
Genome-Wide Discovery of miRNAs with Differential Expression Patterns in Responses to Salinity in the Two Contrasting Wheat Cultivars





Abstract
:1. Introduction
2. Results
2.1. Total ATPase Activity and Total Amino Acid and Soluble Sugar Contents
2.2. MicroRNAs Profiling Response to Salinity Stress
2.3. Identification of Known and Novel miRNAs
2.4. Evolutionary Conservation Analysis of Known miRNAs Families
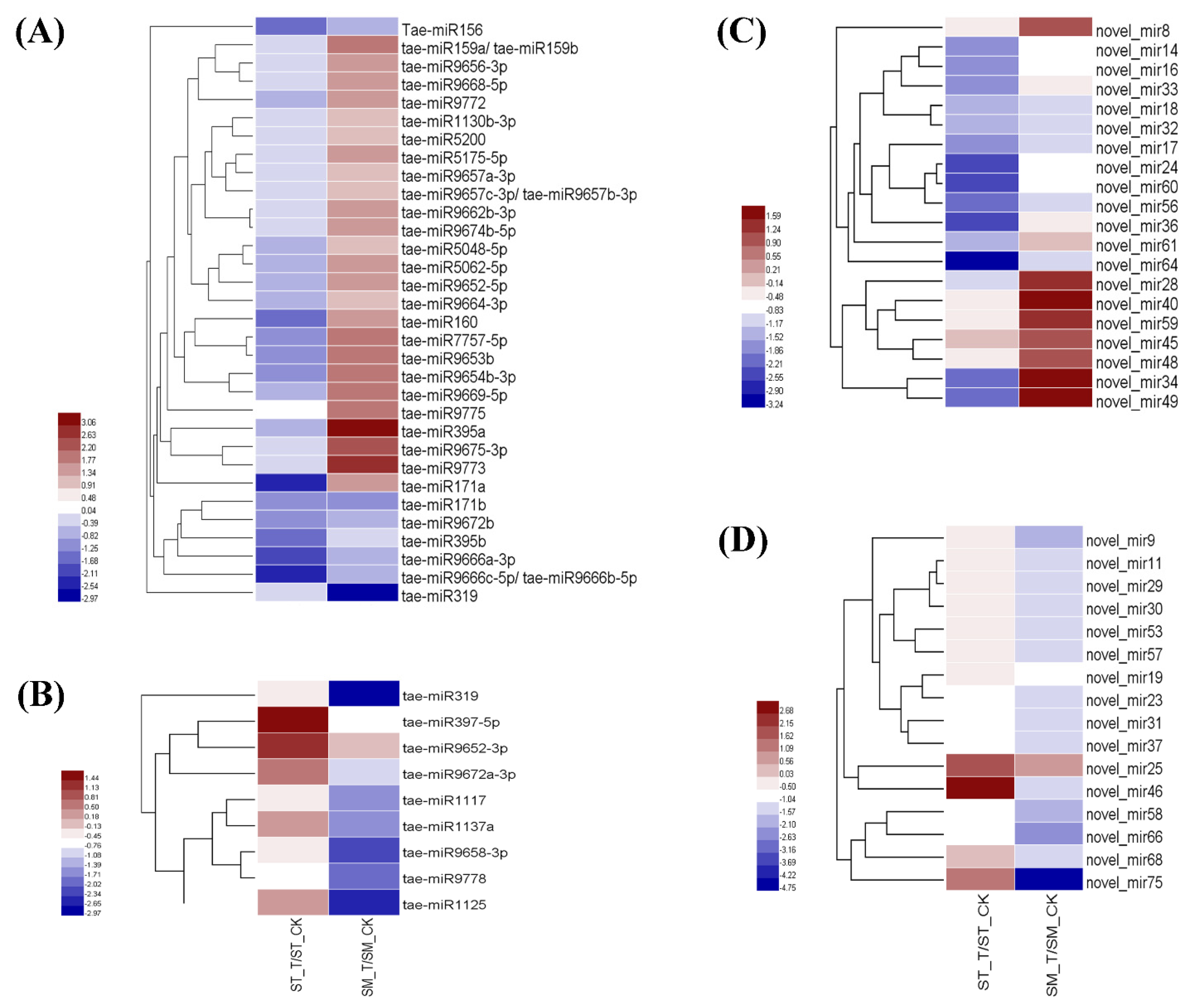
2.5. Identification of miRNAs Related to Salt Stress
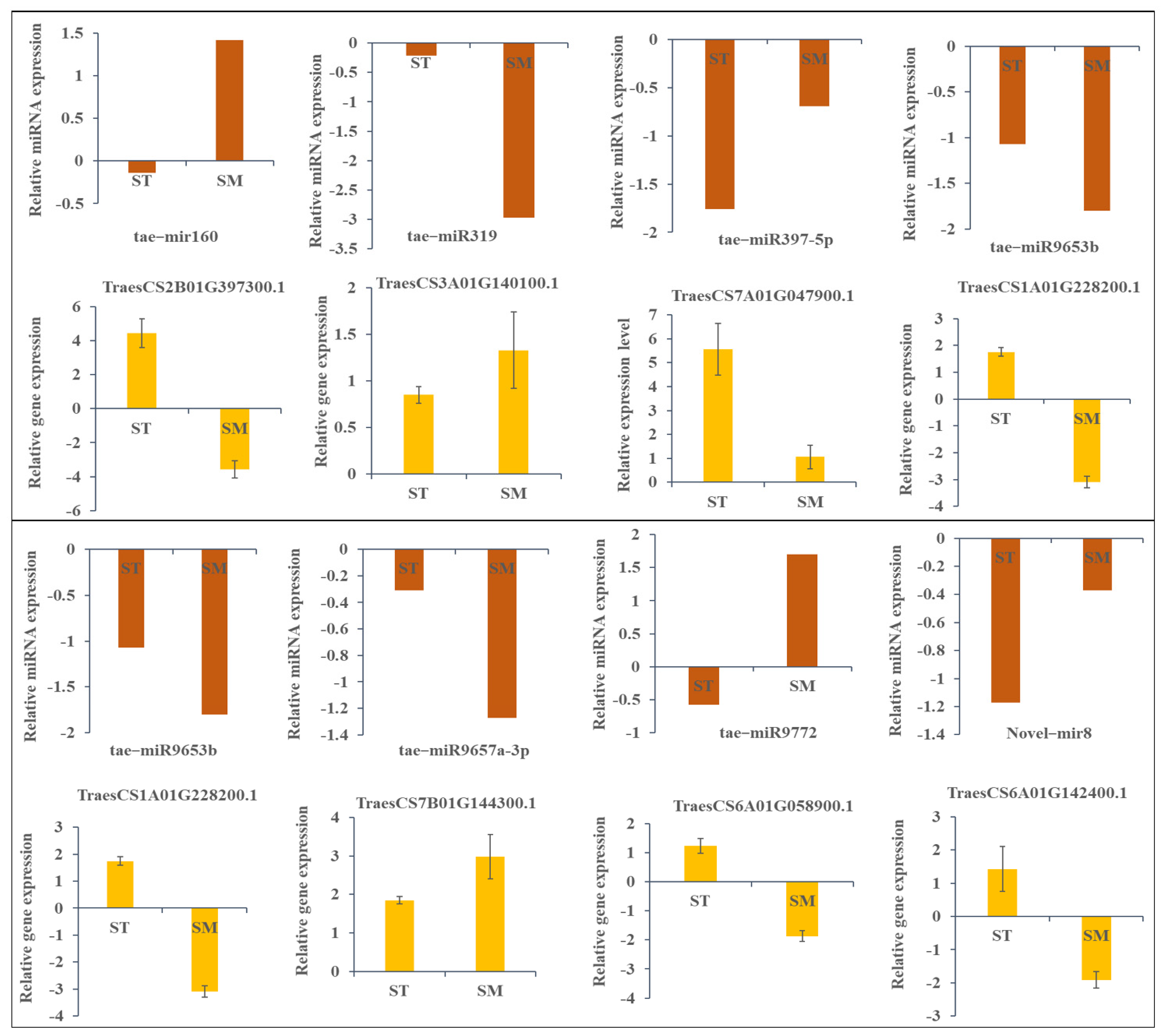
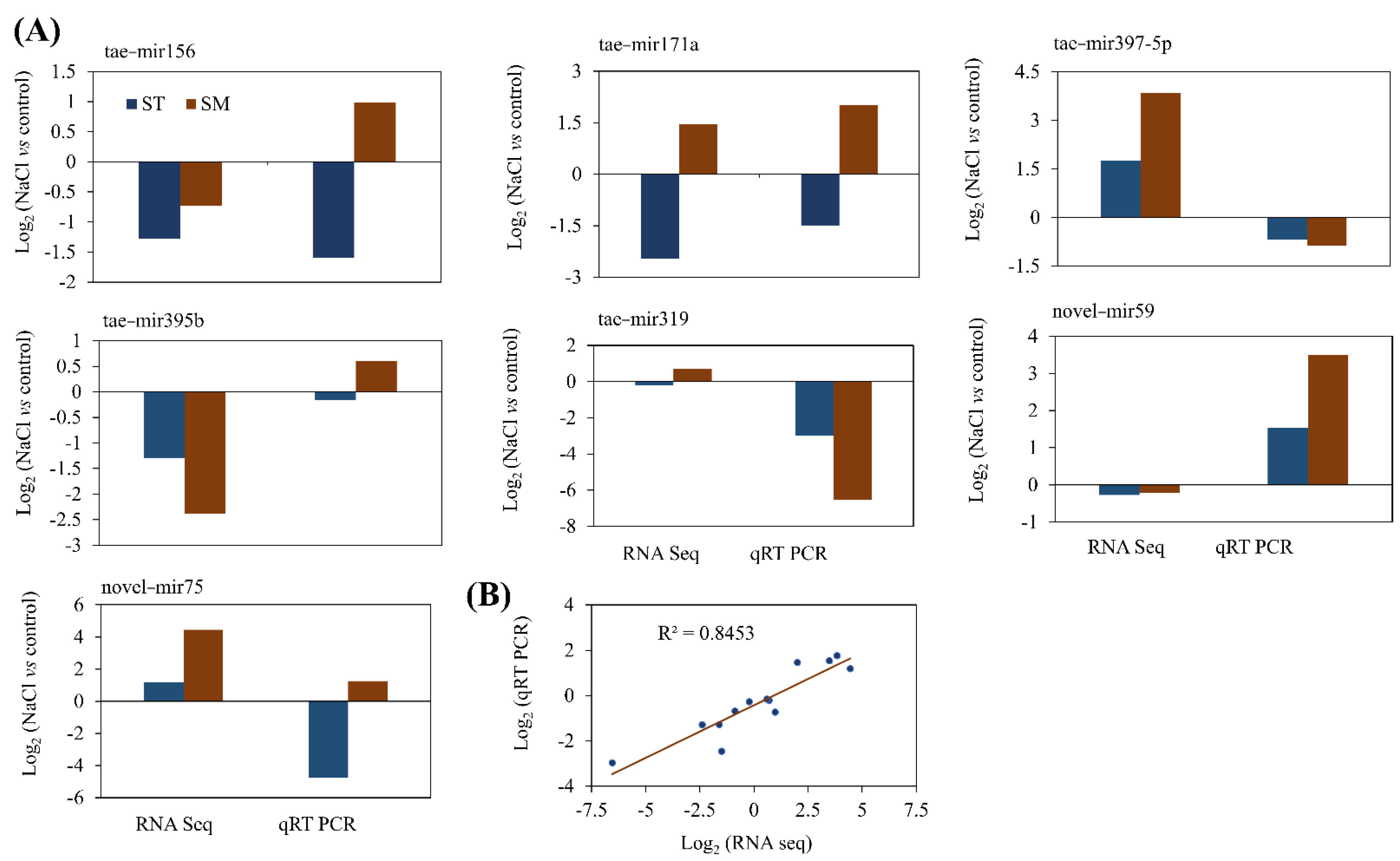
2.6. Validation of Targets mRNA and miRNA with qRT-PCR
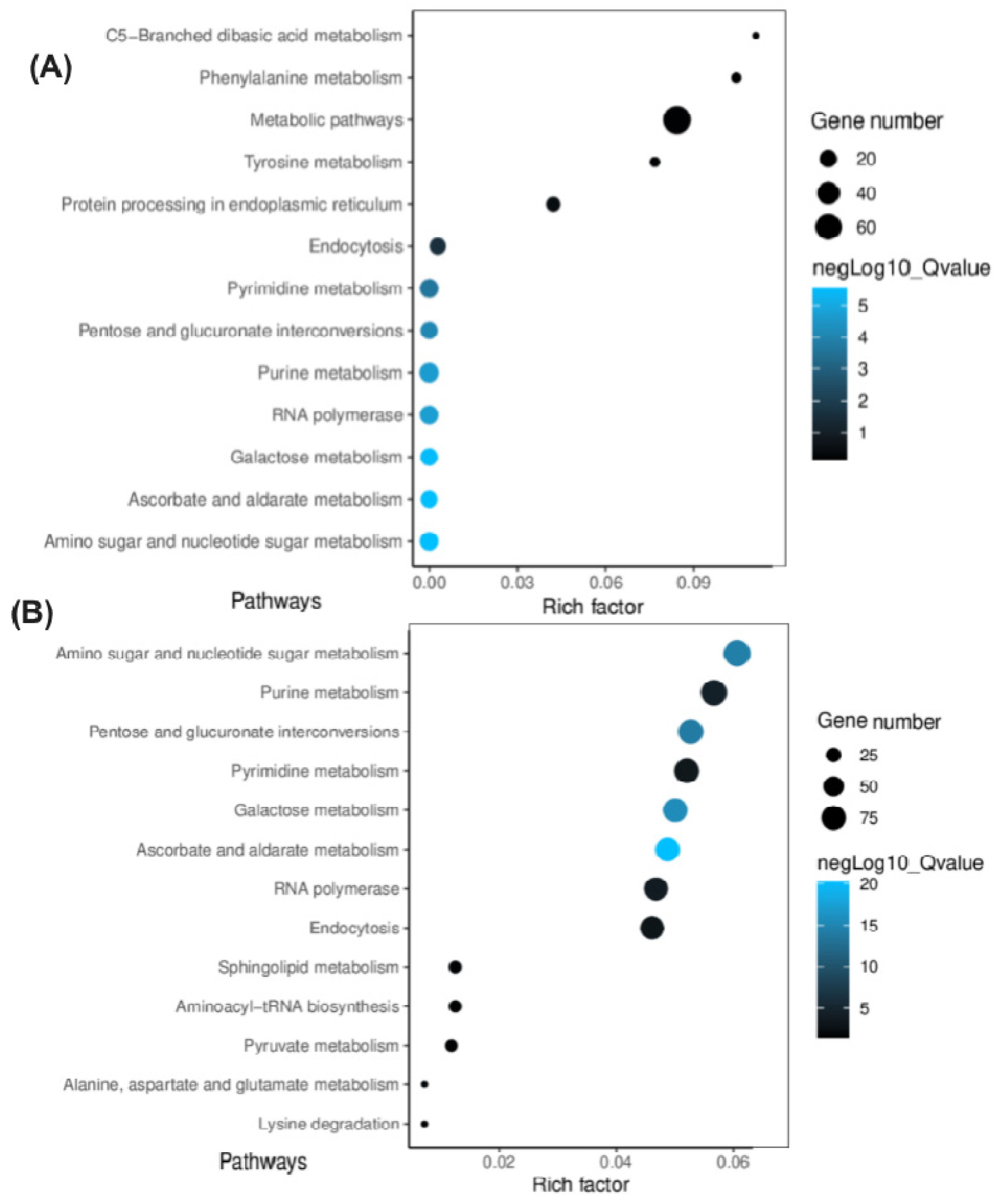
2.7. Prediction of Target Genes and Their Associated Regulatory Pathways
3. Discussion
3.1. Effect of Salinity on Total ATPase Activity, Total Amino Acids and Soluble Sugar Contents
3.2. tae-miR156, tae-miR160 and tae-miR171a/b May Confer Salt Tolerance in Roots of Suntop
3.3. miR319/PCF5 Module May Regulate Salt Tolerance in Suntop
3.4. miR159a-b, and miR9657 May Regulate Salt Tolerance in Suntop
3.5. Novel miRNAs Exclusively Expressed in Suntop
4. Materials and Methods
4.1. Plant Materials and Experimental Design
4.2. Determination of Total Soluble Sugars
4.3. Determination of Total Amino Acids Contents and ATPase Activity
4.4. Small RNA Library Construction and High Throughput Sequencing
4.5. Data Pre-Processing and De Novo Assembly
4.6. Differential Expression of Salinity-Responsive miRNAs
4.7. Target Gene Prediction and GO and KEGG Enrichment Analysis
4.8. mRNAs and miRNAs Validation by qRT-PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Meng, X.; Dobrovolskaya, O.B.; Orlov, Y.L.; Chen, M. Non-coding RNAs and their roles in stress response in plants. Genom. Proteom. Bioinform. 2017, 15, 301–312. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhu, J.-K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [Green Version]
- Manavella, P.A.; Yang, S.W.; Palatnik, J. Keep calm and carry on: miRNA biogenesis under stress. Plant J. 2019, 99, 832–843. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Reinhart, B.J.; Bartel, D.P.; Zamore, P.D. A biochemical framework for RNA silencing in plants. Genes Dev. 2003, 17, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006, 38, S31–S36. [Google Scholar] [CrossRef]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D.G. A Plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Sci. 2006, 312, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Chen, X. MicroRNA biogenesis and function in plants. FEBS Lett. 2005, 579, 5923–5931. [Google Scholar] [CrossRef] [Green Version]
- Kuang, L.; Shen, Q.; Wu, L.; Yu, J.; Fu, L.; Wu, D.; Zhang, G. Identification of microRNAs responding to salt stress in barley by high-throughput sequencing and degradome analysis. Environ. Exp. Bot. 2019, 160, 59–70. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, G.; Sutoh, K.; Zhu, J.K.; Zhang, W. Identification of cold-inducible microRNAs in plants by transcriptome analysis. Biochim. Biophys. Acta—Gene Regul. Mech. 2008, 1779, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Liang, R.; Ge, L.; Li, W.; Xiao, H.; Lin, H.; Ruan, K.; Jin, Y. Identification of drought-induced microRNAs in rice. Biochem. Biophys. Res. Commun. 2007, 354, 585–590. [Google Scholar] [CrossRef]
- Yu, J.; Wu, L.; Fu, L.; Shen, Q.; Kuang, L.; Wu, D.; Zhang, G. Genotypic difference of cadmium tolerance and the associated microRNAs in wild and cultivated barley. Plant Growth Regul. 2019, 87, 389–401. [Google Scholar] [CrossRef]
- Cui, N.; Sun, X.; Sun, M.; Jia, B.; Duanmu, H.; Lv, D.; Duan, X.; Zhu, Y. Overexpression of OsmiR156k leads to reduced tolerance to cold stress in rice (Oryza Sativa). Mol. Breed. 2015, 35, 214. [Google Scholar] [CrossRef]
- Zhang, X.; Zou, Z.; Gong, P.; Zhang, J.; Ziaf, K.; Li, H.; Xiao, F.; Ye, Z. Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol. Lett. 2011, 33, 403–409. [Google Scholar] [CrossRef]
- Sunkar, R. MicroRNAs with macro-effects on plant stress responses. Semin. Cell Dev. Biol. 2010, 21, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Wang, J.; Wang, T.; Wei, L.; Li, J.; Liu, L. Identification of rapeseed microRNAs Involved in early stage seed germination under salt and drought stresses. Front. Plant Sci. 2016, 7, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, M.; Bonnett, D.; Chapman, S.C.; Furbank, R.T.; Manès, Y.; Mather, D.E.; Parry, M.A.J. Raising yield potential of wheat. I. Overview of a consortium approach and breeding strategies. J. Exp. Bot. 2011, 62, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO. World Food Situation, FAO Cereal Supply and Demand Brief. 8 October 2010. Available online: http://www.fao.org/worldfoodsituation/csdb/en/ (accessed on 18 October 2021).
- Munns, R.; James, R.A.; Läuchli, A.; Lauchli, A. Approaches to increasing the salt tolerance of wheat and other cereals. J. Exp. Bot. 2006, 57, 1025–1043. [Google Scholar] [CrossRef] [Green Version]
- Kruszka, K.; Pieczynski, M.; Windels, D.; Bielewicz, D.; Jarmolowski, A.; Szweykowska-Kulinska, Z.; Vazquez, F. Role of microRNAs and other sRNAs of plants in their changing environments. J. Plant Physiol. 2012, 169, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Bita, C.E.; Gerats, T. Plant tolerance to high temperature in a changing environment: Scientific fundamentals and production of heat stress-tolerant crops. Front. Plant Sci. 2013, 4, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; He, F.; Liu, F.; Zheng, X.; Di, C.; Zhou, S.; Gu, H.; Xu, W.; Su, Z. Integration of Cold Signal Transduction Pathway Related to ABA 8′-hydroxylase in Arabidopsis BT—Biotechnology and Sustainable Agriculture 2006 and Beyond; Xu, Z., Li, J., Xue, Y., Yang, W., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 471–474. [Google Scholar]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genomics 2015, 15, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Sun, Y.F.; Song, N.; Wang, X.J.; Feng, H.; Huang, L.L.; Kang, Z.S. Identification of UV-B-induced microRNAs in wheat. Genet. Mol. Res. 2013, 12, 4213–4221. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Wang, L.; Cui, L.; Feng, K.; Liu, F.; Du, X.; Tong, W.; Nie, X.; Ji, W.; Weining, S. Global identification of microRNAs and their targets in barley under salinity stress. PLoS ONE 2015, 10, e0137990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wang, T.; Zhang, Y.; Li, Y. Overexpression of soybean miR172c confers tolerance to water deficit and salt stress, but increases ABA sensitivity in transgenic Arabidopsis thaliana. J. Exp. Bot. 2016, 67, 175–194. [Google Scholar] [CrossRef] [Green Version]
- FAO. FAOSTAT. 2019. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 18 October 2021).
- Zeeshan, M.; Lu, M.; Sehar, S.; Holford, P.; Wu, F. Comparison of biochemical, anatomical, morphological, and physiological responses to salinity stress in wheat and barley genotypes deferring in salinity tolerance. Agronomy 2020, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.-H.; Park, S.; Zhai, J.; Gurazada, S.G.R.; De Paoli, E.; Meyers, B.C.; Green, P.J. Massive analysis of rice small RNAs: Mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 2011, 23, 4185–4207. [Google Scholar] [CrossRef] [Green Version]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Montes, R.A.; de Fátima Rosas-Cárdenas, F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; Marsch-Martínez, N.; Meyers, B.C.; Green, P.J.; de Folter, S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef]
- AMBROS, V. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirdar Mansuri, R.; Shobbar, Z.-S.; Babaeian Jelodar, N.; Ghaffari, M.R.; Nematzadeh, G.-A.; Asari, S. Dissecting molecular mechanisms underlying salt tolerance in rice: A comparative transcriptional profiling of the contrasting genotypes. Rice 2019, 12, 13. [Google Scholar] [CrossRef]
- Xie, K.; Wu, C.; Xiong, L. Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 2006, 142, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; van Staden, J. Partitioning of carbohydrates in salt-sensitive and salt-tolerant soybean callus cultures under salinity stress and its subsequent relief. Plant Growth Regul. 2001, 33, 13–17. [Google Scholar] [CrossRef]
- Bukhari, S.A.H.; Shang, S.; Zhang, M.; Zheng, W.; Zhang, G.; Wang, T.-Z.; Shamsi, I.H.; Wu, F. Genome-wide identification of chromium stress-responsive micro RNAs and their target genes in tobacco (Nicotiana tabacum) roots. Environ. Toxicol. Chem. 2015, 34, 2573–2582. [Google Scholar] [CrossRef]
- Tang, J.; Chu, C. MicroRNAs in crop improvement: Fine-tuners for complex traits. Nat. Plants 2017, 3, 17077. [Google Scholar] [CrossRef]
- Li, C.; Lu, S. Molecular characterization of the SPL gene family in Populus trichocarpa. BMC Plant Biol. 2014, 14, 131. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Hao, M.; Wang, W.; Mei, D.; Tong, C.; Wang, H.; Liu, J.; Fu, L.; Hu, Q. Genomic identification, characterization and differential expression analysis of SBP-box gene family in Brassica napus. BMC Plant Biol. 2016, 16, 196. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.-D.; Yu, L.-J.; Li, Z.-J.; Yan, Y.; Han, R.; Liu, H.; Ma, M. Genome-wide analysis of the SPL family transcription factors and their responses to abiotic stresses in maize. Plant Gene 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.K.; Goel, R.; Kumari, S.; Dahuja, A. Genomic organization, phylogenetic comparison, and expression profiles of the SPL family genes and their regulation in soybean. Dev. Genes Evol. 2017, 227, 101–119. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Xue, D.; Wang, J.; Yan, M.; Liu, G.; Dong, G.; Zeng, D.; Lu, Z.; Zhu, X.; et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010, 42, 541–544. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Kohli, A.; Sreenivasulu, N.; Lakshmanan, P.; Kumar, P.P. The phytohormone crosstalk paradigm takes center stage in understanding how plants respond to abiotic stresses. Plant Cell Rep. 2013, 32, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Sharma, E.; Sharma, R.; Borah, P.; Jain, M.; Khurana, J.P. Emerging Roles of Auxin in Abiotic Stress Responses BT—Elucidation of Abiotic Stress Signaling in Plants: Functional Genomics Perspectives; Pandey, G.K., Ed.; Springer: New York, NY, USA, 2015; pp. 299–328. Volume 1, ISBN 978-1-4939-2211-6. [Google Scholar]
- Ludwig-Müller, J. Auxin conjugates: Their role for plant development and in the evolution of land plants. J. Exp. Bot. 2011, 62, 1757–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenzio, D.L.; Wysocka-Diller, J.; Malamy, J.E.; Pysh, L.; Helariutta, Y.; Freshour, G.; Hahn, M.G.; Feldmann, K.A.; Benfey, P.N. The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the Arabidopsis root. Cell 1996, 86, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Li, D.; Li, Z.; Hu, Q.; Yang, C.; Zhu, L.; Luo, H. Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol. 2013, 161, 1375–1391. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, D.; Yan, J.; Wang, K.; Luo, H.; Zhang, W. MiR319 mediated salt tolerance by ethylene. Plant Biotechnol. J. 2019, 17, 2370–2383. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, B.; Shen, Y.; Wang, H.; Feng, Q.; Shi, H. The Arabidopsis RNA binding protein with K homology motifs, SHINY1, interacts with the C-terminal domain phosphatase-like 1 (CPL1) to repress stress-inducible gene expression. PloS Genet. 2013, 9, e1003625. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [Green Version]
- Zeeshan, M.; Lu, M.; Naz, S.; Sehar, S.; Cao, F.; Wu, F. Resemblance and difference of seedling metabolic and transporter gene expression in high tolerance wheat and barley cultivars in response to salinity stress. Plants 2020, 9, 519. [Google Scholar] [CrossRef]
- Chen, Y.H.; Cao, Y.Y.; Wang, L.J.; Li, L.M.; Yang, J.; Zou, M.X. Identification of MYB transcription factor genes and their expression during abiotic stresses in maize. Biol. Plant. 2018, 62, 222–230. [Google Scholar] [CrossRef]
- Dong, W.; Liu, X.; Li, D.; Gao, T.; Song, Y. Transcriptional profiling reveals that a MYB transcription factor MsMYB4 contributes to the salinity stress response of alfalfa. PLoS ONE 2018, 13, e0204033. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Lv, J.; Qiu, N.; Bai, Y.; Yang, N.; Dong, W. The constitutive expression of alfalfa MsMYB2L enhances salinity and drought tolerance of Arabidopsis thaliana. Plant Physiol. Biochem. PPB 2019, 141, 300–305. [Google Scholar] [CrossRef]
- Hajiebrahimi, A.; Owji, H.; Hemmati, S. Genome-wide identification, functional prediction, and evolutionary analysis of the R2R3-MYB superfamily in Brassica napus. Genome 2017, 60, 797–814. [Google Scholar] [CrossRef]
- He, Y.; Dong, Y.; Yang, X.; Guo, D.; Qian, X.; Yan, F.; Wang, Y.; Li, J.; Wang, Q. Functional activation of a novel R2R3-MYB protein gene, GmMYB68, confers salt-alkali resistance in soybean (Glycine max L.). Genome 2020, 63, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Batistic, O.; Kudla, J. Integration and channeling of calcium signaling through the CBL calcium sensor/CIPK protein kinase network. Planta 2004, 219, 915–924. [Google Scholar] [CrossRef]
- Kolukisaoglu, U.; Weinl, S.; Blazevic, D.; Batistic, O.; Kudla, J. Calcium sensors and their interacting protein kinases: Genomics of the Arabidopsis and rice CBL-CIPK signaling networks. Plant Physiol. 2004, 134, 43–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Ding, S.; Zhang, H.; Du, H.; An, L. CIPK7 is involved in cold response by interacting with CBL1 in Arabidopsis thaliana. Plant Sci. 2011. [Google Scholar] [CrossRef]
- Quan, R.; Lin, H.; Mendoza, I.; Zhang, Y.; Cao, W.; Yang, Y.; Shang, M.; Chen, S.; Pardo, J.M.; Guo, Y. SCABP8/CBL10, a putative calcium sensor, interacts with the protein kinase SOS2 to protect Arabidopsis shoots from salt stress. Plant Cell 2007, 19, 1415–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Q.-S.; Guo, Y.; Quintero, F.J.; Pardo, J.M.; Schumaker, K.S.; Zhu, J.-K. Regulation of Vacuolar Na + /H + Exchange in Arabidopsis thaliana by the Salt-Overly-Sensitive (SOS) Pathway. J. Biol. Chem. 2004, 279, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Hu, W.; Wei, S.; Zhou, S.; Zhang, F.; Han, J.; Chen, L.; Li, Y.; Feng, J.; Fang, B.; et al. TaCIPK29, a CBL-Interacting protein kinase gene from wheat, confers salt stress tolerance in transgenic tobacco. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.J.; Li, H.Z.; Zhou, W.J.; Takeuchi, Y.; Yoneyama, K. Effect of 5-Aminolevulinic acid on development and salt tolerance of potato (Solanum tuberosum L.) microtubers in vitro. Plant Growth Regul. 2006, 49, 27–34. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Schatz, M.C.; Lin, J.; Pop, M.; Salzberg, S.L. Searching for SNPs with cloud computing. Genome Biol. 2009, 10, R134. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.; Huttner, M.; Dueck, A.; Meister, G.; Engelmann, J.C. miRA: Adaptable novel miRNA identification in plants using small RNA sequencing data. BMC Bioinformatics 2015, 16, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
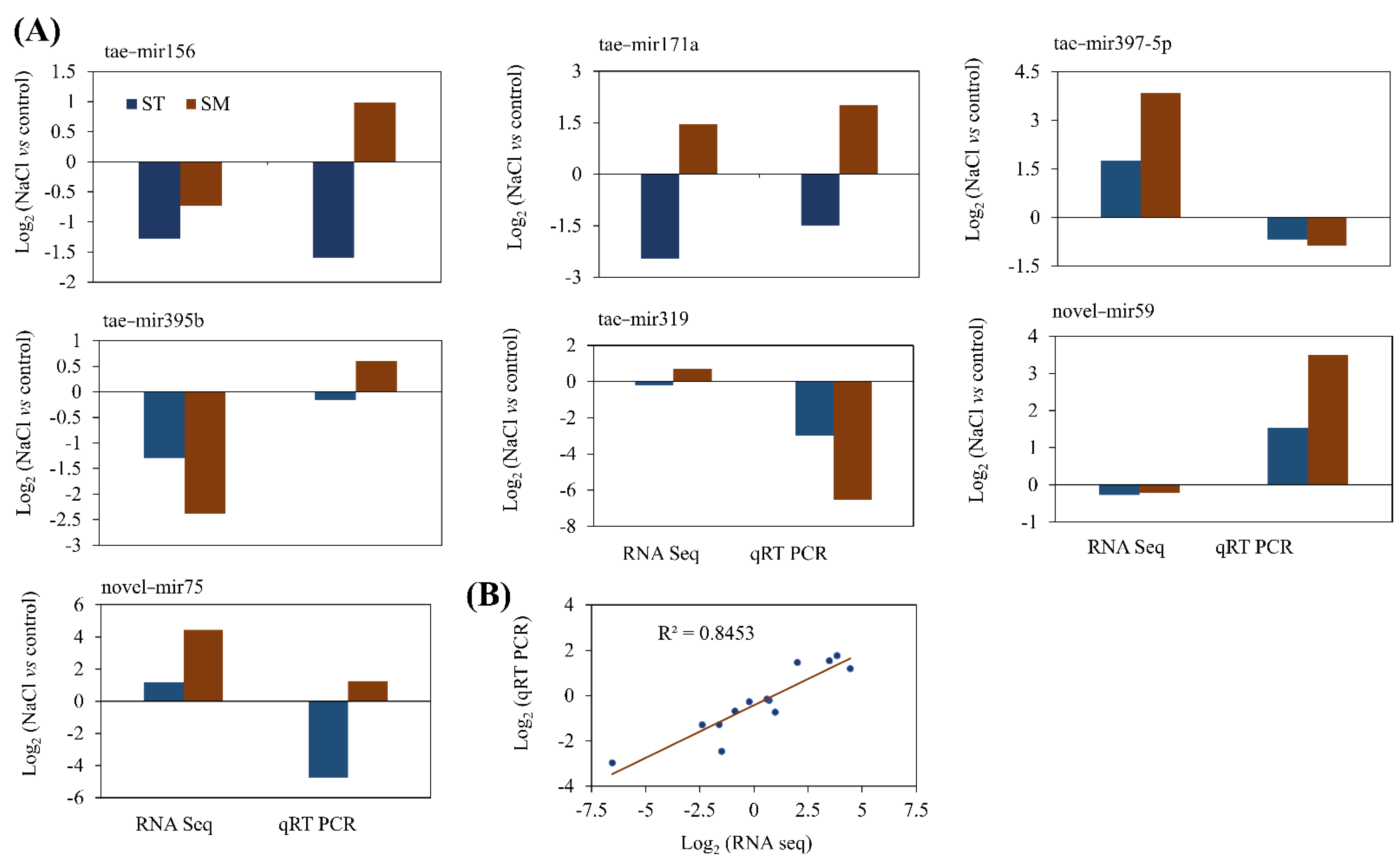{kind=link}
{kind=link}
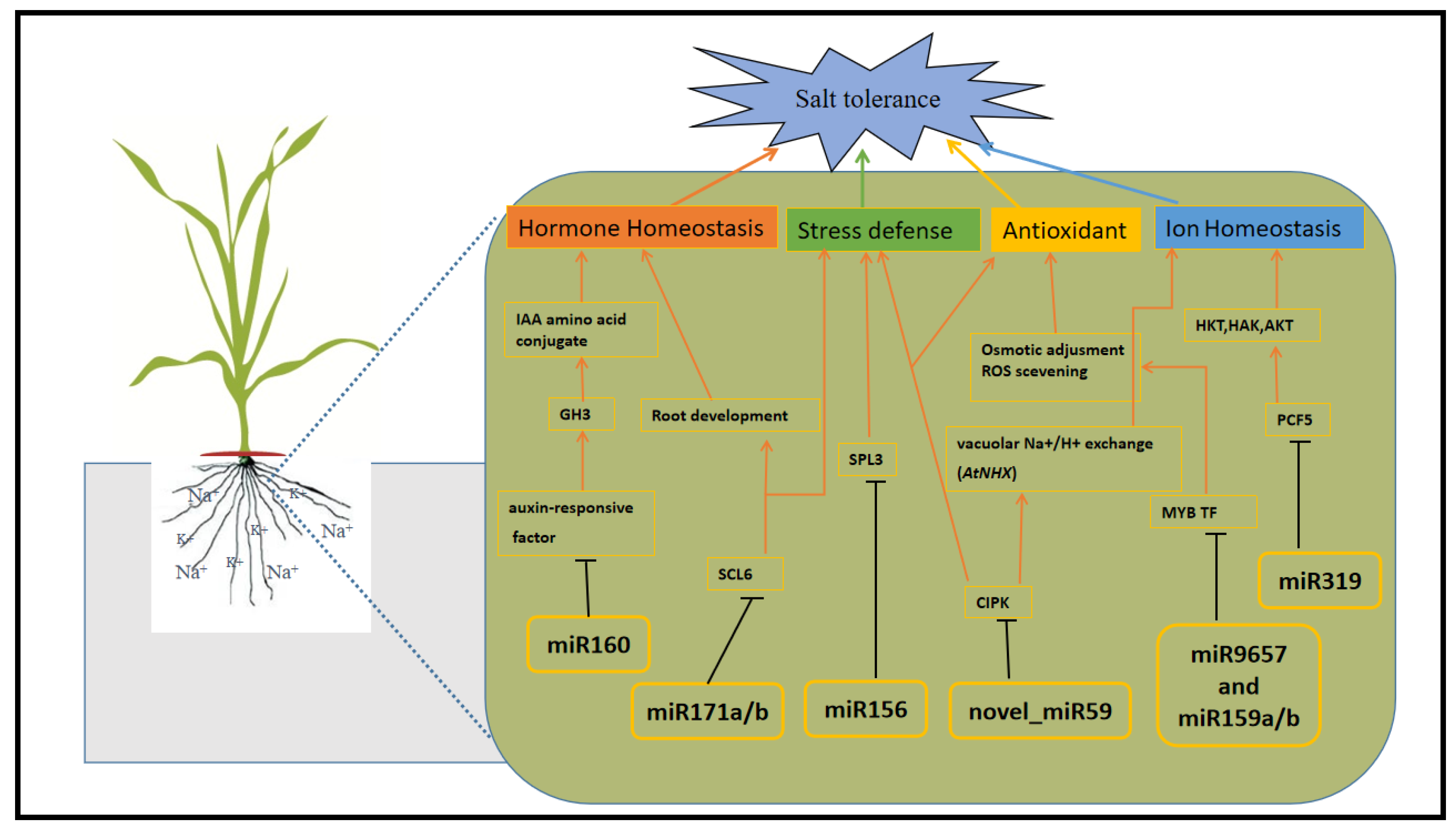{kind=link}
| Library Type | Suntop | Suntop | Sunmate | Sunmate |
|---|---|---|---|---|
| Control | NaCl | Control | NaCl | |
| Total raw reads | 29,114,838.5 | 28,580,752.5 | 28,973,938.5 | 29,753,197 |
| Total clean reads | 27,175,593.5 | 26,292,555 | 25,900,067 | 25,819,464 |
| Clean tag (%) | 95.08 | 90.31 | 89.39 | 86.77 |
| Q20 of clean tag (%) | 99.20 | 99 | 99.30 | 99.30 |
| Mapped reads | 24,908,414 (91.66%) | 24,352,075.5 (92.62%) | 24,230,503.5 (93.55%) | 22,175,264 (85.89%) |
| Total miRNAs reads | 4,180,023 | 5,014,878.5 | 5,127,462.5 | 5,644,374.5 |
| Known miRNAs reads | 529,287 | 452,384.5 | 627,551 | 242,607 |
| Novel miRNAs reads | 3,650,736 | 4,562,494 | 4,499,911.5 | 5,401,767.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeeshan, M.; Qiu, C.-W.; Naz, S.; Cao, F.; Wu, F. Genome-Wide Discovery of miRNAs with Differential Expression Patterns in Responses to Salinity in the Two Contrasting Wheat Cultivars. Int. J. Mol. Sci. 2021, 22, 12556. https://doi.org/10.3390/ijms222212556
Zeeshan M, Qiu C-W, Naz S, Cao F, Wu F. Genome-Wide Discovery of miRNAs with Differential Expression Patterns in Responses to Salinity in the Two Contrasting Wheat Cultivars. International Journal of Molecular Sciences. 2021; 22(22):12556. https://doi.org/10.3390/ijms222212556
Chicago/Turabian StyleZeeshan, Muhammad, Cheng-Wei Qiu, Shama Naz, Fangbin Cao, and Feibo Wu. 2021. "Genome-Wide Discovery of miRNAs with Differential Expression Patterns in Responses to Salinity in the Two Contrasting Wheat Cultivars" International Journal of Molecular Sciences 22, no. 22: 12556. https://doi.org/10.3390/ijms222212556