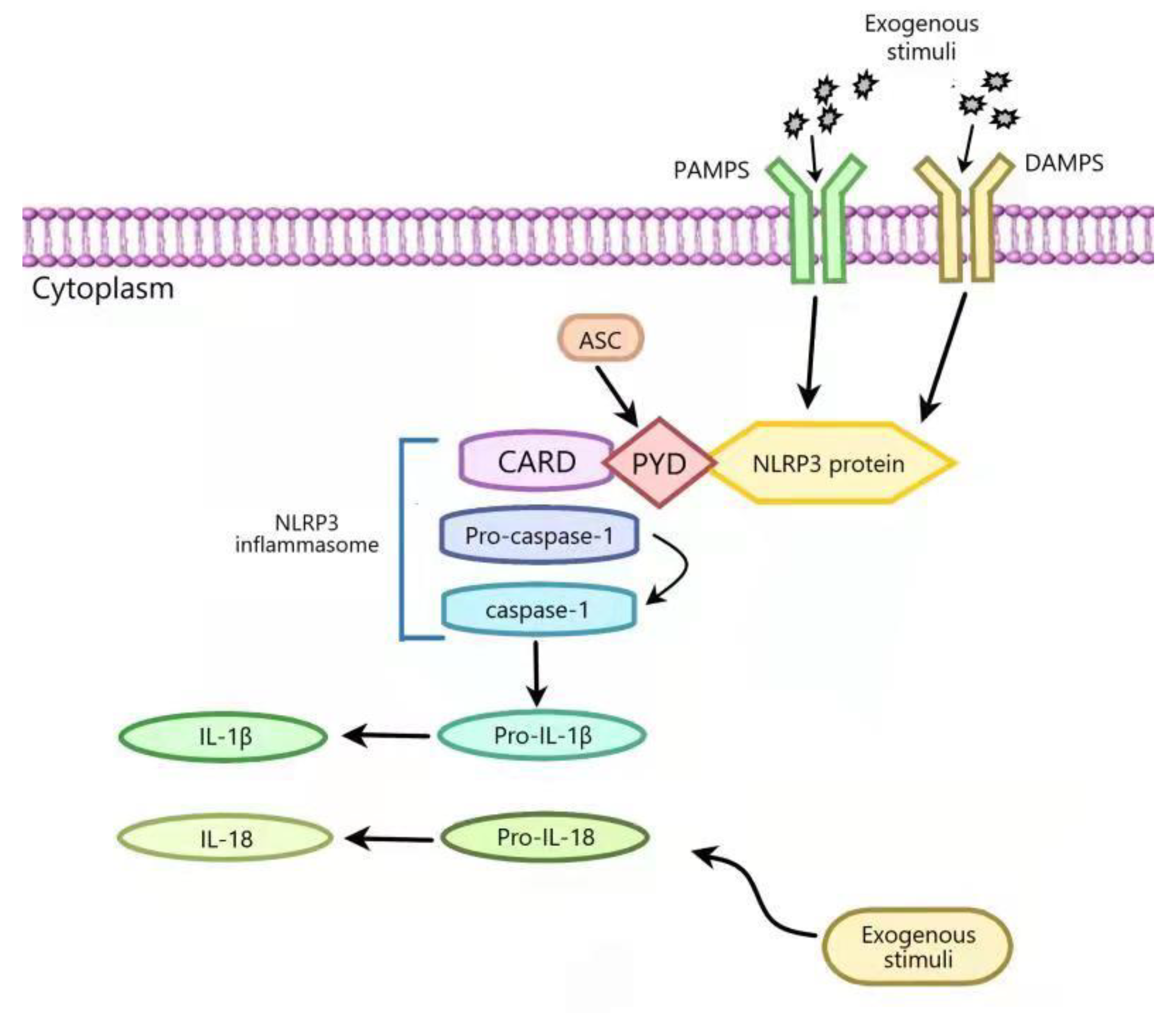
:1. Introduction
2. The Interplay between Autophagy and NLRP3 Inflammasome in Renal Fibrosis
3. The Interplay between Autophagy and NLRP3 Inflammasome in Diabetic Nephropathy
4. The Interplay between Autophagy and NLRP3 Inflammasome in Acute Kidney Injury
5. The Interplay between Autophagy and NLRP3 Inflammasome in Lupus Nephritis
6. The Interplay between Autophagy and NLRP3 Inflammasome in IgA Nephropathy
7. The Interplay between Autophagy and NLRP3 Inflammasome in Uric Acid Nephropathy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Volt, H.; Garcia, J.A.; Doerrier, C.; Diaz-Casado, M.E.; Guerra-Librero, A.; Lopez, L.C.; Escames, G.; Tresguerres, J.A.; Acuna-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Nunez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, D.; Wang, H. Hydrogen sulfide plays an important protective role by influencing autophagy in diseases. Physiol. Res. 2019, 68, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, X.; Wu, T.; Hu, X.; Su, J.; Chen, X. Autophagy in Skin Diseases. Dermatology 2019, 235, 380–389. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and intermittent fasting: The connection for cancer therapy? Clinics 2018, 73 (Suppl. 1), e814s. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Wada, K.; Kabuta, T. Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. J. Biochem. 2017, 161, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Liu, H.; Wang, H. Exogenous Hydrogen Sulfide Plays an Important Role by Regulating Autophagy in Diabetic-Related Diseases. Int. J. Mol. Sci. 2021, 22, 6715. [Google Scholar] [CrossRef]
- Wang, G.H.; Zhang, J.Y.; Huangfu, S.C.; Chen, L.M.; Wang, J.; Wu, D.D.; Xie, X.Z.; Li, Z.Y.; Ji, L.A. Effect of exogenous hydrogen sulfide on lipophagy in mouse primary hepatocytes. Chin. J. Pathophysiol. 2017, 33, 1901–1906. [Google Scholar]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Nastase, M.V.; Zeng-Brouwers, J.; Wygrecka, M.; Schaefer, L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Adv. Drug Deliv. Rev. 2018, 129, 295–307. [Google Scholar] [CrossRef]
- Li, R.; Guo, Y.; Zhang, Y.; Zhang, X.; Zhu, L.; Yan, T. Salidroside Ameliorates Renal Interstitial Fibrosis by Inhibiting the TLR4/NF-kappaB and MAPK Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 1103. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Chen, W.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef]
- Huang, H.; Ni, H.; Ma, K.; Zou, J. ANGPTL2 regulates autophagy through the MEK/ERK/Nrf-1 pathway and affects the progression of renal fibrosis in diabetic nephropathy. Am. J. Transl. Res. 2019, 11, 5472–5486. [Google Scholar]
- Zhang, M.Y.; Chen, H.H.; Tian, J.; Chen, H.J.; Zhu, L.L.; Zhao, P.; Zhang, T. Danggui Shaoyao San Ameliorates Renal Fibrosis via Regulation of Hypoxia and Autophagy. Evid.-Based Complementary Altern. Med. 2019, 2019, 2985270. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.A.; Kim, W.Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-beta and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 78. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, H.; Liu, Z.; Lin, L.; Wang, L.; Xie, M.; Li, D.; Zhang, J.; Zhang, R. Endoplasmic reticulum stress-dependent activation of iNOS/NO-NF-kappaB signaling and NLRP3 inflammasome contributes to endothelial inflammation and apoptosis associated with microgravity. FASEB J. 2020, 34, 10835–10849. [Google Scholar] [CrossRef]
- Yang, Q.; Li, S.; Zhou, Z.; Fu, M.; Yang, X.; Hao, K.; Liu, Y. HDAC6 inhibitor Cay10603 inhibits high glucose-induced oxidative stress, inflammation and apoptosis in retinal pigment epithelial cells via regulating NF-kappaB and NLRP3 inflammasome pathway. Gen. Physiol. Biophys. 2020, 39, 169–177. [Google Scholar] [CrossRef]
- Perry, H.M.; Gorldt, N.; Sung, S.J.; Huang, L.; Rudnicka, K.P.; Encarnacion, I.M.; Bajwa, A.; Tanaka, S.; Poudel, N.; Yao, J.; et al. Perivascular CD73(+) cells attenuate inflammation and interstitial fibrosis in the kidney microenvironment. Am. J. Physiol. Renal Physiol. 2019, 317, F658–F669. [Google Scholar] [CrossRef]
- Nam, S.A.; Kim, W.Y.; Kim, J.W.; Kang, M.G.; Park, S.H.; Lee, M.S.; Kim, H.W.; Yang, C.W.; Kim, J.; Kim, Y.K. Autophagy in FOXD1 stroma-derived cells regulates renal fibrosis through TGF-beta and NLRP3 inflammasome pathway. Biochem. Biophys. Res. Commun. 2019, 508, 965–972. [Google Scholar] [CrossRef]
- Lee, Y.H.; Chen, Y.Y.; Yeh, Y.L.; Wang, Y.J.; Chen, R.J. Stilbene Compounds Inhibit Tumor Growth by the Induction of Cellular Senescence and the Inhibition of Telomerase Activity. Int. J. Mol. Sci. 2019, 20, 2716. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Seo, K.H.; Yokoyama, W. Chemistry of Pterostilbene and Its Metabolic Effects. J. Agric. Food Chem. 2020, 68, 12836–12841. [Google Scholar] [CrossRef]
- Trepiana, J.; Krisa, S.; Portillo, M.P. Activity of Pterostilbene Metabolites against Liver Steatosis in Cultured Hepatocytes. Molecules 2020, 25, 5444. [Google Scholar] [CrossRef]
- Shi, Y.W.; Wang, C.P.; Liu, L.; Liu, Y.L.; Wang, X.; Hong, Y.; Li, Z.; Kong, L.D. Antihyperuricemic and nephroprotective effects of resveratrol and its analogues in hyperuricemic mice. Mol. Nutr. Food Res. 2012, 56, 1433–1444. [Google Scholar] [CrossRef]
- Wang, Y.J.; Chen, Y.Y.; Hsiao, C.M.; Pan, M.H.; Wang, B.J.; Chen, Y.C.; Ho, C.T.; Huang, K.C.; Chen, R.J. Induction of Autophagy by Pterostilbene Contributes to the Prevention of Renal Fibrosis via Attenuating NLRP3 Inflammasome Activation and Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2020, 8, 436. [Google Scholar] [CrossRef]
- Li, S.; Zheng, L.; Zhang, J.; Liu, X.; Wu, Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic. Biol. Med. 2021, 162, 435–449. [Google Scholar] [CrossRef]
- Li, F.; Chen, Y.; Li, Y.; Huang, M.; Zhao, W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-kappaB pathway. Eur. J. Pharmacol. 2020, 886, 173449. [Google Scholar] [CrossRef]
- Jiang, N.; Zhao, H.; Han, Y.; Li, L.; Xiong, S.; Zeng, L.; Xiao, Y.; Wei, L.; Xiong, X.; Gao, P.; et al. HIF-1alpha ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif. 2020, 53, e12909. [Google Scholar] [CrossRef]
- Hwang, S.; Park, J.; Kim, J.; Jang, H.R.; Kwon, G.Y.; Huh, W.; Kim, Y.G.; Kim, D.J.; Oh, H.Y.; Lee, J.E. Tissue expression of tubular injury markers is associated with renal function decline in diabetic nephropathy. J. Diabetes Complicat. 2017, 31, 1704–1709. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, J.; Zhang, W.; Zhang, J.; Yang, J.; Li, K.; He, Y. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: A novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 2013, 45, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Dai, H.; Yuan, J.; Chen, J.; Lin, L.; Zhang, W.; Wang, L.; Zhang, J.; Li, K.; He, Y. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Tschurtschenthaler, M.; Adolph, T.E. The Selective Autophagy Receptor Optineurin in Crohn’s Disease. Front. Immunol. 2018, 9, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Wang, J.; Weng, Q. The diverse role of optineurin in pathogenesis of disease. Biochem. Pharmacol. 2020, 180, 114157. [Google Scholar] [CrossRef]
- Chen, K.; Feng, L.; Hu, W.; Chen, J.; Wang, X.; Wang, L.; He, Y. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. 2019, 33, 4571–4585. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, D.; Wan, X.; Bai, Y.; Yuan, C.; Wang, T.; Yuan, D.; Zhang, C.; Liu, C. Chlorogenic Acid Suppresses miR-155 and Ameliorates Ulcerative Colitis through the NF-kappaB/NLRP3 Inflammasome Pathway. Mol. Nutr. Food Res. 2020, 64, e2000452. [Google Scholar] [CrossRef]
- Chen, S.; Tang, C.; Ding, H.; Wang, Z.; Liu, X.; Chai, Y.; Jiang, W.; Han, Y.; Zeng, H. Maf1 Ameliorates Sepsis-Associated Encephalopathy by Suppressing the NF-kB/NLRP3 Inflammasome Signaling Pathway. Front. Immunol. 2020, 11, 594071. [Google Scholar] [CrossRef]
- Hou, Y.; Lin, S.; Qiu, J.; Sun, W.; Dong, M.; Xiang, Y.; Wang, L.; Du, P. NLRP3 inflammasome negatively regulates podocyte autophagy in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2020, 521, 791–798. [Google Scholar] [CrossRef]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef]
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern. Med. 2017, 167, ITC66–ITC80. [Google Scholar] [CrossRef]
- Nakano, D. Septic acute kidney injury: A review of basic research. Clin. Exp. Nephrol. 2020, 24, 1091–1102. [Google Scholar] [CrossRef]
- Fan, H.; Le, J.W.; Sun, M.; Zhu, J.H. Sirtuin 3 deficiency promotes acute kidney injury induced by sepsis via mitochondrial dysfunction and apoptosis. Iran. J. Basic Med. Sci. 2021, 24, 675–681. [Google Scholar] [CrossRef]
- Gao, Y.; Zeng, Z.; Li, T.; Xu, S.; Wang, X.; Chen, Z.; Lin, C. Polydatin Inhibits Mitochondrial Dysfunction in the Renal Tubular Epithelial Cells of a Rat Model of Sepsis-Induced Acute Kidney Injury. Anesth. Analg. 2015, 121, 1251–1260. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, T.; Lei, X.; Li, Y.; Dai, X.; Cao, Y.; Ding, Q.; Lei, X.; Li, T.; Lin, X. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol. Med. Rep. 2016, 14, 5481–5488. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wei, Q.; Hong, G.; Chen, D.; Liang, J.; He, W.; Chen, M.H. Polydatin induces bone marrow stromal cells migration by activation of ERK1/2. BioMed. Pharmacother. 2016, 82, 49–53. [Google Scholar] [CrossRef]
- Gao, Y.; Dai, X.; Li, Y.; Li, G.; Lin, X.; Ai, C.; Cao, Y.; Li, T.; Lin, B. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J. Transl. Med. 2020, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Wang, R.; Liu, D. Bone Marrow-Derived Mesenchymal Stem Cells Ameliorate Sepsis-Induced Acute Kidney Injury by Promoting Mitophagy of Renal Tubular Epithelial Cells via the SIRT1/Parkin Axis. Front. Endocrinol. 2021, 12, 639165. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Jin, H.; Shao, X.; Zhu, X.; Wu, J.; Zhang, M.; Zhang, Z.; Shen, J.; et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 2021, 17, 2975–2990. [Google Scholar] [CrossRef]
- Bae, E.; Kim, J.H.; Jung, M.H.; Jang, S.J.; Lee, T.W.; Jung, S.; Lee, S.; Jang, H.N.; Chang, S.H.; Park, D.J. Paricalcitol Attenuates Contrast-Induced Acute Kidney Injury by Regulating Mitophagy and Senescence. Oxid. Med. Cell Longev. 2020, 2020, 7627934. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef]
- Dai, X.G.; Xu, W.; Li, T.; Lu, J.Y.; Yang, Y.; Li, Q.; Zeng, Z.H.; Ai, Y.H. Involvement of phosphatase and tensin homolog-induced putative kinase 1-Parkin-mediated mitophagy in septic acute kidney injury. Chin. Med. J. 2019, 132, 2340–2347. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [Green Version]
- Mok, C.C.; Kwok, R.C.; Yip, P.S. Effect of renal disease on the standardized mortality ratio and life expectancy of patients with systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 2154–2160. [Google Scholar] [CrossRef]
- Hanly, J.G.; O'Keeffe, A.G.; Su, L.; Urowitz, M.B.; Romero-Diaz, J.; Gordon, C.; Bae, S.C.; Bernatsky, S.; Clarke, A.E.; Wallace, D.J.; et al. The frequency and outcome of lupus nephritis: Results from an international inception cohort study. Rheumatology 2016, 55, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Flanc, R.S.; Roberts, M.A.; Strippoli, G.F.; Chadban, S.J.; Kerr, P.G.; Atkins, R.C. Treatment of diffuse proliferative lupus nephritis: A meta-analysis of randomized controlled trials. Am. J. Kidney Dis. 2004, 43, 197–208. [Google Scholar] [CrossRef]
- Liu, T.; Liu, H.; Wang, P.; Hu, Y.; Yang, R.; Liu, F.; Kim, H.G.; Dong, Z.; Liu, K. Honokiol Inhibits Melanoma Growth by Targeting Keratin 18 in vitro and in vivo. Front. Cell Dev. Biol. 2020, 8, 603472. [Google Scholar] [CrossRef]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, Y.M.; Lee, C.K.; Jung, J.K.; Han, S.B.; Hong, J.T. Therapeutic applications of compounds in the Magnolia family. Pharmacol. Ther. 2011, 130, 157–176. [Google Scholar] [CrossRef]
- Shen, J.L.; Man, K.M.; Huang, P.H.; Chen, W.C.; Chen, D.C.; Cheng, Y.W.; Liu, P.L.; Chou, M.C.; Chen, Y.H. Honokiol and magnolol as multifunctional antioxidative molecules for dermatologic disorders. Molecules 2010, 15, 6452–6465. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Hsu, W.H.; Wu, C.Y.; Shang, H.S.; Liu, F.C.; Chen, A.; Hua, K.F.; Ka, S.M. Accelerated, severe lupus nephritis benefits from treatment with honokiol by immunoregulation and differentially regulating NF-kappaB/NLRP3 inflammasome and sirtuin 1/autophagy axis. FASEB J. 2020, 34, 13284–13299. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Wang, H.; Li, X. The Role of the Interplay between Autophagy and NLRP3 Inflammasome in Metabolic Disorders. Front. Cell Dev. Biol. 2021, 9, 634118. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhan, N.; Jin, Y.; Ling, H.; Xiao, C.; Xie, Z.; Zhong, H.; Yu, X.; Tang, R.; Ma, J.; et al. Tofacitinib restores the balance of gammadeltaTreg/gammadeltaT17 cells in rheumatoid arthritis by inhibiting the NLRP3 inflammasome. Theranostics 2021, 11, 1446–1457. [Google Scholar] [CrossRef]
- Ruan, S.; Zhai, L.; Wu, S.; Zhang, C.; Guan, Q. SCFAs promote intestinal double-negative T cells to regulate the inflammatory response mediated by NLRP3 inflammasome. Aging 2021, 13, 21470–21482. [Google Scholar] [CrossRef]
- Bhandarkar, S.S.; Bromberg, J.; Carrillo, C.; Selvakumar, P.; Sharma, R.K.; Perry, B.N.; Govindarajan, B.; Fried, L.; Sohn, A.; Reddy, K.; et al. Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth in vitro and in vivo. Clin. Cancer Res. 2008, 14, 5743–5748. [Google Scholar] [CrossRef] [Green Version]
- de la Puente, P.; Azab, F.; Muz, B.; Luderer, M.; Arbiser, J.; Azab, A.K. Tris DBA palladium overcomes hypoxia-mediated drug resistance in multiple myeloma. Leuk. Lymphoma 2016, 57, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Hua, K.F.; Chu, C.L.; Yang, S.R.; Arbiser, J.L.; Yang, S.S.; Lin, Y.C.; Liu, F.C.; Yang, S.M.; Ka, S.M.; et al. Tris DBA Ameliorates Accelerated and Severe Lupus Nephritis in Mice by Activating Regulatory T Cells and Autophagy and Inhibiting the NLRP3 Inflammasome. J. Immunol. 2020, 204, 1448–1461. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Julian, B.A.; Rizk, D.V. IgA Nephropathy: An Interesting Autoimmune Kidney Disease. Am. J. Med. Sci. 2021, 361, 176–194. [Google Scholar] [CrossRef]
- Wu, C.Y.; Hua, K.F.; Yang, S.R.; Tsai, Y.S.; Yang, S.M.; Hsieh, C.Y.; Wu, C.C.; Chang, J.F.; Arbiser, J.L.; Chang, C.T.; et al. Tris DBA ameliorates IgA nephropathy by blunting the activating signal of NLRP3 inflammasome through SIRT1- and SIRT3-mediated autophagy induction. J. Cell Mol. Med. 2020, 24, 13609–13622. [Google Scholar] [CrossRef]
- Park, N.J.; Bong, S.K.; Lee, S.; Jung, Y.; Jegal, H.; Kim, J.; Kim, S.K.; Kim, Y.K.; Kim, S.N. Compound K improves skin barrier function by increasing SPINK5 expression. J. Ginseng. Res. 2020, 44, 799–807. [Google Scholar] [CrossRef]
- Luo, Z.B.; Rahman, S.U.; Xuan, M.F.; Han, S.Z.; Li, Z.Y.; Yin, X.J.; Kang, J.D. The protective role of ginsenoside compound K in porcine oocyte meiotic maturation failed caused by benzo(a)pyrene during in vitro maturation. Theriogenology 2020, 157, 96–109. [Google Scholar] [CrossRef]
- Wu, C.Y.; Hua, K.F.; Hsu, W.H.; Suzuki, Y.; Chu, L.J.; Lee, Y.C.; Takahata, A.; Lee, S.L.; Wu, C.C.; Nikolic-Paterson, D.J.; et al. IgA Nephropathy Benefits from Compound K Treatment by Inhibiting NF-kappaB/NLRP3 Inflammasome and Enhancing Autophagy and SIRT1. J. Immunol 2020, 205, 202–212. [Google Scholar] [CrossRef]
- Wu, X.; Liu, L.; Xie, H.; Liao, J.; Zhou, X.; Wan, J.; Yu, K.; Li, J.; Zhang, Y. Tanshinone IIA prevents uric acid nephropathy in rats through NF-kappaB inhibition. Planta Med. 2012, 78, 866–873. [Google Scholar] [CrossRef]
- Hou, S.X.; Zhu, W.J.; Pang, M.Q.; Jeffry, J.; Zhou, L.L. Protective effect of iridoid glycosides from Paederia scandens (LOUR.) MERRILL (Rubiaceae) on uric acid nephropathy rats induced by yeast and potassium oxonate. Food Chem. Toxicol. 2014, 64, 57–64. [Google Scholar] [CrossRef]
- Hu, J.; Wu, H.; Wang, D.; Yang, Z.; Dong, J. LncRNA ANRIL promotes NLRP3 inflammasome activation in uric acid nephropathy through miR-122-5p/BRCC3 axis. Biochimie 2019, 157, 102–110. [Google Scholar] [CrossRef]
- Hu, J.; Wu, H.; Wang, D.; Yang, Z.; Zhuang, L.; Yang, N.; Dong, J. Weicao capsule ameliorates renal injury through increasing autophagy and NLRP3 degradation in UAN rats. Int. J. Biochem. Cell Biol. 2018, 96, 1–8. [Google Scholar] [CrossRef]
- Bao, J.; Shi, Y.; Tao, M.; Liu, N.; Zhuang, S.; Yuan, W. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy. Clin. Sci. 2018, 132, 2299–2322. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Renal fibrosis (RF) | Reduction in UUO-induced damage due to mitochondrial and oxidative stress | [17] |
| Renal fibrosis | Pteronene inhibition of TGF-β-mediated NLRP3 inflammasome activation by promoting autophagy | [26] |
| Diabetic nephropathy (DN) | Optineurin inhibition of HG-induced NLRP3 inflammasome by promoting mitophagy of RTECs | [35] |
| Diabetic nephropathy | The restoration of podocyte autophagy by inactivation of NLRP3 inflammasome | [38] |
| Sepsis-induced acute kidney injury (SI-AKI) | Polydatin inhibition of NLRP3 inflammasome activation by promoting Parkin-dependent mitophagy via activation of the sirtuin 1 pathway | [46] |
| Contrast-induced acute kidney injury (CI-AKI) | PINK1–Parkin-mediated mitophagy inhibition of exposure-induced apoptosis by suppressing NLRP3 inflammasome | [52] |
| Accelerated and severe lupus nephritis (ASLN) | Honokiol inhibition of NLRP3 inflammasome activation by promoting autophagy | [63] |
| ASLN | Tris DBA inhibition of NLRP3 inflammasome activation by promoting autophagy by reducing mitochondrial ROS or inhibiting the JNK/ERK/p38 MAPK pathway | [68] |
| IgA nephropathy (IgAN) | Tris DBA inhibition of NLRP3 inflammasome activation by promoting autophagy by reducing mitochondrial ROS or inhibiting the JNK/ERK/p38 MAPK pathway | [70] |
| IgAN | Compound K inhibition of NLRP3 inflammasome activation through sirtuin 1-dependent autophagy induction | [73] |
| Uric acid nephropathy (UAN) | Weicao inhibition of NLRP3 inflammasome by inducing autophagy | [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Fu, X.; Wang, Q.; Liu, H.; Wang, H.; Wu, D. The Complex Interplay between Autophagy and NLRP3 Inflammasome in Renal Diseases. Int. J. Mol. Sci. 2021, 22, 12766. https://doi.org/10.3390/ijms222312766
Ding Y, Fu X, Wang Q, Liu H, Wang H, Wu D. The Complex Interplay between Autophagy and NLRP3 Inflammasome in Renal Diseases. International Journal of Molecular Sciences. 2021; 22(23):12766. https://doi.org/10.3390/ijms222312766
Chicago/Turabian StyleDing, Yong, Xiaodi Fu, Qimeng Wang, Huiyang Liu, Honggang Wang, and Dongdong Wu. 2021. "The Complex Interplay between Autophagy and NLRP3 Inflammasome in Renal Diseases" International Journal of Molecular Sciences 22, no. 23: 12766. https://doi.org/10.3390/ijms222312766
APA StyleDing, Y., Fu, X., Wang, Q., Liu, H., Wang, H., & Wu, D. (2021). The Complex Interplay between Autophagy and NLRP3 Inflammasome in Renal Diseases. International Journal of Molecular Sciences, 22(23), 12766. https://doi.org/10.3390/ijms222312766