
Dietary Intake of 17α-Ethinylestradiol Promotes HCC Progression in Humanized Male Mice Expressing Sex Hormone-Binding Globulin


Abstract
:1. Introduction
2. Results
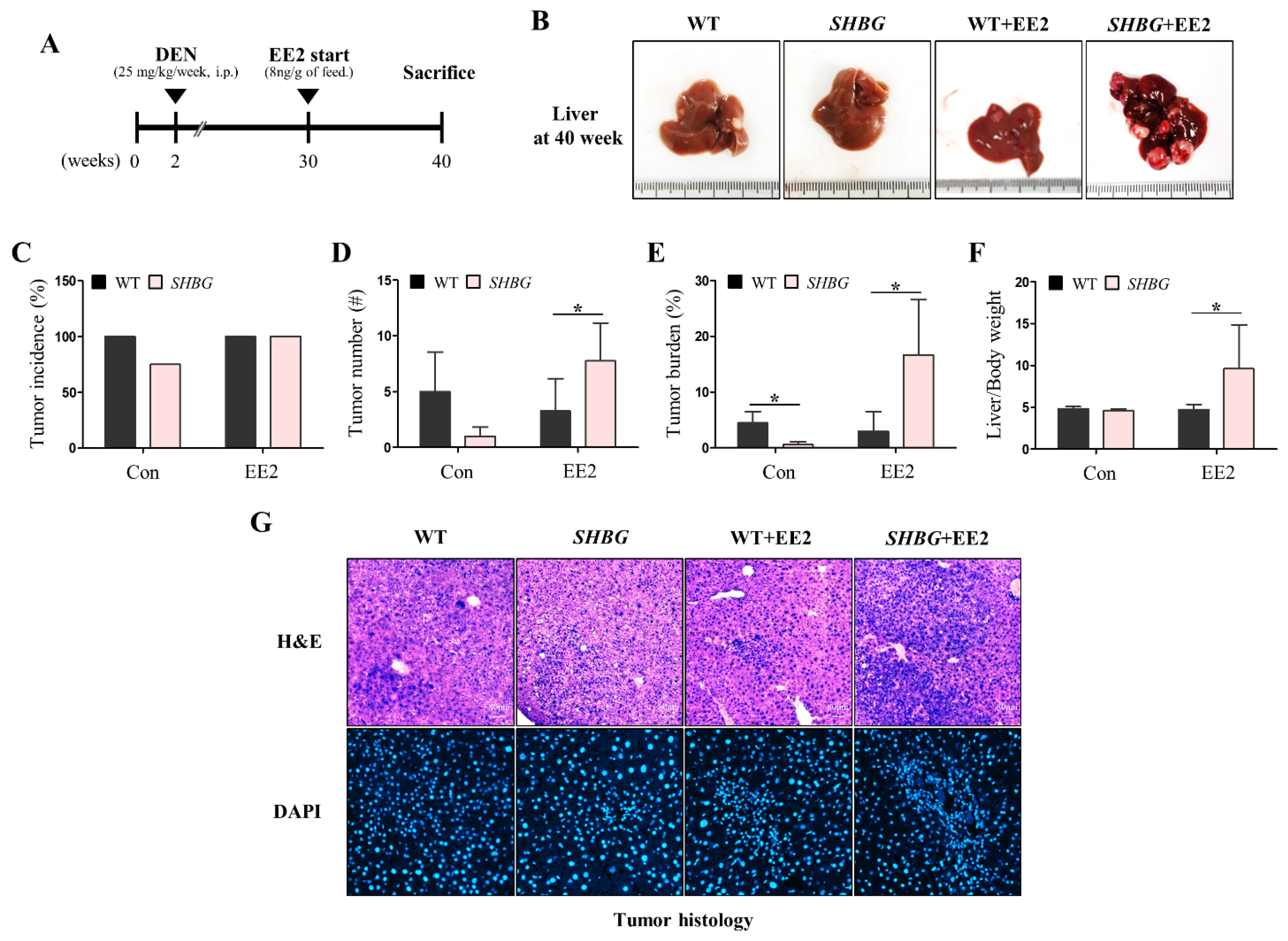
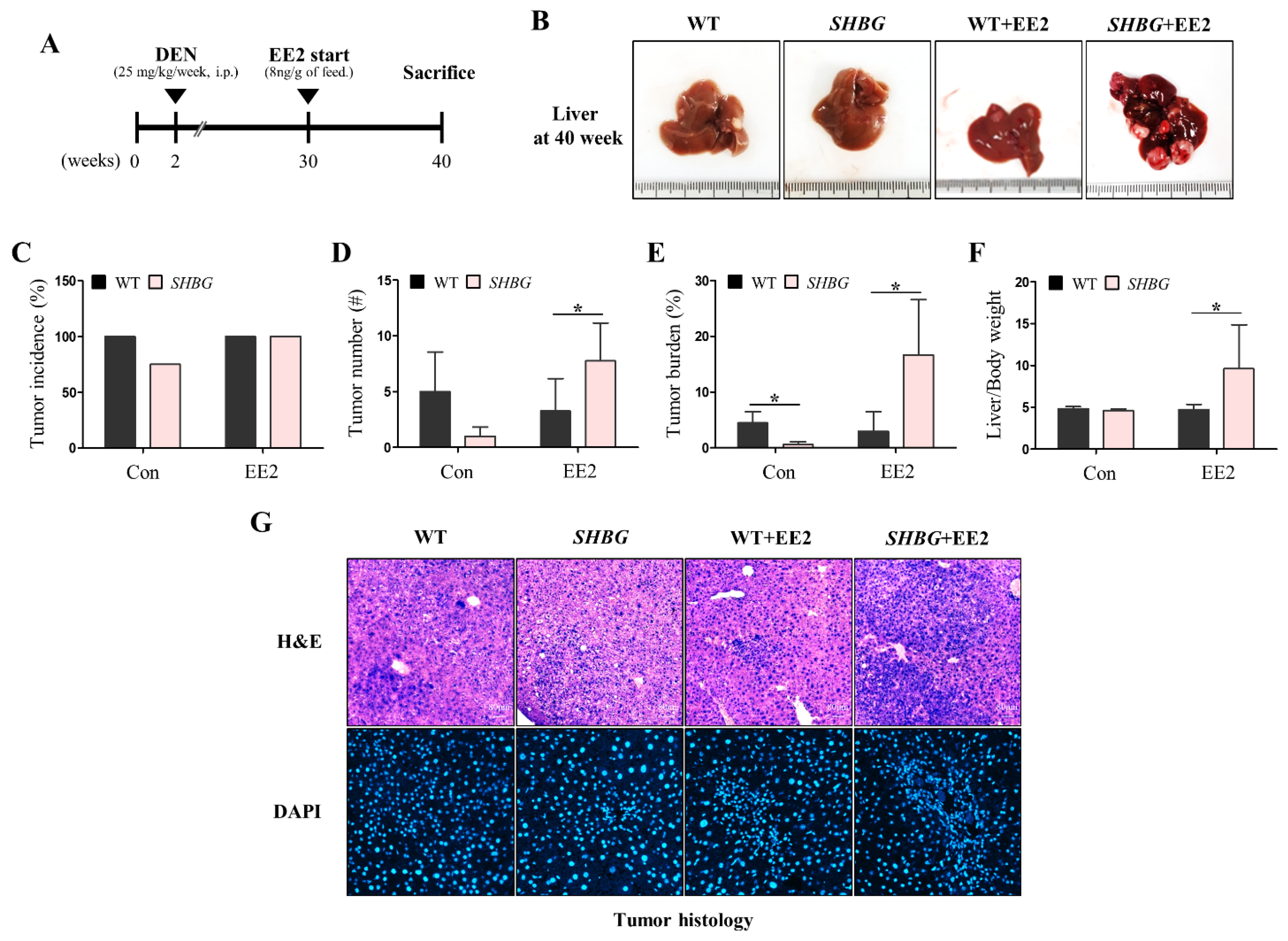
2.1. EE2 Promotes HCC Progression in SHBG Mice
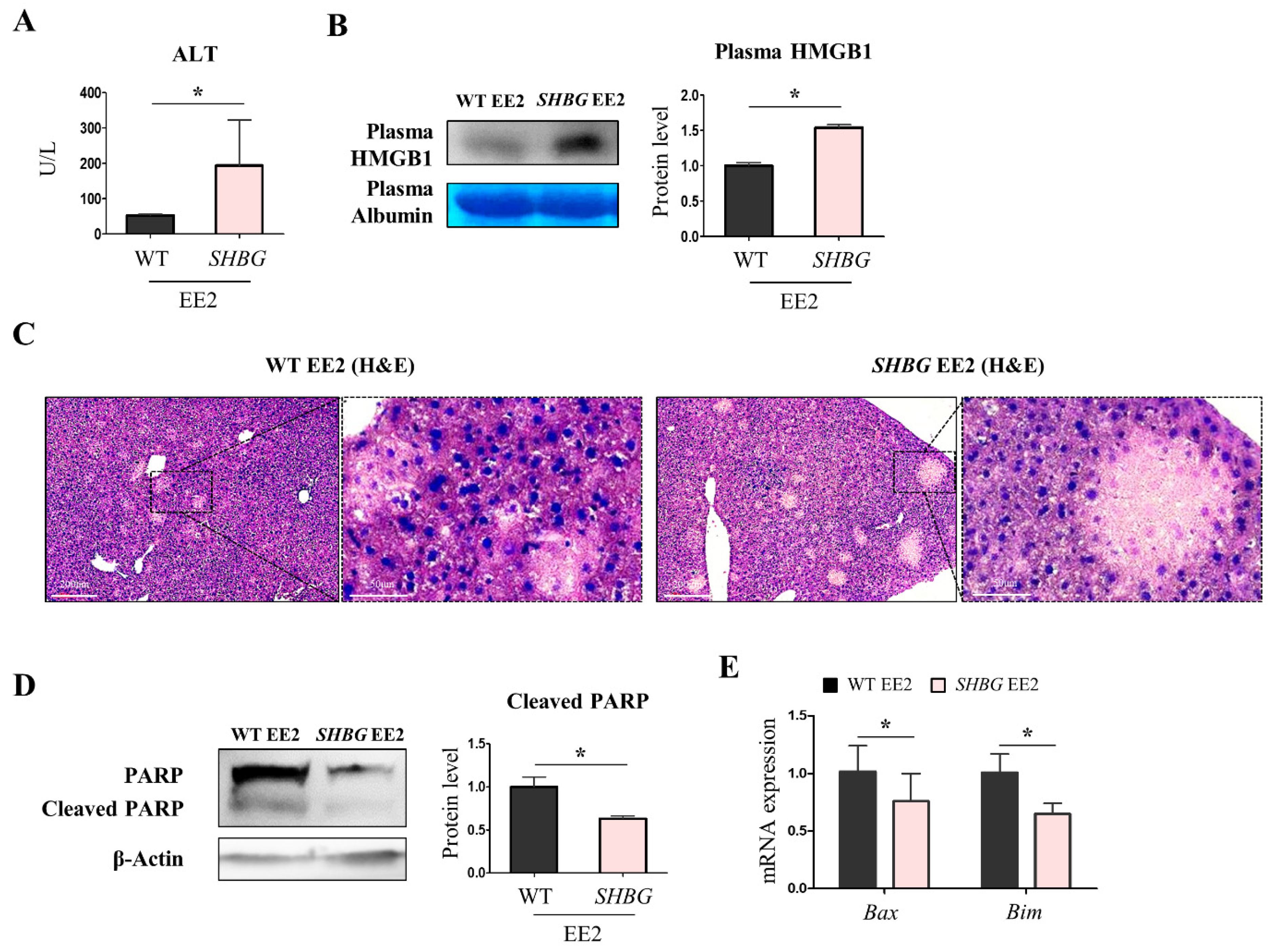
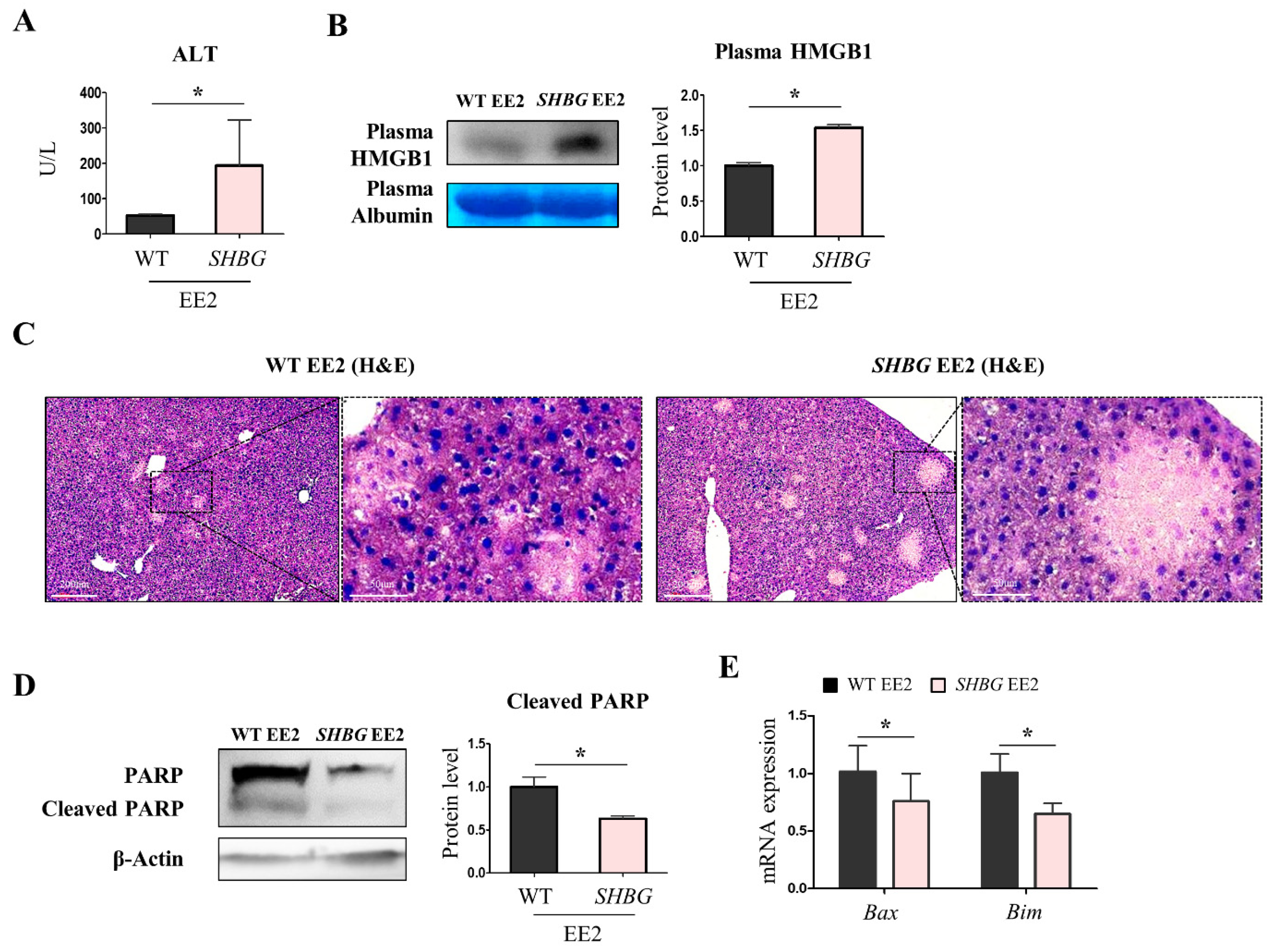
2.2. EE2 Exacerbates Hepatic Necrosis in SHBG Mice
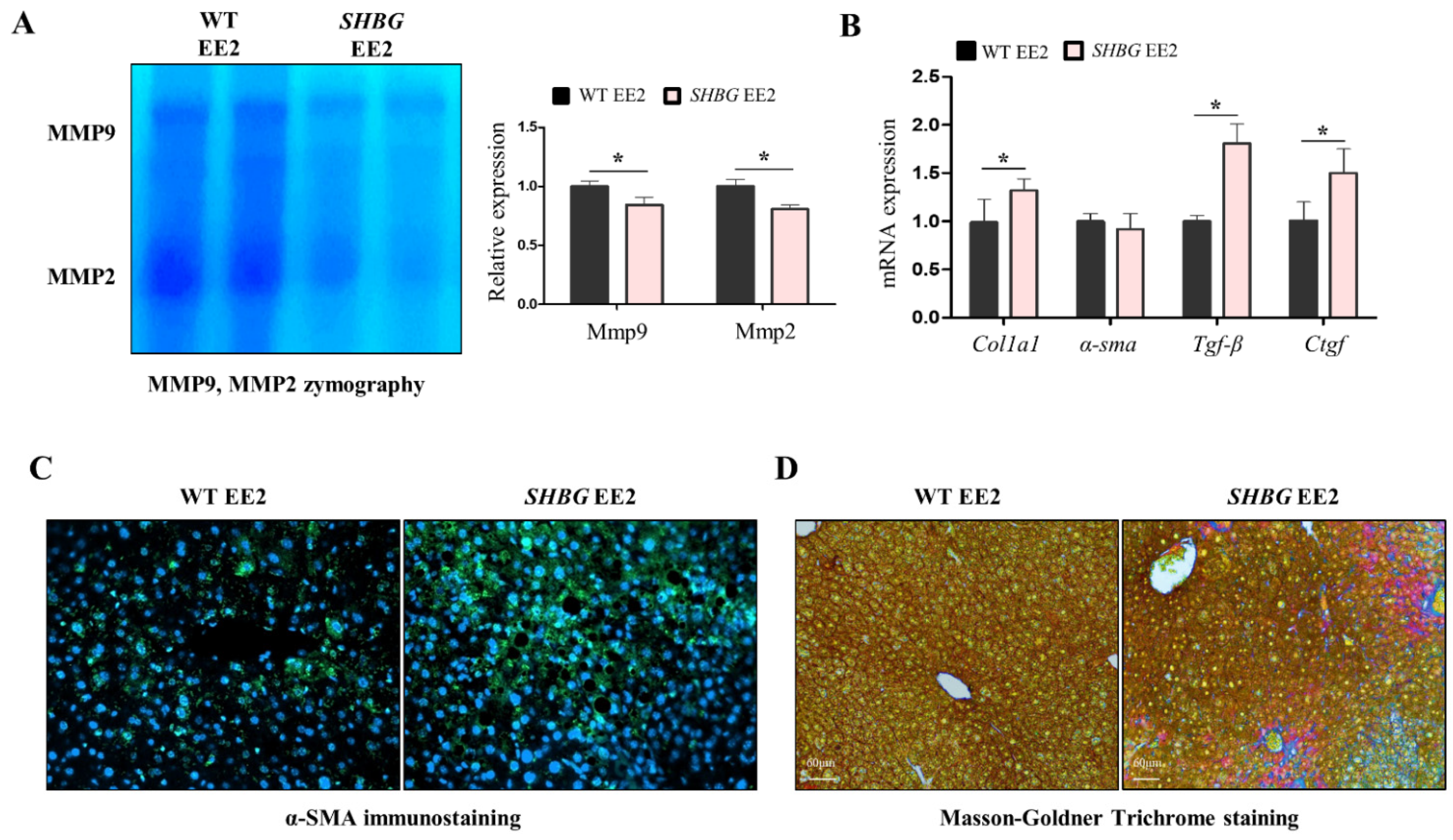
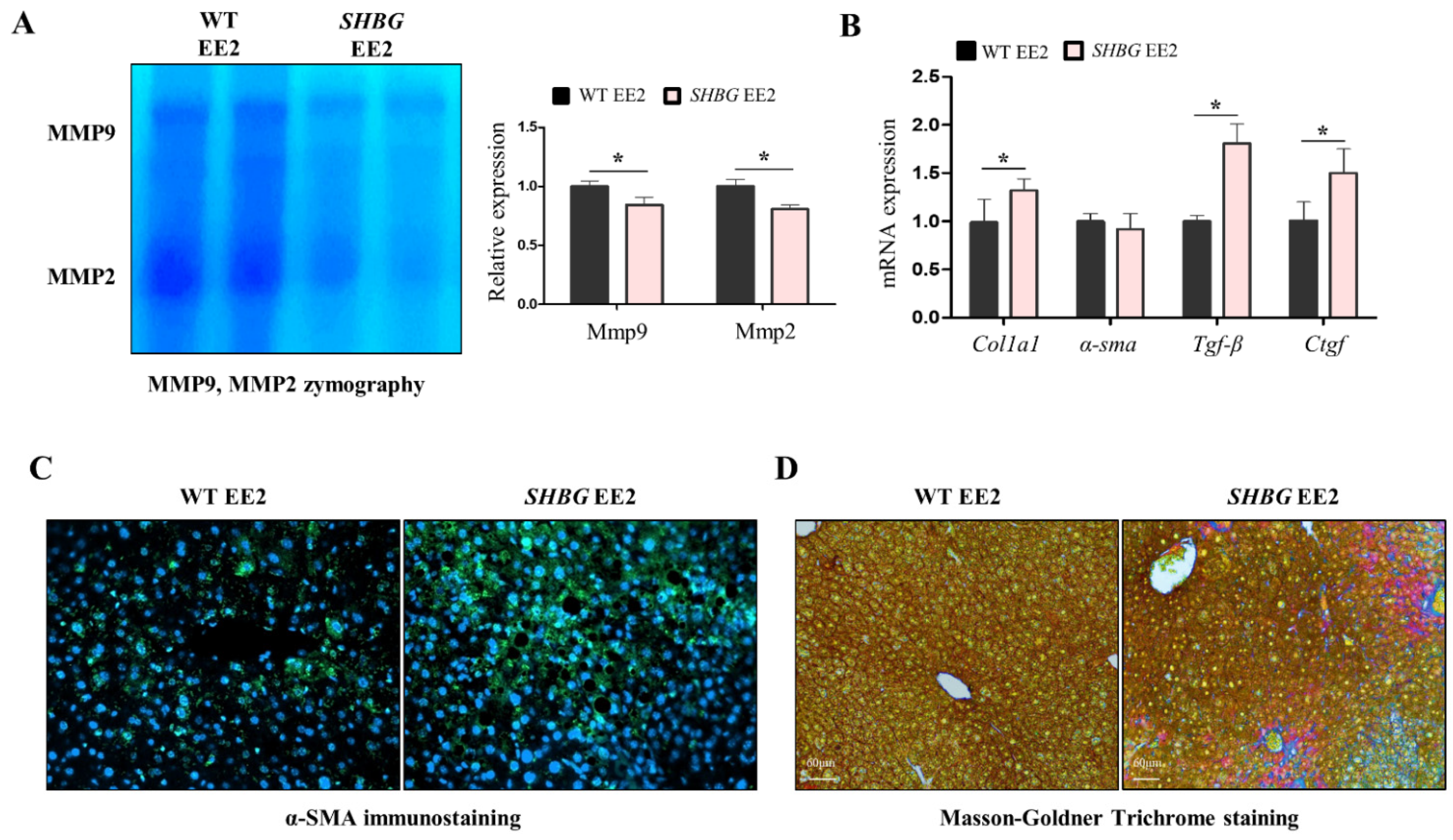
2.3. EE2 Increases Hepatic Fibrosis in SHBG Mice
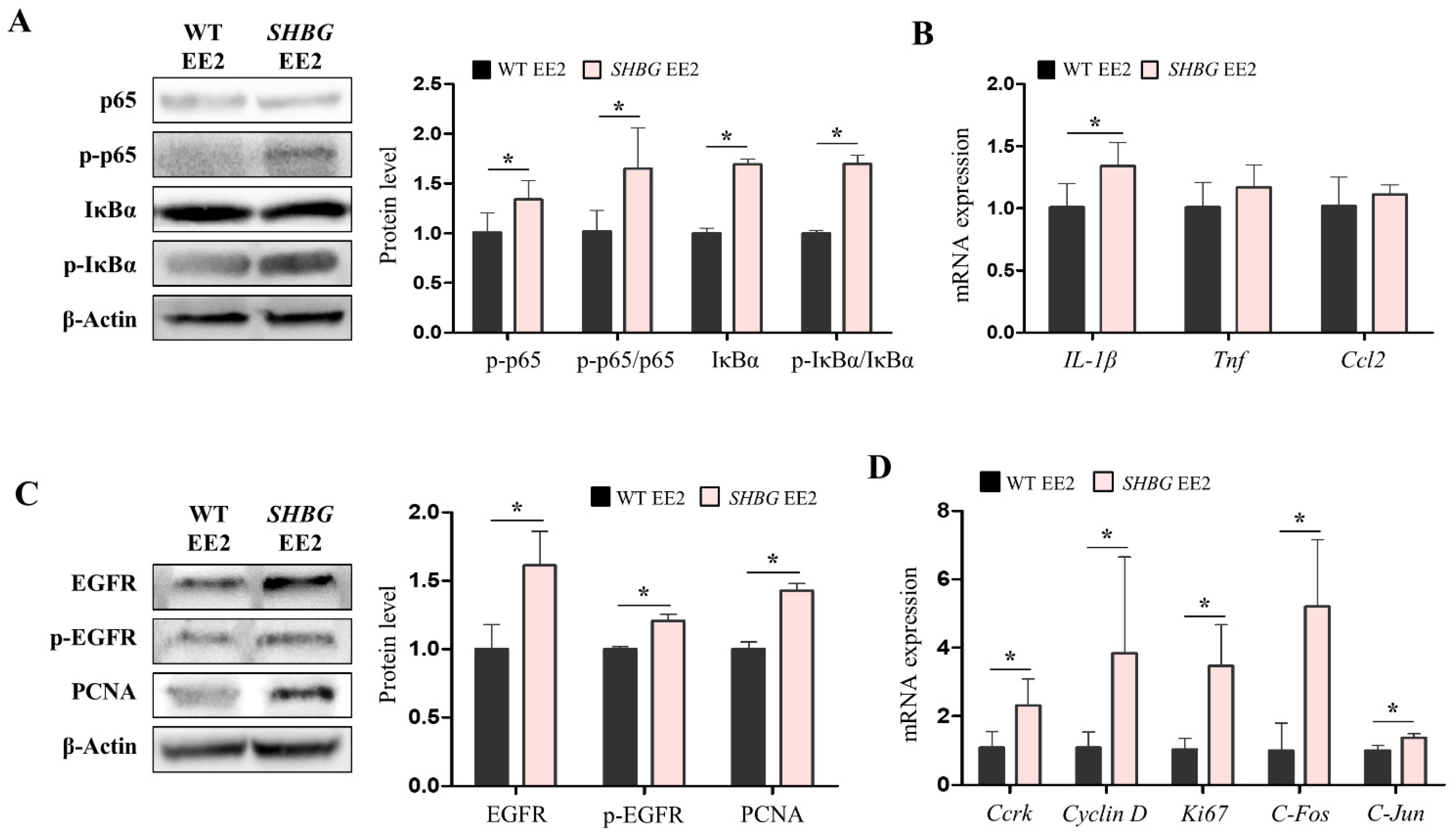
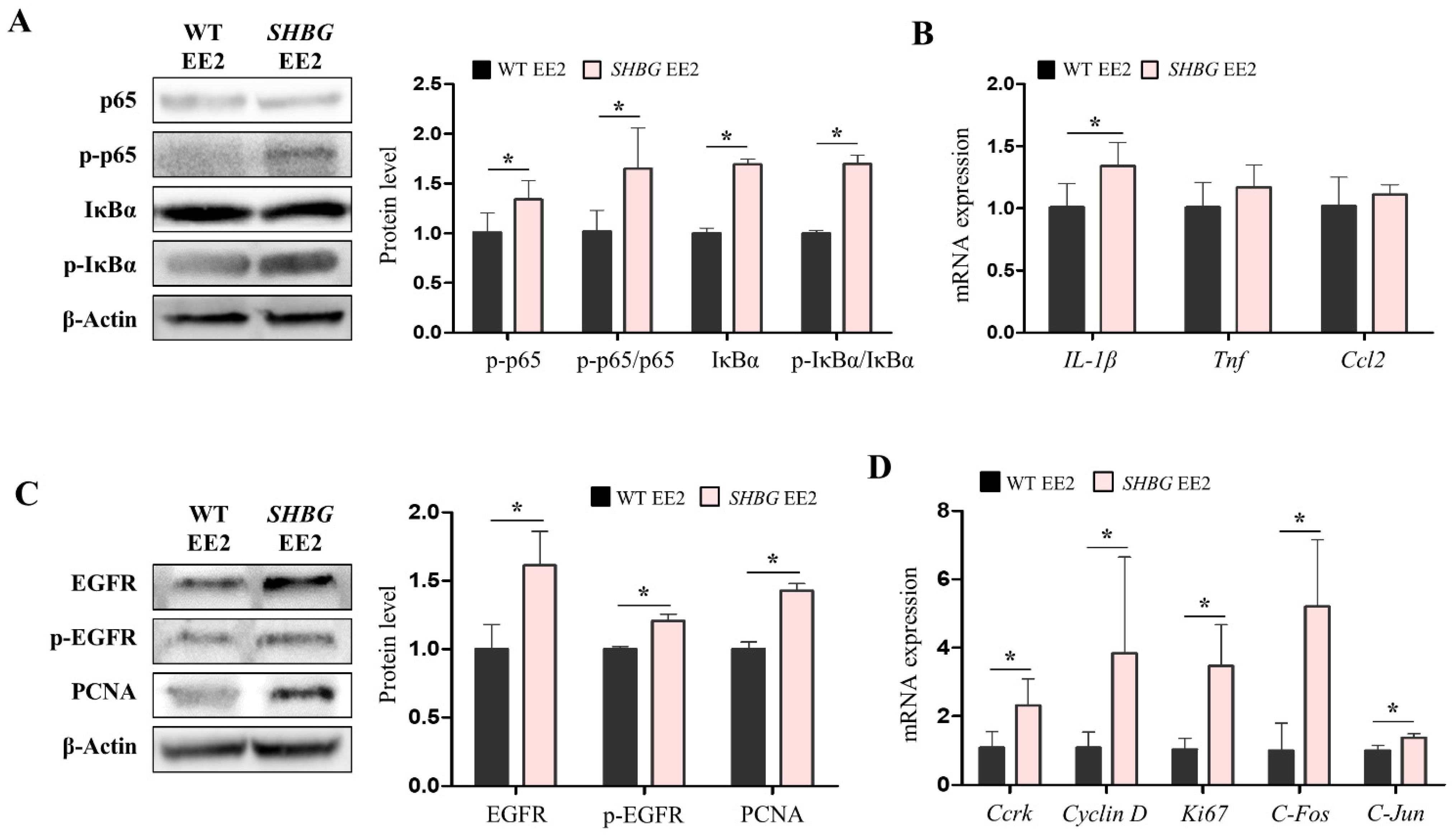
2.4. EE2 Increases Hepatic Pro-Inflammatory and Proliferation Responses in SHBG Mice
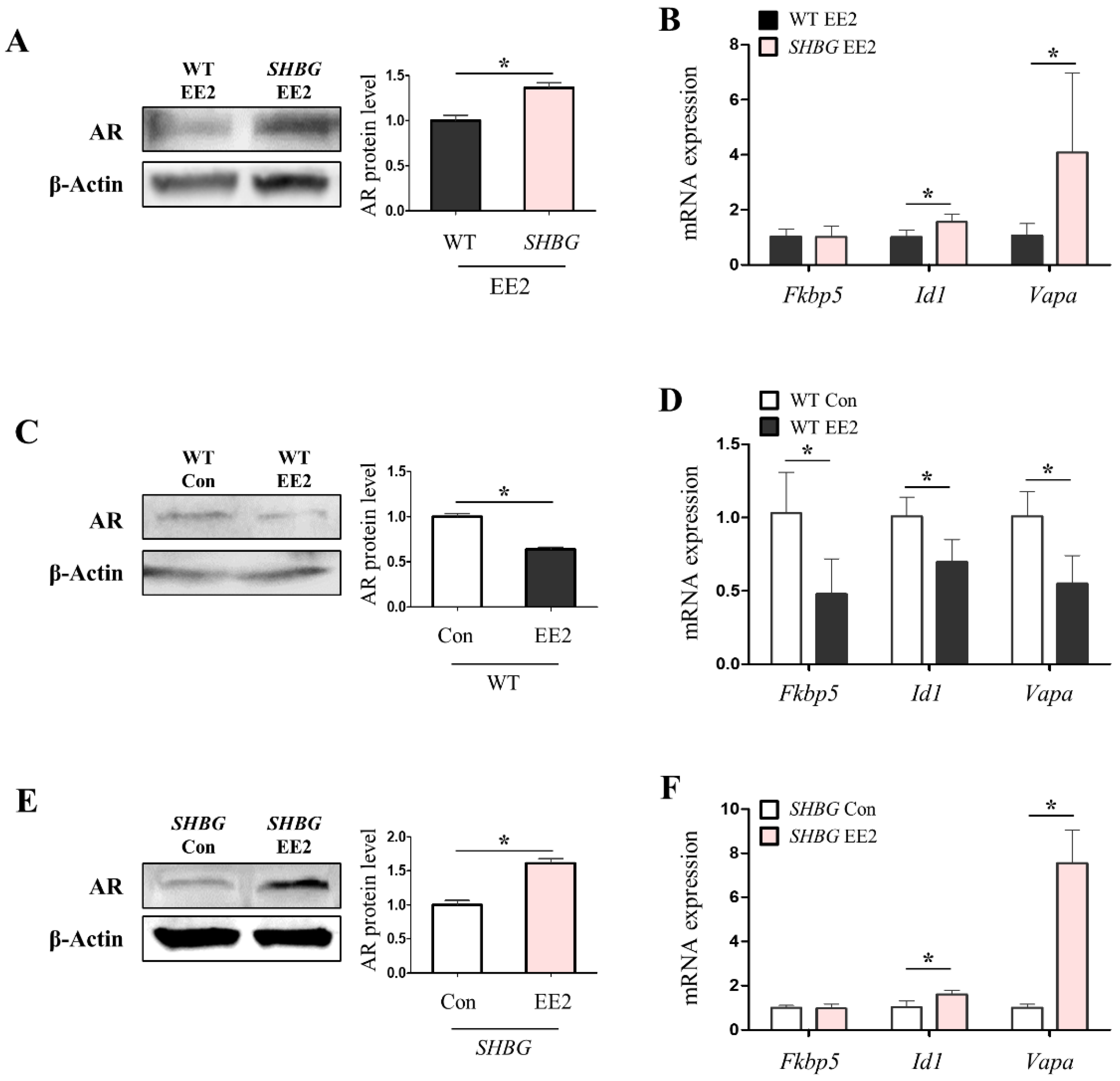
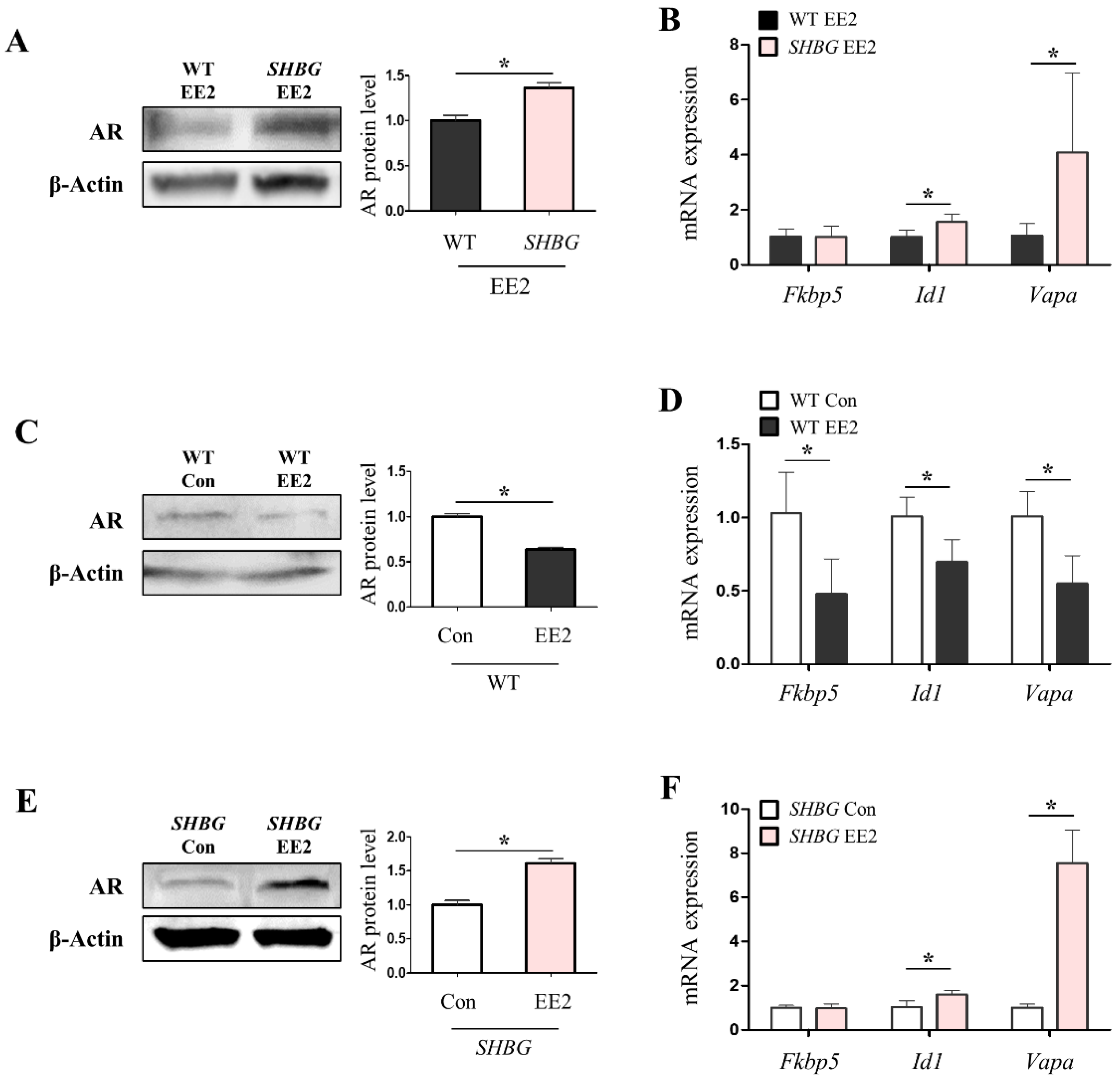
2.5. The Presence of Plasma SHBG Accentuates Androgenic Effects
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. RNA Isolation, Reverse Transcription and qRT-PCR
4.3. Western Blotting
4.4. Cell Culture
4.5. Histology
4.6. Plasma ALT
4.7. MMP9 and MMP2 Measurement
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, X.; Yang, Y.; Zhang, Y.; Wang, H.-Y.; Zheng, X.S. Significance and mechanism of androgen receptor overexpression and androgen receptor/mechanistic target of rapamycin cross-talk in hepatocellular carcinoma. Hepatology 2018, 67, 2271–2286. [Google Scholar] [CrossRef]
- Keng, V.W.; Largaespada, D.A.; Villanueva, A. Why men are at higher risk for hepatocellular carcinoma? J. Hepatol. 2012, 57, 453–454. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.L. Plasma steroid-binding proteins: Primary gatekeepers of steroid hormone action. J. Endocrinol. 2016, 230, R13–R25. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.L. Diverse Roles for Sex Hormone-Binding Globulin in Reproduction. Biol. Reprod. 2011, 85, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, M.; Hammond, G.; Blokland, M.; Jardí, F.; Antonio, L.; Dubois, V.; Khalil, R.; Sterk, S.S.; Gielen, E.; Decallonne, B.; et al. Sex hormone-binding globulin regulation of androgen bioactivity in vivo: Validation of the free hormone hypothesis. Sci. Rep. 2016, 6, 35539. [Google Scholar] [CrossRef] [Green Version]
- Hong, E.-J.; Sahu, B.; Jänne, O.A.; Hammond, G. Cytoplasmic Accumulation of Incompletely Glycosylated SHBG Enhances Androgen Action in Proximal Tubule Epithelial Cells. Mol. Endocrinol. 2011, 25, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Lee, Y.H.; Yang, H.; Lee, H.W.; Lee, G.-S.; An, B.-S.; Jeung, E.-B.; Park, B.-K.; Hong, E.-J. Sex hormone-binding globulin suppresses NAFLD-triggered hepatocarcinogenesis after menopause. Carcinogenesis 2019, 40, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Endrikat, J.; Muller, U.; Dusterberg, B. A twelve-month comparative clinical investigation of two low-dose oral contraceptives containing 20 micrograms ethinylestradiol/75 micrograms gestodene and 30 micrograms ethinylestradiol/75 micrograms gestodene, with respect to efficacy, cycle control, and tolerance. Contraception 1997, 55, 131–137. [Google Scholar]
- Campen, D.; Maronpot, R.; Lucier, G. Dose-response relationships in promotion of rat hepatocarcinogenesis by 17α-ethinylestradiol. J. Toxicol. Environ. Health Part A Curr. Issues 1990, 29, 257–268. [Google Scholar] [CrossRef]
- Yager, J.D.; Campbell, H.A.; Longnecker, D.S.; Roebuck, B.D.; Benoit, M.C. Enhancement of hepatocarcinogenesis in female rats by ethinyl estradiol and mestranol but not estradiol. Cancer Res. 1984, 44, 3862–3869. [Google Scholar]
- Ferrara, B.E.; Rutland, E.D. Liver tumor in long-term user of oral contraceptives. Postgrad. Med. 1988, 84, 107–109. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Sahasrabuddhe, V.V.; Campbell, P.T.; Graubard, B.I.; Chen, J.; Schwartz, L.M.; Petrick, J.L.; Alavanja, M.C.; Andreotti, G.; Boggs, D.A.; et al. Reproductive factors, exogenous hormone use and risk of hepatocellular carcinoma among US women: Results from the Liver Cancer Pooling Project. Br. J. Cancer 2015, 112, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, J.W.; Lycklama, A.A.B.; Nijeholt, A.; Jonas, U. Oestrogen in the treatment of prostatic carcinoma. What is the safe and effective dose of ethinyloestradiol? Br. J. Urol. 1989, 63, 76–79. [Google Scholar] [CrossRef]
- Hayashi, T.; Sekii, Y.; Katayama, K.; Kamoto, A.; Kakuta, Y.; Mori, N.; Itatani, H.; Yoshioka, T. The efficacy of ethinylestradiol for castration-resistant prostate cancer. Nihon Hinyokika Gakkai Zasshi 2014, 105, 37–42. [Google Scholar] [PubMed]
- Izumi, K.; Kadono, Y.; Shima, T.; Konaka, H.; Mizokami, A.; Koh, E.; Namiki, M. Ethinylestradiol improves prostate-specific antigen levels in pretreated castration-resistant prostate cancer patients. Anticancer Res. 2010, 30, 5201–5205. [Google Scholar] [PubMed]
- Hasegawa, Y.; Tomita, M.; Asano, K.; Hasegawa, N.; Ikemoto, I.; Onodera, S.; Ohishi, Y.; Ishikawa, T. Primary, nonviral, hepatocellular carcinoma in patients with prostate cancer treated by hormone therapy: 2 case reports. Nihon Hinyokika Gakkai Zasshi 2005, 96, 503–506. [Google Scholar] [PubMed] [Green Version]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallach, D.; Kovalenko, A. Keeping inflammation at bay. eLife 2014, 3, e02583. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef]
- Giannitrapani, L.; Soresi, M.; La Spada, E.; Cervello, M.; D’Alessandro, N.; Montalto, G. Sex Hormones and Risk of Liver Tumor. Ann. N. Y. Acad. Sci. 2006, 1089, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sakai, H.; Hashizume, M.; Hirohata, T. Serum testosterone:estradiol ratio and the development of hepatocellular carcinoma among male cirrhotic patients. Cancer Res. 2000, 60, 5106–5110. [Google Scholar] [PubMed]
- Stanczyk, F.Z.; Archer, D.F.; Bhavnani, B.R. Ethinyl estradiol and 17beta-estradiol in combined oral contraceptives: Pharmacokinetics, pharmacodynamics and risk assessment. Contraception 2013, 87, 706–727. [Google Scholar] [CrossRef] [PubMed]
- Blair, R.M.; Fang, H.; Branham, W.S.; Hass, B.S.; Dial, S.L.; Moland, C.L.; Tong, W.; Shi, L.; Perkins, R.; Sheehan, D.M. The Estrogen Receptor Relative Binding Affinities of 188 Natural and Xenochemicals: Structural Diversity of Ligands. Toxicol. Sci. 2000, 54, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Hee, L.; Kettner, L.O.; Vejtorp, M. Continuous use of oral contraceptives: An overview of effects and side-effects. Acta Obstet. Gynecol. Scand. 2012, 92, 125–136. [Google Scholar] [CrossRef]
- Jänne, M.; Hogeveen, K.N.; Deol, H.K.; Hammond, G.L. Expression and regulation of human sex hormone-binding globulin transgenes in mice during development. Endocrinology 1999, 140, 4166–4174. [Google Scholar]
- Lee, S.R.; Hong, E.-J. Sex hormone-binding globulin impedes hepatocellular carcinoma growth via MERTK regulation. Genes Dis. 2021, in press. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Heo, J.; Jo, S.; Ryu, J.; Kim, G.; Yon, J.-M.; Lee, M.; Lee, G.-S.; An, B.-S.; et al. Loss of PGRMC1 Delays the Progression of Hepatocellular Carcinoma via Suppression of Pro-Inflammatory Immune Responses. Cancers 2021, 13, 2438. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Jeong, E.K.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Regulation of Tumor Progression by Programmed Necrosis. Oxidative Med. Cell. Longev. 2018, 2018, 3537471. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, A.R.; Andersen, K.J.; Hamilton-Dutoit, S.; Nyengaard, J.R.; Mortensen, F.V. Correlation between liver cell necrosis and circulating alanine aminotransferase after ischaemia/reperfusion injuries in the rat liver. Int. J. Exp. Pathol. 2016, 97, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-P.; Chang, H.-L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.-Y.; Wu, A.T.H.; Yeh, C.-T.; Tsai, S.-H.; Lee, W.-H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786. [Google Scholar] [CrossRef] [Green Version]
- Rachfal, A.W.; Brigstock, D.R. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol. Res. 2003, 26, 1–9. [Google Scholar] [CrossRef]
- Saile, B.; Matthes, N.; Knittel, T.; Ramadori, G. Transforming growth factor beta and tumor necrosis factor alpha inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology 1999, 30, 196–202. [Google Scholar] [CrossRef]
- Carpino, G.; Morini, S.; Corradini, S.G.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Muda, A.O.; Berloco, P.; Rossi, M. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef]
- Ringelhan, M.; McKeating, J.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160274. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.-X.; Ling, Y.; Wang, H.-Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar] [PubMed]
- Juríková, M.; Danihel, L.; Polák, Š.; Varga, I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Nigh, E.; Lee, R.J.; Ye, H.; Thompson, M.A.; Saudou, F.; Pestell, R.G.; Greenberg, M.E. Fos Family Members Induce Cell Cycle Entry by Activating Cyclin D1. Mol. Cell. Biol. 1998, 18, 5609–5619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Tian, Y.; Wan, H.; Tan, G. Cell cycle-related kinase in carcinogenesis. Oncol. Lett. 2012, 4, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Wisdom, R.; Johnson, R.S.; Moore, C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999, 18, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Hu, J.; Yu, X.; Guan, W.; Lu, H.; Yu, Y.; Yu, Y.; Zang, G.; Tang, Z. The Role of IL-1 Family Members and Kupffer Cells in Liver Regeneration. BioMed Res. Int. 2016, 2016, 6495793. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.; Langley, M.S.; Robinson, P.A.; Nummi, S.; Lund, L. Serum steroid binding protein concentrations, distribution of progestogens, and bioavailability of testosterone during treatment with contraceptives containing desogestrel or levonorgestrel. Fertil. Steril. 1984, 42, 44–51. [Google Scholar] [CrossRef]
- Mashchak, C.; Lobo, R.A.; Dozono-Takano, R.; Eggena, P.; Nakamura, R.M.; Brenner, P.F.; Mishell, D.R. Comparison of pharmacodynamic properties of various estrogen formulations. Am. J. Obstet. Gynecol. 1982, 144, 511–518. [Google Scholar] [CrossRef]
- Laurent, M.R.; Helsen, C.; Antonio, L.; Schollaert, D.; Joniau, S.; Vos, M.J.; Decallonne, B.; Hammond, G.; Vanderschueren, D.; Claessens, F. Effects of sex hormone-binding globulin (SHBG) on androgen bioactivity in vitro. Mol. Cell. Endocrinol. 2016, 437, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Davison, S.L.; Bell, R.; Donath, S.; Montalto, J.G.; Davis, S.R. Androgen Levels in Adult Females: Changes with Age, Menopause, and Oophorectomy. J. Clin. Endocrinol. Metab. 2005, 90, 3847–3853. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Branham, W.S.; Ng, H.W.; Moland, C.L.; Dial, S.L.; Fang, H.; Perkins, R.; Sheehan, D.; Tong, W. Human Sex Hormone-Binding Globulin Binding Affinities of 125 Structurally Diverse Chemicals and Comparison with Their Binding to Androgen Receptor, Estrogen Receptor, and α-Fetoprotein. Toxicol. Sci. 2014, 143, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Onishi, T.; Shibahara, T.; Masui, S.; Sugino, Y.; Higashi, S.; Sasaki, T. Efficacy of Ethinylestradiol Re-challenge for Metastatic Castration-resistant Prostate Cancer. Anticancer Res. 2016, 36, 2999–3004. [Google Scholar]
- Jänne, M.; Deol, H.K.; Power, S.G.; Yee, S.P.; Hammond, G.L. Human sex hormone-binding globulin gene expression in transgenic mice. Mol. Endocrinol. 1998, 12, 123–136. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Upper Primer (5′–3′) | Lower Primer (5′–3′) | Species |
|---|---|---|---|
| IL-1β | GAA ATG CCA CCT TTT GAC AGT G | CTG GAT GCT CTC ATC AGG ACA | Mouse |
| Tnf | CCT GTA GCC CAC GTC GTA G | GGG AGT AGA CAA GGT ACA ACC C | Mouse |
| Ccl2 | TTA AAA ACC TGG ATC GGA ACC AA | GCA TTA GCT TCA GAT TTA CGG GT | Mouse |
| Id1 | TAC GAC ATG AAC GGC TGC TA | GTG GTC CCG ACT TCA GAC TC | Mouse |
| Fkbp5 | CAA AGC CTC AGA GTC GTT CC | GGA TTG ACT GCC AAC ACC TT | Mouse |
| Vapa | CAC CAG GGA TTG CTT CAA CT | AGT CGC TTG CAC TCT TCC AT | Mouse |
| Ccrk | GCT CAA AGG TGT TGC GTT TT | GTC AAC GCC CTG GTC ATA CT | Mouse |
| C-Jun | CCT TCT ACG ACG ATG CCC TC | GGT TCA AGG TCA TGC TCT GTT T | Mouse |
| C-Fos | CGG GTT TCA ACG CCG ACT A | TTG GCA CTA GAG ACG GAC AGA | Mouse |
| Cyclin D | GCG TAC CCT GAC ACC AAT CTC | CTC CTC TTC GCA CTT CTG CTC | Mouse |
| Ki67 | ATC ATT GAC CGC TCC TTT AGG T | GCT CGC CTT GAT GGT TCC T | Mouse |
| Ctgf | GGG CCT CTT CTG CGA TTT C | ATC CAG GCA AGT GCA TTG GTA | Mouse |
| Tgfβ1 | GAC GTC ACT GGA GTT GTA CG | GGT TCA TGT CAT GGA TGG TG | Mouse |
| α-Sma | GCT ATT CAG GCT GTG CTG TC | GGT AGT CGG TGA GAT CTC GG | Mouse |
| Col1a1 | ATG TGC CAC TCT GAC TGG AA | TCC ATC GGT CAT GCT CTC TC | Mouse |
| Bim | GAC AGA ACC GCA AGG TAA TCC | ACT TGT CAC AAC TCA TGG GTG | Mouse |
| Rplp0 | GCA GCA GAT CCG CAT GTC GCT CCG | GAG CTG GCA CAG TGA CCT CAC ACG G | Mouse |
| Bax | TGA AGA CAG GGG CCT TTT TG | AAT TCG CCG GAG ACA CTC | Mouse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.R.; Jeong, S.H.; Heo, J.H.; Jo, S.L.; Ko, J.-W.; Kwun, H.-J.; Hong, E.-J. Dietary Intake of 17α-Ethinylestradiol Promotes HCC Progression in Humanized Male Mice Expressing Sex Hormone-Binding Globulin. Int. J. Mol. Sci. 2021, 22, 12557. https://doi.org/10.3390/ijms222212557
Lee SR, Jeong SH, Heo JH, Jo SL, Ko J-W, Kwun H-J, Hong E-J. Dietary Intake of 17α-Ethinylestradiol Promotes HCC Progression in Humanized Male Mice Expressing Sex Hormone-Binding Globulin. International Journal of Molecular Sciences. 2021; 22(22):12557. https://doi.org/10.3390/ijms222212557
Chicago/Turabian StyleLee, Sang R., Su Hee Jeong, Jun H. Heo, Seong Lae Jo, Je-Won Ko, Hyo-Jung Kwun, and Eui-Ju Hong. 2021. "Dietary Intake of 17α-Ethinylestradiol Promotes HCC Progression in Humanized Male Mice Expressing Sex Hormone-Binding Globulin" International Journal of Molecular Sciences 22, no. 22: 12557. https://doi.org/10.3390/ijms222212557
APA StyleLee, S. R., Jeong, S. H., Heo, J. H., Jo, S. L., Ko, J.-W., Kwun, H.-J., & Hong, E.-J. (2021). Dietary Intake of 17α-Ethinylestradiol Promotes HCC Progression in Humanized Male Mice Expressing Sex Hormone-Binding Globulin. International Journal of Molecular Sciences, 22(22), 12557. https://doi.org/10.3390/ijms222212557