
Obstructive Sleep Apnea as an Acceleration Trigger of Cellular Senescence Processes through Telomere Shortening

 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sleep Quality and Telomere Length
3. OSA and Telomere Length
3.1. Telomere Lengthening in OSA
3.2. Telomere Shortening in OSA
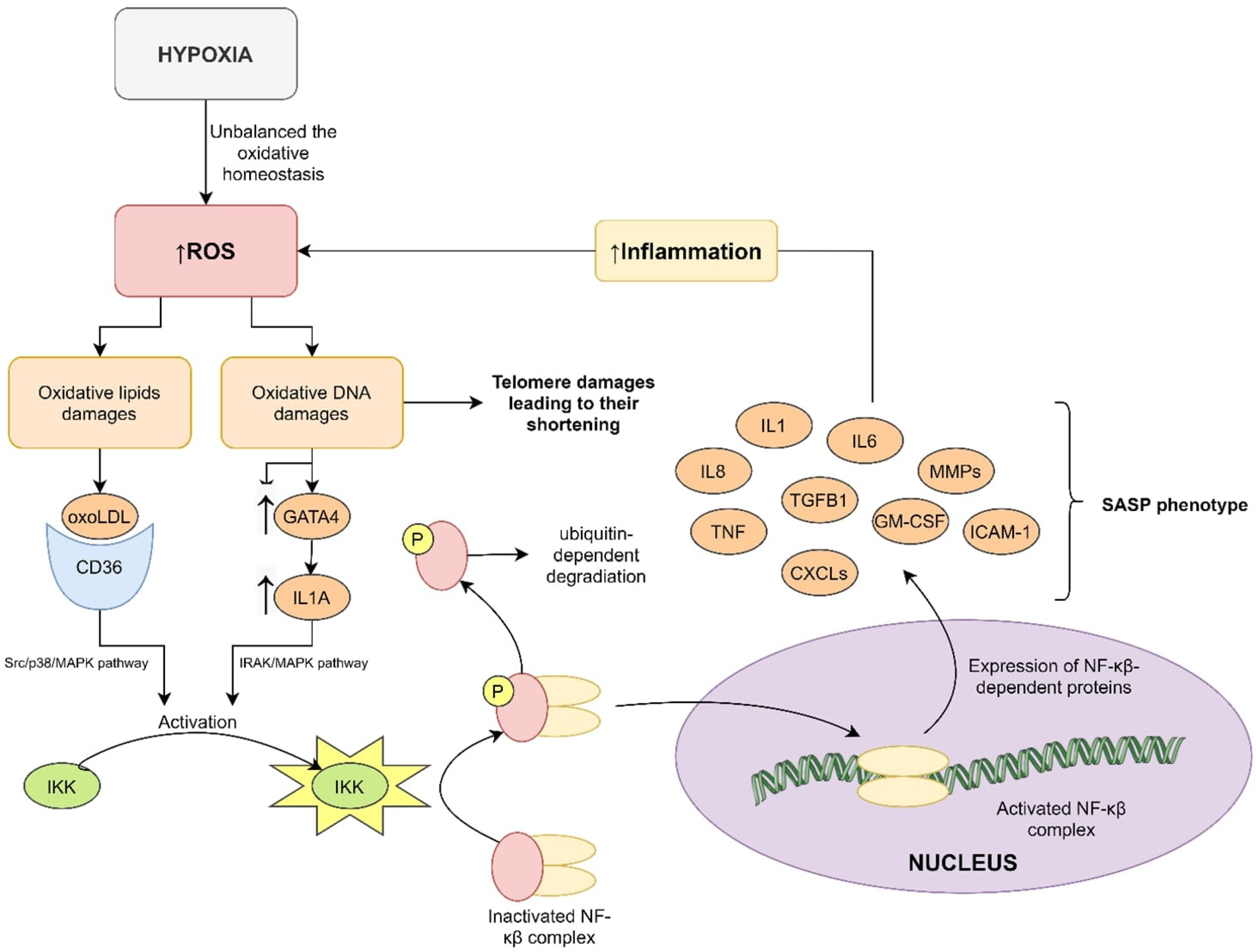
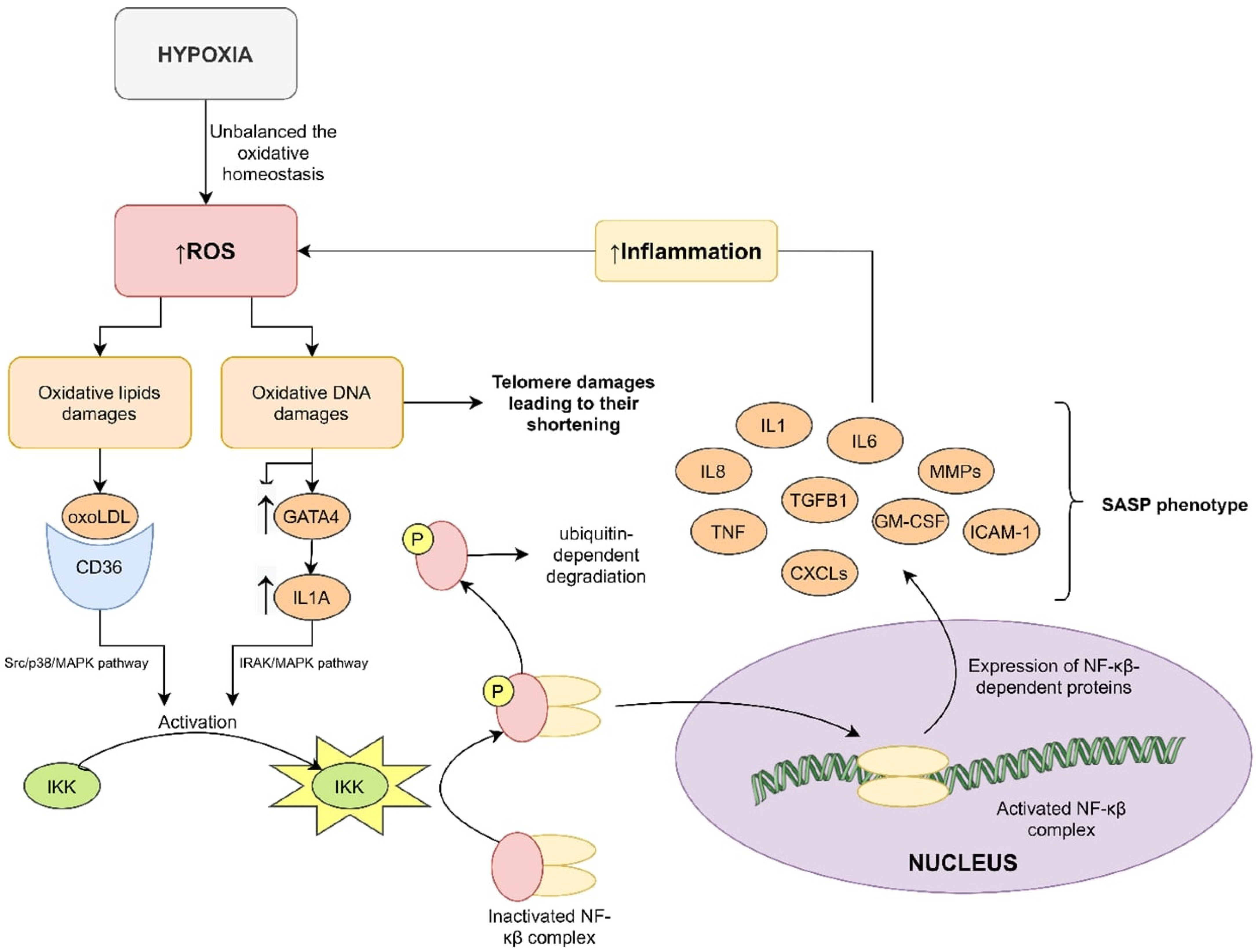
4. Mechanism of OSA Influence of Cellular Senescence
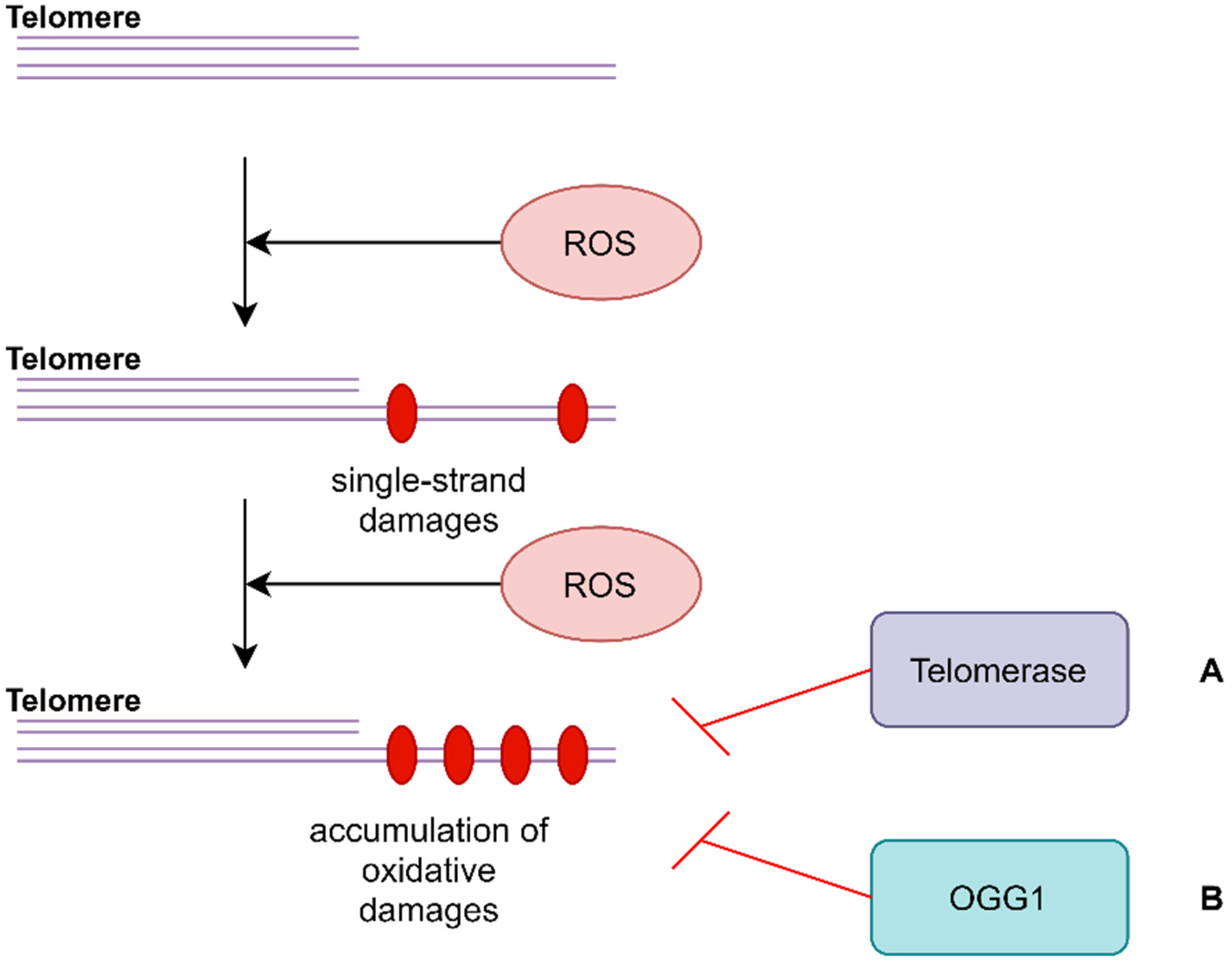
4.1. Oxidative Stress and Telomere Length
4.2. Inflammation and Telomere Length
4.3. Hypoxia and Telomere Length
5. Circadian Clock Disturbances and Accelerated Aging
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Lévy, P.; Kohler, M.; McNicholas, W.T.; Barbé, F.; McEvoy, R.D.; Somers, V.K.; Lavie, L.; Pépin, J.L. Obstructive sleep apnoea syndrome. Nat. Rev. Dis. Primers 2015, 1, 15015. [Google Scholar] [CrossRef]
- Mokros, Ł.; Kuczynski, W.; Gabryelska, A.; Franczak, Ł.; Spałka, J.; Białasiewicz, P. High Negative Predictive Value of Normal Body Mass Index for Obstructive Sleep Apnea in the Lateral Sleeping Position. J. Clin. Sleep Med. 2018, 14, 985–990. [Google Scholar] [CrossRef]
- Bradley, T.D.; Floras, J.S. Obstructive sleep apnoea and its cardiovascular consequences. Lancet 2009, 373, 82–93. [Google Scholar] [CrossRef]
- Gabryelska, A.; Łukasik, Z.M.; Makowska, J.S.; Białasiewicz, P. Obstructive Sleep Apnea: From Intermittent Hypoxia to Cardiovascular Complications via Blood Platelets. Front. Neurol. 2018, 9, 635. [Google Scholar] [CrossRef]
- Gabryelska, A.; Karuga, F.F.; Szmyd, B.; Białasiewicz, P. HIF-1α as a Mediator of Insulin Resistance, T2DM, and Its Complications: Potential Links with Obstructive Sleep Apnea. Front. Physiol. 2020, 11, 1035. [Google Scholar] [CrossRef] [PubMed]
- Gabryelska, A.; Chrzanowski, J.; Sochal, M.; Kaczmarski, P.; Turkiewicz, S.; Ditmer, M.; Karuga, F.F.; Czupryniak, L.; Białasiewicz, P. Nocturnal Oxygen Saturation Parameters as Independent Risk Factors for Type 2 Diabetes Mellitus among Obstructive Sleep Apnea Patients. J. Clin. Med. 2021, 10, 3770. [Google Scholar] [CrossRef] [PubMed]
- Hunyor, I.; Cook, K.M. Models of intermittent hypoxia and obstructive sleep apnea: Molecular pathways and their contribution to cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R669–R687. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Ji, S. Cellular senescence: Molecular mechanisms and pathogenicity. J. Cell. Physiol. 2018, 233, 9121–9135. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Evans, M.K. Techniques to Induce and Quantify Cellular Senescence. J. Vis. Exp. 2017, 123, e55533. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell. Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, K.J.; Vasu, V.; Griffin, D.K. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mander, B.A.; Winer, J.R.; Walker, M.P. Sleep and Human Aging. Neuron 2017, 94, 19–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prather, A.A.; Puterman, E.; Lin, J.; O’Donovan, A.; Krauss, J.; Tomiyama, A.J.; Epel, E.S.; Blackburn, E.H. Shorter leukocyte telomere length in midlife women with poor sleep quality. J. Aging Res. 2011, 2011, 721390. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Schernhammer, E.; Qi, L.; Gao, X.; De Vivo, I.; Han, J. Associations between rotating night shifts, sleep duration, and telomere length in women. PLoS ONE 2011, 6, e23462. [Google Scholar] [CrossRef] [Green Version]
- Cribbet, M.R.; Carlisle, M.; Cawthon, R.M.; Uchino, B.N.; Williams, P.G.; Smith, T.W.; Gunn, H.E.; Light, K.C. Cellular aging and restorative processes: Subjective sleep quality and duration moderate the association between age and telomere length in a sample of middle-aged and older adults. Sleep 2014, 37, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Jackowska, M.; Hamer, M.; Carvalho, L.A.; Erusalimsky, J.D.; Butcher, L.; Steptoe, A. Short sleep duration is associated with shorter telomere length in healthy men: Findings from the Whitehall II cohort study. PLoS ONE 2012, 7, e47292. [Google Scholar] [CrossRef] [Green Version]
- Garland, S.N.; Palmer, C.; Donelson, M.; Gehrman, P.; Johnson, F.B.; Mao, J.J. A nested case-controlled comparison of telomere length and psychological functioning in breast cancer survivors with and without insomnia symptoms. Rejuvenation Res. 2014, 17, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, S.; Bhattacharjee, R.; Khalyfa, A.; Kheirandish-Gozal, L.; Gozal, D. Leukocyte telomere length and plasma catestatin and myeloid-related protein 8/14 concentrations in children with obstructive sleep apnea. Chest 2010, 138, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Polonis, K.; Sompalli, S.; Becari, C.; Xie, J.; Covassin, N.; Schulte, P.J.; Druliner, B.R.; Johnson, R.A.; Narkiewicz, K.; Boardman, L.A.; et al. Telomere Length and Risk of Major Adverse Cardiac Events and Cancer in Obstructive Sleep Apnea Patients. Cells 2019, 8, 381. [Google Scholar] [CrossRef] [Green Version]
- Polonis, K.; Somers, V.K.; Becari, C.; Covassin, N.; Schulte, P.J.; Druliner, B.R.; Johnson, R.A.; Narkiewicz, K.; Boardman, L.A.; Singh, P. Moderate-to-severe obstructive sleep apnea is associated with telomere lengthening. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1022–H1030. [Google Scholar] [CrossRef] [PubMed]
- Barceló, A.; Piérola, J.; López-Escribano, H.; de la Peña, M.; Soriano, J.B.; Alonso-Fernández, A.; Ladaria, A.; Agustí, A. Telomere shortening in sleep apnea syndrome. Respir. Med. 2010, 104, 1225–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savolainen, K.; Eriksson, J.G.; Kajantie, E.; Lahti, M.; Räikkönen, K. The history of sleep apnea is associated with shorter leukocyte telomere length: The Helsinki Birth Cohort Study. Sleep Med. 2014, 15, 209–212. [Google Scholar] [CrossRef]
- Kwon, A.M.; Baik, I.; Thomas, R.J.; Shin, C. The association between leukocyte telomere lengths and sleep instability based on cardiopulmonary coupling analysis. Sleep Breath. 2015, 19, 963–968. [Google Scholar] [CrossRef]
- Boyer, L.; Audureau, E.; Margarit, L.; Marcos, E.; Bizard, E.; Le Corvoisier, P.; Macquin-Mavier, I.; Derumeaux, G.; Damy, T.; Drouot, X.; et al. Telomere Shortening in Middle-Aged Men with Sleep-disordered Breathing. Ann. Am. Thorac. Soc. 2016, 13, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Kwak, J.W.; Lim, S.J.; Park, Y.K.; Yang, H.S.; Kim, H.J. Oxidative Stress-induced Telomere Length Shortening of Circulating Leukocyte in Patients with Obstructive Sleep Apnea. Aging Dis. 2016, 7, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.M.; Thomas, R.J.; Yoon, D.W.; Lee, S.K.; Baik, I.; Shin, C. Interaction between Obstructive Sleep Apnea and Shortened Telomere Length on Brain White Matter Abnormality. Sleep 2016, 39, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Riestra, P.; Gebreab, S.Y.; Xu, R.; Khan, R.J.; Quarels, R.; Gibbons, G.; Davis, S.K. Obstructive sleep apnea risk and leukocyte telomere length in African Americans from the MH-GRID study. Sleep Breath. 2017, 21, 751–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempaku, P.F.; Mazzotti, D.R.; Hirotsu, C.; Andersen, M.L.; Xavier, G.; Maurya, P.K.; Rizzo, L.B.; Brietzke, E.; Belangero, S.I.; Bittencourt, L.; et al. The effect of the severity of obstructive sleep apnea syndrome on telomere length. Oncotarget 2016, 7, 69216–69224. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.E.; Irwin, M.R.; Seeman, T.E.; Diez-Roux, A.V.; Prather, A.A.; Olmstead, R.; Epel, J.; Lin, R.; Redline, S. Obstructive sleep apnea, nighttime arousals, and leukocyte telomere length: The Multi-Ethnic Study of Atherosclerosis. Sleep 2019, 42, zsz089. [Google Scholar] [CrossRef]
- Pinilla, L.; Santamaria-Martos, F.; Benítez, I.D.; Zapater, A.; Targa, A.; Mediano, O.; Masa, J.F.; Masdeu, M.J.; Minguez, O.; Aguilà, M.; et al. Association of Obstructive Sleep Apnea with the Aging Process. Ann. Am. Thorac. Soc. 2021, 18, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Wang, H.Y.; Liaw, S.F.; Chiu, C.H.; Lin, M.W. Effect of oral appliance on circulating leukocyte telomere length and SIRT1 in obstructive sleep apnea. Clin. Oral Investig. 2019, 23, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Marin, J.M.; Qiao, Z.; Rubio, D.S.; Kheirandish-Gozal, L.; Gozal, D. Plasma exosomes in OSA patients promote endothelial senescence: Effect of long-term adherent continuous positive airway pressure. Sleep 2020, 43, zsz217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.X.; Gozal, D. Reactive oxygen species and the brain in sleep apnea. Respir. Physiol. Neurobiol. 2010, 174, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef]
- Oikawa, S.; Tada-Oikawa, S.; Kawanishi, S. Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 2001, 40, 4763–4768. [Google Scholar] [CrossRef]
- Ahmed, W.; Lingner, J. Impact of oxidative stress on telomere biology. Differentiation 2018, 99, 21–27. [Google Scholar] [CrossRef]
- Xie, J.; Jiang, J.; Shi, K.; Zhang, T.; Zhu, T.; Chen, H.; Chen, R.; Qi, L.; Ding, W.; Yi, Q.; et al. DNA damage in peripheral blood lymphocytes from patients with OSAHS. Sleep Breath. 2014, 18, 775–780. [Google Scholar] [CrossRef]
- Iyama, T.; Wilson, D.M., III. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Hills, M.; Conomos, D.; Stutz, M.D.; Dagg, R.A.; Lau, L.M.; Reddel, R.R.; Pickett, H.A. Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 2014, 42, 1733–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lima, F.F.; Mazzotti, D.R.; Tufik, S.; Bittencourt, L. The role inflammatory response genes in obstructive sleep apnea syndrome: A review. Sleep Breath. 2016, 20, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The NF-kappaB regulatory network. Cardiovasc. Toxicol. 2006, 6, 111–130. [Google Scholar] [CrossRef]
- Saretzki, G. Telomeres, Telomerase and Ageing. In Biochemistry and Cell Biology of Ageing: Part I Biomedical Science; Harris, J., Korolchuk, V., Eds.; Springer: Singapore, 2018; Volume 90. [Google Scholar]
- Zhang, J.; Rane, G.; Dai, X.; Shanmugam, M.K.; Arfuso, F.; Samy, R.P.; Lai, M.K.; Kappei, D.; Kumar, A.P.; Sethi, G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res. Rev. 2016, 25, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Poulsen, O.; Haddad, G.G. Intermittent hypoxia induces murine macrophage foam cell formation by IKK-β-dependent NF-κB pathway activation. J. Appl. Physiol. 2016, 121, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Peng, Y.J.; Nanduri, J. Hypoxia-inducible factors and obstructive sleep apnea. J. Clin. Investig. 2020, 130, 5042–5051. [Google Scholar] [CrossRef]
- Gabryelska, A.; Szmyd, B.; Szemraj, J.; Stawski, R.; Sochal, M.; Białasiewicz, P. Patients with obstructive sleep apnea present with chronic upregulation of serum HIF-1α protein. J. Clin. Sleep Med. 2020, 16, 1761–1768. [Google Scholar] [CrossRef]
- Gabryelska, A.; Stawski, R.; Sochal, M.; Szmyd, B.; Białasiewicz, P. Influence of one-night CPAP therapy on the changes of HIF-1α protein in OSA patients: A pilot study. J. Sleep Res. 2020, 4, e12995. [Google Scholar] [CrossRef] [PubMed]
- Gabryelska, A.; Szmyd, B.; Panek, M.; Szemraj, J.; Kuna, P.; Białasiewicz, P. Serum hypoxia-inducible factor-1α protein level as a diagnostic marker of obstructive sleep apnea. Pol. Arch. Intern. Med. 2020, 130, 158–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Chen, X.; Jiao, Q.; Qiu, Z.; Shen, C.; Zhang, G.; Sun, Z.; Zhang, H.; Luo, Q.Y. HIF-1α-Mediated Telomerase Reverse Transcriptase Activation Inducing Autophagy Through Mammalian Target of Rapamycin Promotes Papillary Thyroid Carcinoma Progression During Hypoxia Stress. Thyroid 2021, 31, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Yatabe, N.; Kyo, S.; Maida, Y.; Nishi, H.; Nakamura, M.; Kanaya, T.; Tanaka, M.; Isaka, K.; Ogawa, S.; Inoue, M. HIF-1-mediated activation of telomerase in cervical cancer cells. Oncogene 2004, 23, 3708–3715. [Google Scholar] [CrossRef] [Green Version]
- Lou, F.; Chen, X.; Jalink, M.; Zhu, Q.; Ge, N.; Zhao, S.; Fang, X.; Fan, Y.; Björkholm, M.; Liu, Z.; et al. The opposing effect of hypoxia-inducible factor-2alpha on expression of telomerase reverse transcriptase. Mol. Cancer Res. 2007, 5, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Nakada, T.; Kyo, S.; Inoue, M.; Shay, J.W.; Isaka, K. Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT). Mol. Cell. Biol. 2004, 24, 6076–6083. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, A.; Zara, S.; Rapino, M.; Zingariello, M.; di Giacomo, V.; Antonucci, A. p53 and telomerase control rat myocardial tissue response to hypoxia and ageing. Eur. J. Histochem. 2009, 53, e25. [Google Scholar] [CrossRef]
- Yeo, E.J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Y.; Lv, Y.; Le, Z.; Xin, Y.; Zhang, P.; Liu, Y. TERT alleviates irradiation-induced late rectal injury by reducing hypoxia-induced ROS levels through the activation of NF-κB and autophagy. Int. J. Mol. Med. 2016, 38, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.K.; Yamada, R.G.; Ukai, H.; Baggs, J.E.; Miraglia, L.J.; Kobayashi, T.J.; Welsh, D.K.; Kay, S.A.; Ueda, H.R.; Hogenesch, J.B. Feedback repression is required for mammalian circadian clock function. Nat. Genet. 2006, 38, 312–319. [Google Scholar] [CrossRef]
- Siepka, S.M.; Yoo, S.H.; Park, J.; Lee, C.; Takahashi, J.S. Genetics and neurobiology of circadian clocks in mammals. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Chelliah, Y.; Shan, Y.; Taylor, C.A.; Yoo, S.H.; Partch, C.; Green, C.B.; Zhang, H.; Takahashi, J.S. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science 2012, 337, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego, M.; Virshup, D.M. Post-translational modifications regulate the ticking of the circadian clock. Nat. Rev. Mol. Cell. Biol. 2007, 8, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.L.; Kwon, H.; Portolano, N.; Parkin, G.; Venkatraman Girija, U.; Basran, J.; Fielding, A.J.; Fairall, L.; Svistunenko, D.A.; Moody, P.C.; et al. Heme binding to human CLOCK affects interactions with the E-box. Proc. Natl. Acad. Sci. USA 2019, 116, 19911–19916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partch, C.L.; Green, C.B.; Takahashi, J.S. Molecular architecture of the mammalian circadian clock. Trends Cell. Biol. 2014, 24, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Welsh, D.K.; Takahashi, J.S.; Kay, S.A. Suprachiasmatic nucleus: Cell autonomy and network properties. Annu. Rev. Physiol. 2010, 72, 551–577. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.K.; Kornblum, I.; Vivek, K.; Koike, N.; Nussbaum, J.; et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 2013, 152, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Reischl, S.; Vanselow, K.; Westermark, P.O.; Thierfelder, N.; Maier, B.; Herzel, H.; Kramer, A. Beta-TrCP1-mediated degradation of PERIOD2 is essential for circadian dynamics. J. Biol. Rhythms. 2007, 22, 375–386. [Google Scholar] [CrossRef]
- Busino, L.; Bassermann, F.; Maiolica, A.; Lee, C.; Nolan, P.M.; Godinho, S.I.; Draetta, G.F.; Pagano, M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science 2007, 316, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Siepka, S.M.; Yoo, S.H.; Park, J.; Song, W.; Kumar, V.; Hu, Y.; Lee, C.; Takahashi, J.S. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell 2007, 129, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabryelska, A.; Sochal, M.; Turkiewicz, S.; Białasiewicz, P. Relationship between HIF-1 and Circadian Clock Proteins in Obstructive Sleep Apnea Patients-Preliminary Study. J. Clin. Med. 2020, 9, 1599. [Google Scholar] [CrossRef] [PubMed]
- Hood, S.; Amir, S. The aging clock: Circadian rhythms and later life. J. Clin. Investig. 2017, 127, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Kunieda, T.; Minamino, T.; Katsuno, T.; Tateno, K.; Nishi, J.; Miyauchi, H.; Orimo, M.; Oskada, S.; Komuro, I. Cellular senescence impairs circadian expression of clock genes in vitro and in vivo. Circ. Res. 2006, 98, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.; Kretzschmar, D.; Rakshit, K.; Chow, E.; Giebultowicz, J.M. The circadian clock gene period extends healthspan in aging Drosophila melanogaster. Aging 2009, 1, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Zhu, Q.; Mirek, E.; Na, L.; Raduwan, H.; Anthony, T.G.; Belden, W.J. BMAL1 associates with chromosome ends to control rhythms in TERRA and telomeric heterochromatin. PLoS ONE 2019, 14, e0223803. [Google Scholar] [CrossRef] [Green Version]
- Hemmeryckx, B.; Hohensinner, P.; Swinnen, M.; Heggermont, W.; Wojta, J.; Lijnen, H.R. Antioxidant Treatment Improves Cardiac Dysfunction in a Murine Model of Premature Aging. J. Cardiovasc. Pharmacol. 2016, 68, 374–382. [Google Scholar] [CrossRef]
- Khapre, R.V.; Kondratova, A.A.; Susova, O.; Kondratov, R.V. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle 2011, 10, 4162–4169. [Google Scholar] [CrossRef] [Green Version]
- Bordoni, V.; Tartaglia, E.; Refolo, G.; Sacchi, A.; Grassi, G.; Antinori, A.; Fimia, G.M.; Agrati, C. Per2 Upregulation in Circulating Hematopoietic Progenitor Cells During Chronic HIV Infection. Front. Cell. Infect. Microbiol. 2020, 10, 362. [Google Scholar] [CrossRef]
- Chen, W.D.; Wen, M.S.; Shie, S.S.; Lo, Y.L.; Wo, H.T.; Wang, C.C.; Hsieh, I.C.; Lee, T.H.; Wang, C.Y. The circadian rhythm controls telomeres and telomerase activity. Biochem. Biophys. Res. Commun. 2014, 451, 408–414. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turkiewicz, S.; Ditmer, M.; Sochal, M.; Białasiewicz, P.; Strzelecki, D.; Gabryelska, A. Obstructive Sleep Apnea as an Acceleration Trigger of Cellular Senescence Processes through Telomere Shortening. Int. J. Mol. Sci. 2021, 22, 12536. https://doi.org/10.3390/ijms222212536
Turkiewicz S, Ditmer M, Sochal M, Białasiewicz P, Strzelecki D, Gabryelska A. Obstructive Sleep Apnea as an Acceleration Trigger of Cellular Senescence Processes through Telomere Shortening. International Journal of Molecular Sciences. 2021; 22(22):12536. https://doi.org/10.3390/ijms222212536
Chicago/Turabian StyleTurkiewicz, Szymon, Marta Ditmer, Marcin Sochal, Piotr Białasiewicz, Dominik Strzelecki, and Agata Gabryelska. 2021. "Obstructive Sleep Apnea as an Acceleration Trigger of Cellular Senescence Processes through Telomere Shortening" International Journal of Molecular Sciences 22, no. 22: 12536. https://doi.org/10.3390/ijms222212536
APA StyleTurkiewicz, S., Ditmer, M., Sochal, M., Białasiewicz, P., Strzelecki, D., & Gabryelska, A. (2021). Obstructive Sleep Apnea as an Acceleration Trigger of Cellular Senescence Processes through Telomere Shortening. International Journal of Molecular Sciences, 22(22), 12536. https://doi.org/10.3390/ijms222212536