
The Genetic, Environmental, and Immunopathological Complexity of Autoantibody-Negative Rheumatoid Arthritis


Abstract
1. Introduction
2. Genetic Susceptibility
3. Environmental and Lifestyle Factors
3.1. Smoking, Other Pulmonary Irritants, and Respiratory Diseases
3.2. Microbial Dysbiosis and Mucosal Inflammation
3.3. Body Weight and Diet
3.4. Hormonal Factors
3.5. Mental Health, Post-Traumatic Stress Disorders, and Depression
3.6. Drugs
4. Immunopathogenesis
4.1. Autoantibodies
4.2. Cytokine Networks
4.3. Synovial Pathology
4.4. Extra-Articular Involvement
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van der Helm-van Mil, A.H.; Verpoort, K.N.; Breedveld, F.C.; Toes, R.E.; Huizinga, T.W. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R949–R958. [Google Scholar] [CrossRef] [PubMed]
- Westerlind, H.; Rönnelid, J.; Hansson, M.; Alfredsson, L.; Mathsson-Alm, L.; Serre, G.; Cornillet, M.; Holmdahl, R.; Jakobsson, P.; Skriner, K.; et al. Anti–Citrullinated Protein Antibody Specificities, Rheumatoid Factor Isotypes, and Incident Cardiovascular Events in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 1658–1667. [Google Scholar] [CrossRef]
- Matthijssen, X.M.E.; Niemantsverdriet, E.; Huizinga, T.W.J.; van der Helm-van Mil, A.H.M. Enhanced treatment strategies and distinct disease outcomes among autoantibody-positive and -negative rheumatoid arthritis patients over 25 years: A longitudinal cohort study in the Netherlands. PLoS Med. 2020, 17, e1003296. [Google Scholar] [CrossRef]
- Boeters, D.M.; Gaujoux-Viala, C.; Constantin, A.; van der Helm-van Mil, A.H. The 2010 ACR/EULAR criteria are not sufficiently accurate in the early identification of autoantibody-negative rheumatoid arthritis: Results from the Leiden-EAC and ESPOIR cohorts. Semin. Arthritis Rheum. 2017, 47, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Courvoisier, D.S.; Chatzidionysiou, K.; Mongin, D.; Lauper, K.; Mariette, X.; Morel, J.; Gottenberg, J.-E.; Bergstra, S.A.; Suarez, M.P.; Codreanu, C.; et al. The impact of seropositivity on the effectiveness of biologic anti-rheumatic agents: Results from a collaboration of 16 registries. Rheumatology 2020, 60, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Malmström, V.; Catrina, A.I.; Klareskog, L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat. Rev. Immunol. 2017, 17, 60–75. [Google Scholar] [CrossRef]
- Scherer, H.U.; Häupl, T.; Burmester, G.R. The etiology of rheumatoid arthritis. J. Autoimmun. 2020, 110, 102400. [Google Scholar] [CrossRef]
- McGonagle, D.; Watad, A.; Savic, S. Mechanistic immunological based classification of rheumatoid arthritis. Autoimmun. Rev. 2018, 17, 1115–1123. [Google Scholar] [CrossRef]
- Pouw, J.; Leijten, E.; Van Laar, J.; Boes, M. Revisiting B cell tolerance and autoantibodies in seropositive and seronegative autoimmune rheumatic disease (AIRD). Clin. Exp. Immunol. 2021, 203, 160–173. [Google Scholar] [CrossRef]
- Van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.M.; Huizinga, T.W.J.; Thomson, W.; Worthinton, J.; van der Helm-van Mil, A.H.M.; de Vries, R.R.P. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef]
- Hensvold, A.H.; Magnusson, P.; Joshua, V.; Hansson, M.; Israelsson, L.; Ferreira, R.C.; Jakobsson, P.-J.; Holmdahl, R.; Hammarström, L.; Malmström, V.; et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: An epidemiological investigation in twins. Ann. Rheum. Dis. 2015, 74, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Frisell, T.; Holmqvist, M.; Källberg, H.; Klareskog, L.; Alfredsson, L.; Askling, J. Familial Risks and Heritability of Rheumatoid Arthritis: Role of Rheumatoid Factor/Anti-Citrullinated Protein Antibody Status, Number and Type of Affected Relatives, Sex, and Age. Arthritis Rheum. 2013, 65, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Frisell, T.; Hellgren, K.; Alfredsson, L.; Raychaudhuri, S.; Klareskog, L.; Askling, J. Familial aggregation of arthritis-related diseases in seropositive and seronegative rheumatoid arthritis: A register-based case-control study in Sweden. Ann. Rheum. Dis. 2016, 75, 183–189. [Google Scholar] [CrossRef]
- Kirino, Y.; Remmers, E.F. Genetic architectures of seropositive and seronegative rheumatic diseases. Nat. Rev. Rheumatol. 2015, 11, 401–414. [Google Scholar] [CrossRef]
- Padyukov, L.; Seielstad, M.; Ong, R.T.H.; Ding, B.; Ronnelid, J.; Seddighzadeh, M.; Alfredsson, L.; Klareskog, L.; Epidemiological Investigation of Rheumatoid Arthritis (EIRA) Study Group. A genome-wide association study suggests contrasting associations in ACPA-positive versus ACPA-negative rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 259–265. [Google Scholar] [CrossRef]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012, 44, 1336–1440. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Diogo, D.; Eyre, S.; Kallberg, H.; Zhernakova, A.; Bowes, J.; Padyukov, L.; Okada, Y.; González-Gay, M.A.; Dahlqvist, S.R.; et al. Fine Mapping Seronegative and Seropositive Rheumatoid Arthritis to Shared and Distinct HLA Alleles by Adjusting for the Effects of Heterogeneity. Am. J. Hum. Genet. 2014, 94, 522–532. [Google Scholar] [CrossRef]
- Bossini-Castillo, L.; de Kovel, C.; Kallberg, H.; van ‘t Slot, R.; Italiaander, A.; Coenen, M.; Tak, P.P.; Posthumus, M.D.; Wijmenga, C.; Huizinga, T.; et al. A genome-wide association study of rheumatoid arthritis without antibodies against citrullinated peptides. Ann. Rheum. Dis. 2015, 74, e15. [Google Scholar] [CrossRef]
- Terao, C.; Brynedal, B.; Chen, Z.; Jiang, X.; Westerlind, H.; Hansson, M.; Jakobsson, P.-J.; Lundberg, K.; Skriner, K.; Serre, G.; et al. Distinct HLA associations with rheumatoid arthritis subsets defined by serological subphenotype. Am. J. Hum. Genet. 2019, 105, 616–624. [Google Scholar] [CrossRef]
- Irigoyen, P.; Lee, A.T.; Wener, M.H.; Li, W.; Kern, M.; Batliwalla, F.; Lum, R.F.; Massarotti, E.; Weisman, M.; Bombardier, C.; et al. Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: Contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum. 2005, 52, 3813–3818. [Google Scholar] [CrossRef]
- Verpoort, K.N.; van Gaalen, F.A.; van der Helm-van Mil, A.H.; Schreuder, G.M.T.; Breedveld, F.C.; Huizinga, T.W.J.; de Vries, R.R.P.; Toes, R.E.M. Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum. 2005, 52, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; van Wijk, F. CD8(+) T cells in human autoimmune arthritis: The unusual suspects. Nat. Rev. Rheumatol. 2016, 12, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Kelkka, T.; Savola, P.; Bhattacharya, D.; Huuhtanen, J.; Lönnberg, T.; Kankainen, M.; Paalanen, K.; Tyster, M.; Lepistö, M.; Ellonen, P.; et al. Adult-Onset Anti-Citrullinated Peptide Antibody-Negative Destructive Rheumatoid Arthritis Is Characterized by a Disease-Specific CD8+ T Lymphocyte Signature. Front. Immunol. 2020, 11, 578848. [Google Scholar] [CrossRef] [PubMed]
- Scally, S.W.; Petersen, J.; Law, S.C.; Dudek, N.L.; Nel, H.J.; Loh, K.L.; Wijeyewickrema, L.; Eckle, S.; Van Heemst, J.; Pike, R.; et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 2013, 210, 2569–2582. [Google Scholar] [CrossRef] [PubMed]
- Hinks, A.; Bowes, J.; Cobb, J.; Ainsworth, H.C.; Marion, M.C.; Comeau, M.; Sudman, M.; Han, B.; Becker, M.L.; Bohnsack, J.F.; et al. Fine-mapping the MHC locus in juvenile idiopathic arthritis (JIA) reveals genetic heterogeneity corresponding to distinct adult inflammatory arthritic diseases. Ann. Rheum. Dis. 2017, 76, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Bowes, J.; Lunt, M.; Eyre, S.; Barton, A.; Worthington, J. Genetic markers of rheumatoid arthritis susceptibility in anti-citrullinated peptide antibody negative patients. Ann. Rheum. Dis. 2012, 71, 1984–1990. [Google Scholar] [CrossRef]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Terao, C.; Ohmura, K.; Kochi, Y.; Ikari, K.; Okada, Y.; Shimizu, M.; Nishina, N.; Suzuki, A.; Myouzen, K.; Kawaguchi, T.; et al. Anti-citrullinated peptide/protein antibody (ACPA)-negative RA shares a large proportion of susceptibility loci with ACPA-positive RA: A meta-analysis of genome-wide association study in a Japanese population. Arthritis Res. Ther. 2015, 17, 104. [Google Scholar] [CrossRef]
- Viatte, S.; Massey, J.; Bowes, J.; Duffus, K.; Eyre, S.; Barton, A.; Worthington, J. Replication of Associations of Genetic Loci Outside the HLA Region with Susceptibility to Anti–Cyclic Citrullinated Peptide–Negative Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Ugidos, N.; Mena, J.; Baquero, S.; Alloza, I.; Azkargorta, M.; Elortza, F.; Vandenbroeck, K. Interactome of the Autoimmune Risk Protein ANKRD55. Front. Immunol. 2019, 10, 2067. [Google Scholar] [CrossRef]
- Wei, W.-H.; Viatte, S.; Merriman, T.R.; Barton, A.; Worthington, J. Genotypic variability based association identifies novel non-additive loci DHCR7 and IRF4 in sero-negative rheumatoid arthritis. Sci. Rep. 2017, 7, 5261. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishimura, K.; Tamaki, K.; Tsuji, G.; Nakazawa, T.; Morinobu, A.; Kumagai, S. Impact of smoking as a risk factor for developing rheumatoid arthritis: A meta-analysis of observational studies. Ann. Rheum. Dis. 2010, 69, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Di Giuseppe, D.; Discacciati, A.; Orsini, N.; Wolk, A. Cigarette smoking and risk of rheumatoid arthritis: A dose-response meta-analysis. Arthritis Res. Ther. 2014, 16, R61. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Rönnelid, J.; Klareskog, L.; Alfredsson, L. Complex Relationships of Smoking, HLA-DRB1 Genes, and Serologic Profiles in Patients with Early Rheumatoid Arthritis: Update from a Swedish Population-Based Case-Control Study. Arthritis Rheumatol. 2019, 71, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Ikari, K.; Hashimoto, M.; Ohmura, K.; Tanaka, M.; Ito, H.; Taniguchi, A.; Yamanaka, H.; Mimori, T.; Terao, C. Shared epitope defines distinct associations of cigarette smoking with levels of anticitrullinated protein antibody and rheumatoid factor. Ann. Rheum. Dis. 2019, 78, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, C.; Rodriguez-Rodriguez, L.; Lopez-Mejias, R.; Nuño, L.; Triguero-Martinez, A.; Perez-Pampin, E.; Corrales, A.; Villalba, A.; Lopez-Golan, Y.; Abasolo, L.; et al. A predominant involvement of the triple seropositive patients and others with rheumatoid factor in the association of smoking with rheumatoid arthritis. Sci. Rep. 2020, 10, 3355. [Google Scholar] [CrossRef]
- Bang, S.-Y.; Lee, K.-H.; Cho, S.-K.; Lee, H.-S.; Lee, K.W.; Bae, S.-C. Smoking increases rheumatoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody status. Arthritis Rheum. 2010, 62, 369–377. [Google Scholar] [CrossRef]
- Myasoedova, E.; Davis, J.; Matteson, E.L.; Crowson, C.S. Is the epidemiology of rheumatoid arthritis changing? Results from a population-based incidence study, 1985–2014. Ann. Rheum. Dis. 2020, 79, 440–444. [Google Scholar] [CrossRef]
- Hart, J.E.; Laden, F.; Puett, R.C.; Costenbader, K.H.; Karlson, E.W. Exposure to Traffic Pollution and Increased Risk of Rheumatoid Arthritis. Environ. Health Perspect. 2009, 117, 1065–1069. [Google Scholar] [CrossRef]
- De Roos, A.J.; Koehoorn, M.; Tamburic, L.; Davies, H.W.; Brauer, M. Proximity to traffic, ambient air pollution, and community noise in relation to incident rheumatoid arthritis. Environ. Health Perspect. 2014, 122, 1075–1080. [Google Scholar] [CrossRef]
- Ilar, A.; Gustavsson, P.; Wiebert, P.; Alfredsson, L. Occupational exposure to organic dusts and risk of developing rheumatoid arthritis: Findings from a Swedish population-based case–control study. RMD Open 2019, 5, e001049. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.W.; Alfredsson, L.; Costenbader, K.H.; Kamen, D.L.; Nelson, L.M.; Norris, J.M.; De Roos, A.J. Epidemiology of environmental exposures and human autoimmune diseases: Findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J. Autoimmun. 2012, 39, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Ilar, A.; Klareskog, L.; Saevarsdottir, S.; Wiebert, P.; Askling, J.; Gustavsson, P.; Alfredsson, L. Occupational exposure to asbestos and silica and risk of developing rheumatoid arthritis: Findings from a Swedish population-based case-control study. RMD Open 2019, 5, e000978. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Chen, Z.; Klareskog, L.; Alfredsson, L.; Bengtsson, C.; Jiang, X. Amount of smoking, duration of smoking cessation and their interaction with silica exposure in the risk of rheumatoid arthritis among males: Results from the Swedish Epidemiological Investigation of Rheumatoid Arthritis (EIRA) study. Ann. Rheum. Dis. 2018, 77, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef] [PubMed]
- Kronzer, V.L.; Westerlind, H.; Alfredsson, L.; Crowson, C.S.; Nyberg, F.; Tornling, G.; Klareskog, L.; Holmqvist, M.; Askling, J. Respiratory Diseases as Risk Factors for Seropositive and Seronegative Rheumatoid Arthritis and in Relation to Smoking. Arthritis Rheumatol. 2021, 73, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The Microbiome in Infectious Disease and Inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef]
- Wilson, T.M.; Trent, B.; Kuhn, K.A.; Demoruelle, M.K. Microbial Influences of Mucosal Immunity in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2020, 22, 83. [Google Scholar] [CrossRef]
- Gómez-Bañuelos, E.; Mukherjee, A.; Darrah, E.; Andrade, F. Rheumatoid Arthritis-Associated Mechanisms of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans. J. Clin. Med. 2019, 8, 1309. [Google Scholar] [CrossRef]
- Cheng, Z.; Do, T.; Mankia, K.; Meade, J.; Hunt, L.; Clerehugh, V.; Speirs, A.; Tugnait, A.; Emery, P.; Devine, D. Dysbiosis in the oral microbiomes of anti-CCP positive individuals at risk of developing rheumatoid arthritis. Ann. Rheum. Dis. 2021, 80, 162–168. [Google Scholar] [CrossRef]
- Rooney, C.M.; Mankia, K.; Mitra, S.; Moura, I.B.; Emery, P.; Wilcox, M.H. Perturbations of the gut microbiome in anti-CCP positive individuals at risk of developing rheumatoid arthritis. Rheumatology 2021, 60, 3380–3387. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Wu, H.-J.J.; Mauro, D.; Schett, G.; Ciccia, F. The gut–joint axis in rheumatoid arthritis. Nat. Rev. Rheumatol. 2021, 17, 224–237. [Google Scholar] [CrossRef]
- Rogier, R.; Ederveen, T.H.A.; Boekhorst, J.; Wopereis, H.; Scher, J.U.; Manasson, J.; Frambach, S.J.C.M.; Knol, J.; Garssen, J.; Van Der Kraan, P.M.; et al. Aberrant intestinal microbiota due to IL-1 receptor antagonist deficiency promotes IL-17- and TLR4-dependent arthritis. Microbiome 2017, 5, 63. [Google Scholar] [CrossRef]
- Hammad, D.B.M.; Hider, S.L.; Liyanapathirana, V.C.; Tonge, D.P. Molecular Characterization of Circulating Microbiome Signatures in Rheumatoid Arthritis. Front. Cell. Infect. Microbiol. 2020, 9, 440. [Google Scholar] [CrossRef]
- Pedersen, M.; Jacobsen, S.; Klarlund, M.; Pedersen, B.V.; Wiik, A.; Wohlfahrt, J.; Frisch, M. Environmental risk factors differ between rheumatoid arthritis with and without auto-antibodies against cyclic citrullinated peptides. Arthritis Res. Ther. 2006, 8, R133. [Google Scholar] [CrossRef]
- Lahiri, M.; Luben, R.; Morgan, C.; Bunn, D.K.; Marshall, T.; Lunt, M.; Verstappen, S.; Symmons, D.; Khaw, K.-T.; Wareham, N.; et al. Using lifestyle factors to identify individuals at higher risk of inflammatory polyarthritis (results from the European Prospective Investigation of Cancer-Norfolk and the Norfolk Arthritis Register—the EPIC-2-NOAR Study). Ann. Rheum. Dis. 2014, 73, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Wesley, A.; Bengtsson, C.; Elkan, A.-C.; Klareskog, L.; Alfredsson, L.; Wedrén, S.; Epidemiological Investigation of Rheumatoid Arthritis Study Group. Association between body mass index and anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis: Results from a population-based case-control study. Arthritis Care Res. 2013, 65, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, Q.; Yu, F.; Wang, Z.; Chen, S.; Jin, Z.; Cai, Q.; Liu, Y.; He, J. Body Mass Index and Risk of Rheumatoid Arthritis: A Meta-Analysis of Observational Studies. Medicine 2016, 95, e2859. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Hiraki, L.T.; Sparks, J.; Malspeis, S.; Chen, C.-Y.; Awosogba, J.A.; Arkema, E.; Costenbader, K.H.; Karlson, E.W. Being overweight or obese and risk of developing rheumatoid arthritis among women: A prospective cohort study. Ann. Rheum. Dis. 2014, 73, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Turk, S.A.; Van Schaardenburg, D.; Boers, M.; De Boer, S.; Fokker, C.; Lems, W.F.; Nurmohamed, M.T. An unfavorable body composition is common in early arthritis patients: A case control study. PLoS ONE 2018, 13, e0193377. [Google Scholar] [CrossRef]
- Giles, J.T.; Ling, S.M.; Ferrucci, L.; Bartlett, S.; Andersen, R.E.; Towns, M.; Muller, D.; Fontaine, K.R.; Bathon, J.M. Abnormal body composition phenotypes in older rheumatoid arthritis patients: Association with disease characteristics and pharmacotherapies. Arthritis Rheum. 2008, 59, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Hasseli, R.; Ohl, S.; Lange, U.; Frommer, K.; Müller-Ladner, U. Adipokines and Autoimmunity in Inflammatory Arthritis. Cells 2021, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hazlewood, G.; Kaplan, G.G.; Eksteen, B.; Barnabe, C. Impact of Obesity on Remission and Disease Activity in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Arthritis Care Res. 2017, 69, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Rönnelid, J.; Saevarsdottir, S.; Padyukov, L.; Alfredsson, L. The importance of differences; On environment and its interactions with genes and immunity in the causation of rheumatoid arthritis. J. Intern. Med. 2020, 287, 514–533. [Google Scholar] [CrossRef]
- Salliot, C.; Nguyen, Y.; Boutron-Ruault, M.-C.; Seror, R. Environment and Lifestyle: Their Influence on the Risk of RA. J. Clin. Med. 2020, 9, 3109. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.W.; Demoruelle, M.K.; Deane, K.D.; Weisman, M.H.; Buckner, J.H.; Gregersen, P.K.; Mikuls, T.R.; O’Dell, J.R.; Keating, R.M.; Fingerlin, T.; et al. Omega-3 fatty acids are associated with a lower prevalence of autoantibodies in shared epitope-positive subjects at risk for rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.W.; Bemis, E.A.; Demoruelle, M.K.; Striebich, C.C.; Brake, S.; Feser, M.L.; Moss, L.; Clare-Salzler, M.; Holers, V.M.; Deane, K.D.; et al. The association between omega-3 fatty acid biomarkers and inflammatory arthritis in an anti-citrullinated protein antibody positive population. Rheumatology 2017, 56, 2229–2236. [Google Scholar] [CrossRef]
- Hu, Y.; Sparks, J.; Malspeis, S.; Costenbader, K.H.; Hu, F.B.; Karlson, E.W.; Lu, B. Long-term dietary quality and risk of developing rheumatoid arthritis in women. Ann. Rheum. Dis. 2017, 76, 1357–1364. [Google Scholar] [CrossRef]
- Källberg, H.; Jacobsen, S.; Bengtsson, C.; Pedersen, M.; Padyukov, L.; Garred, P.; Frisch, M.; Karlson, E.W.; Klareskog, L.; Alfredsson, L. Alcohol consumption is associated with decreased risk of rheumatoid arthritis: Results from two Scandinavian case–control studies. Ann. Rheum. Dis. 2009, 68, 222–227. [Google Scholar] [CrossRef]
- Silman, A.; Kay, A.; Brennan, P. Timing of Pregnancy in Relation to the Onset of Rheumatoid Arthritis. Arthritis Rheum. 1992, 35, 152–155. [Google Scholar] [CrossRef]
- Lansink, M.; De Boer, A.; Dijkmans, B.A.; Vandenbroucke, J.P.; Hazes, J.M. The onset of rheumatoid arthritis in relation to pregnancy and childbirth. Clin. Exp. Rheumatol. 1993, 11, 171–174. [Google Scholar]
- Förger, F.; Villiger, P.M. Immunological adaptations in pregnancy that modulate rheumatoid arthritis disease activity. Nat. Rev. Rheumatol. 2020, 16, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, H.; Lam, S.; Smith, C.; Giles, I. Does Rheumatoid Arthritis Really Improve During Pregnancy? A Systematic Review and Metaanalysis. J. Rheumatol. 2019, 46, 245–250. [Google Scholar] [CrossRef]
- de Man, Y.A.; Bakker-Jonges, L.E.; Goorbergh, C.M.; Tillemans, S.P.; Hooijkaas, H.; Hazes, J.M.; Dolhain, R.J.E.M. Women with rheumatoid arthritis negative for anti-cyclic citrullinated peptide and rheumatoid factor are more likely to improve during pregnancy, whereas in autoantibody-positive women autoantibody levels are not influenced by pregnancy. Ann. Rheum. Dis. 2010, 69, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Wallenius, M.; Skomsvoll, J.F.; Irgens, L.M.; Salvesen, K.; Koldingsnes, W.; Mikkelsen, K.; Kaufmann, C.; Kvien, T.K. Postpartum onset of rheumatoid arthritis and other chronic arthritides: Results from a patient register linked to a medical birth registry. Ann. Rheum. Dis. 2010, 69, 332–336. [Google Scholar] [CrossRef]
- Orellana, C.; Wedrén, S.; Källberg, H.; Holmqvist, M.; Karlsson, E.W.; Alfredsson, L.; Bengtsson, C.; EIRA Study Group. Parity and the risk of developing rheumatoid arthritis: Results from the Swedish Epidemiological Investigation of Rheumatoid Arthritis study. Ann. Rheum. Dis. 2014, 73, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Guo, P.; Sun, Q.-M.; Liu, H.; Chen, Y.; Huang, Y.; Cai, X.-J. Number of parity and the risk of rheumatoid arthritis in women: A dose-response meta-analysis of observational studies. J. Obstet. Gynaecol. Res. 2017, 43, 1428–1440. [Google Scholar] [CrossRef]
- Chen, W.M.; Subesinghe, S.; Muller, S.; Hider, S.L.; Mallen, C.; Scott, I.C. The association between gravidity, parity and the risk of developing rheumatoid arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2020, 50, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, K.A.; Gammill, H.S.; Madeleine, M.M.; Dugowson, C.E.; Nelson, J.L. Parity and HLA alleles in risk of rheumatoid arthritis. Chimerism 2011, 2, 11–15. [Google Scholar] [CrossRef]
- Bengtsson, C.; Malspeis, S.; Orellana, C.; Sparks, J.A.; Costenbader, K.H.; Karlson, E.W. Association Between Menopausal Factors and the Risk of Seronegative and Seropositive Rheumatoid Arthritis: Results from the Nurses’ Health Studies. Arthritis Care Res. 2017, 69, 1676–1684. [Google Scholar] [CrossRef]
- Pikwer, M.; Bergström, U.; Nilsson, J.; Jacobsson, L.; Turesson, C. Early menopause is an independent predictor of rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.E.; Huang, W.-T.; Pope, J.; Haraoui, B.; Boire, G.; Thorne, J.C.; Hitchon, C.A.; Tin, D.; Keystone, E.C.; Bykerk, V.P.; et al. Effect of Age at Menopause on Disease Presentation in Early Rheumatoid Arthritis: Results from the Canadian Early Arthritis Cohort. Arthritis Care Res. 2015, 67, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Alpízar-Rodríguez, D.; Mueller, R.B.; Möller, B.; Dudler, J.; Ciurea, A.; Zufferey, P.; Kyburz, D.; Walker, U.A.; Von Mühlenen, I.; Roux-Lombard, P.; et al. Female hormonal factors and the development of anti-citrullinated protein antibodies in women at risk of rheumatoid arthritis. Rheumatology 2017, 56, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, C.; Bondt, A.; Harre, U.; Raufer, J.; Pfeifle, R.; Camponeschi, A.; Wuhrer, M.; Seeling, M.; Mårtensson, I.-L.; Nimmerjahn, F.; et al. Estrogen induces St6gal1 expression and increases IgG sialylation in mice and patients with rheumatoid arthritis: A potential explanation for the increased risk of rheumatoid arthritis in postmenopausal women. Arthritis Res. Ther. 2018, 20, 84. [Google Scholar] [CrossRef]
- Dominguez, P.; Han, J.; Li, T.; Ascherio, A.; Qureshi, A. Depression and the risk of psoriasis in US women. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1163–1167. [Google Scholar] [CrossRef]
- Lewinson, R.T.; Vallerand, I.A.; Lowerison, M.W.; Parsons, L.M.; Frolkis, A.D.; Kaplan, G.G.; Bulloch, A.G.; Swain, M.G.; Patten, S.; Barnabe, C. Depression Is Associated with an Increased Risk of Psoriatic Arthritis among Patients with Psoriasis: A Population-Based Study. J. Investig. Dermatol. 2017, 137, 828–835. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Pan, A.; Higuchi, L.M.; de Silva, P.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Association between depressive symptoms and incidence of Crohn’s disease and ulcerative colitis: Results from the Nurses’ Health Study. Clin. Gastroenterol. Hepatol. 2013, 11, 57–62. [Google Scholar] [CrossRef]
- Lu, M.-C.; Guo, H.-R.; Lin, M.-C.; Livneh, H.; Lai, N.-S.; Tsai, T.-Y. Bidirectional associations between rheumatoid arthritis and depression: A nationwide longitudinal study. Sci. Rep. 2016, 6, 20647. [Google Scholar] [CrossRef]
- Vallerand, I.; Lewinson, R.T.; Frolkis, A.D.; Lowerison, M.W.; Kaplan, G.G.; Swain, M.G.; Bulloch, A.G.M.; Patten, S.B.; Barnabe, C. Depression as a risk factor for the development of rheumatoid arthritis: A population-based cohort study. RMD Open 2018, 4, e000670. [Google Scholar] [CrossRef]
- Sparks, J.A.; Malspeis, S.; Hahn, J.; Wang, J.; Roberts, A.L.; Kubzansky, L.D.; Costenbader, K.H. Depression and Subsequent Risk for Incident Rheumatoid Arthritis Among Women. Arthritis Care Res. 2021, 73, 78–89. [Google Scholar] [CrossRef]
- Lee, Y.C.; Agnew-Blais, J.; Malspeis, S.; Keyes, K.M.; Costenbader, K.H.; Kubzansky, L.D.; Roberts, A.L.; Koenen, K.C.; Karlson, E.W. Post-Traumatic Stress Disorder and Risk for Incident Rheumatoid Arthritis. Arthritis Care Res. 2016, 68, 292–298. [Google Scholar] [CrossRef]
- Bookwalter, D.B.; Roenfeldt, K.A.; LeardMann, C.A.; Kong, S.Y.; Riddle, M.S.; Rull, R. Posttraumatic stress disorder and risk of selected autoimmune diseases among US military personnel. BMC Psychiatry 2020, 20, 23. [Google Scholar] [CrossRef]
- Miller-Archie, S.A.; Izmirly, P.M.; Berman, J.R.; Brite, J.; Walker, D.J.; Dasilva, R.C.; Petrsoric, L.J.; Cone, J.E. Systemic autoimmune disease among adults exposed to the September 11, 2001, terrorist attack. Arthritis Rheumatol. 2020, 72, 849–859. [Google Scholar] [CrossRef]
- Nerurkar, L.; Siebert, S.; McInnes, I.B.; Cavanagh, J. Rheumatoid arthritis and depression: An inflammatory perspective. Lancet Psychiatry 2019, 6, 164–173. [Google Scholar] [CrossRef]
- Michot, J.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.H.; Calabrese, C.; Cappelli, L.C. Rheumatic immune-related adverse events from cancer immunotherapy. Nat. Rev. Rheumatol. 2018, 14, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jaquith, J.M.; McCarthy-Fruin, K.; Zhu, X.; Zhou, X.; Li, Y.; Crowson, C.; Davis, J.M.; Thanarajasingam, U.; Zeng, H. Immune checkpoint inhibitor-induced inflammatory arthritis: A novel clinical entity with striking similarities to seronegative rheumatoid arthritis. Clin. Rheumatol. 2020, 39, 3631–3637. [Google Scholar] [CrossRef]
- Richter, M.D.; Crowson, C.; Kottschade, L.A.; Finnes, H.D.; Markovic, S.N.; Thanarajasingam, U. Rheumatic Syndromes Associated with Immune Checkpoint Inhibitors: A Single-Center Cohort of Sixty-One Patients. Arthritis Rheumatol. 2019, 71, 468–475. [Google Scholar] [CrossRef]
- Braaten, T.J.; Brahmer, J.R.; Forde, P.M.; Le, D.; Lipson, E.J.; Naidoo, J.; Schollenberger, M.; Zheng, L.; Bingham, C.O.; Shah, A.A.; et al. Immune checkpoint inhibitor-induced inflammatory arthritis persists after immunotherapy cessation. Ann. Rheum. Dis. 2020, 79, 332–338. [Google Scholar] [CrossRef]
- Cappelli, L.C.; Brahmer, J.R.; Forde, P.M.; Le, D.T.; Lipson, E.J.; Naidoo, J.; Zheng, L.; Bingham, C.O., 3rd; Shah, A.A. Clinical presentation of immune checkpoint inhibitor-induced inflammatory arthritis differs by immunotherapy regimen. Semin. Arthritis Rheum. 2018, 48, 553–557. [Google Scholar] [CrossRef]
- Rönnelid, J.; Hansson, M.; Mathsson-Alm, L.; Cornillet, M.; Reed, E.; Jakobsson, P.-J.; Alfredsson, L.; Holmdahl, R.; Skriner, K.; Serre, G.; et al. Anticitrullinated protein/peptide antibody multiplexing defines an extended group of ACPA-positive rheumatoid arthritis patients with distinct genetic and environmental determinants. Ann. Rheum. Dis. 2017, 77, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Trouw, L.; Rispens, T.; Toes, R. Beyond citrullination: Other post-translational protein modifications in rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Carubbi, F.; Alunno, A.; Gerli, R.; Giacomelli, R. Post-Translational Modifications of Proteins: Novel Insights in the Autoimmune Response in Rheumatoid Arthritis. Cells 2019, 8, 657. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Manzo, A.; Montecucco, C.; Caporali, R.F. The Clinical Value of Autoantibodies in Rheumatoid Arthritis. Front. Med. 2018, 5, 339. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Bogliolo, L.; Montecucco, C.; Manzo, A. B cell autoimmunity and bone damage in rheumatoid arthritis. Reumatismo 2016, 68, 117–125. [Google Scholar] [CrossRef]
- Ajeganova, S.; van Steenbergen, H.W.; Verheul, M.K.; Forslind, K.; Hafström, I.; Toes, R.E.; Huizinga, T.W.J.; Svensson, B.; Trouw, L.A.; van der Helm-van Mil, A.H.M. The association between anti-carbamylated protein (anti-CarP) antibodies and radiographic progression in early rheumatoid arthritis: A study exploring replication and the added value to ACPA and rheumatoid factor. Ann. Rheum. Dis. 2017, 76, 112–118. [Google Scholar] [CrossRef]
- Truchetet, M.E.; Dublanc, S.; Barnetche, T.; Vittecoq, O.; Mariette, X.; Richez, C.; Blanco, P.; Mahler, M.; Contin-Bordes, C.; Schaeverbeke, T.; et al. Association of the Presence of Anti-Carbamylated Protein Antibodies in Early Arthritis with a Poorer Clinical and Radiologic Outcome: Data from the French ESPOIR Cohort. Arthritis Rheumatol. 2017, 69, 2292–2302. [Google Scholar] [CrossRef]
- Lamacchia, C.; Courvoisier, D.S.; Jarlborg, M.; Bas, S.; Roux-Lombard, P.; Möller, B.; Ciurea, A.; Finckh, A.; Bentow, C.; Martinez-Prat, L.; et al. Predictive value of anti-CarP and anti-PAD3 antibodies alone or in combination with RF and ACPA for the severity of rheumatoid arthritis. Rheumatology 2021, 60, 4598–4608. [Google Scholar] [CrossRef]
- Regueiro, C.; Casares-Marfil, D.; Lundberg, K.; Knevel, R.; Acosta-Herrera, M.; Rodriguez-Rodriguez, L.; Lopez-Mejias, R.; Perez-Pampin, E.; Triguero-Martinez, A.; Nuño, L.; et al. HLA–B*08 Identified as the Most Prominently Associated Major Histocompatibility Complex Locus for Anti–Carbamylated Protein Antibody–Positive/Anti–Cyclic Citrullinated Peptide–Negative Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 73, 963–969. [Google Scholar] [CrossRef]
- Curran, A.M.; Naik, P.; Giles, J.T.; Darrah, E. PAD enzymes in rheumatoid arthritis: Pathogenic effectors and autoimmune targets. Nat. Rev. Rheumatol. 2020, 16, 301–315. [Google Scholar] [CrossRef]
- Li, K.; Mo, W.; Wu, L.; Wu, X.; Luo, C.; Xiao, X.; Jia, X.; Yang, H.; Fei, Y.; Chen, H.; et al. Novel autoantibodies identified in ACPA-negative rheumatoid arthritis. Ann. Rheum. Dis. 2021, 80, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Daigo, K.; Inforzato, A.; Barajon, I.; Garlanda, C.; Bottazzi, B.; Meri, S.; Mantovani, A. Pentraxins in the activation and regulation of innate immunity. Immunol. Rev. 2016, 274, 202–217. [Google Scholar] [CrossRef]
- McInnes, I.B.; Buckley, C.D.; Isaacs, J. Cytokines in rheumatoid arthritis—Shaping the immunological landscape. Nat. Rev. Rheumatol. 2015, 12, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; McInnes, I.B.; Neurath, M.F. Reframing Immune-Mediated Inflammatory Diseases through Signature Cytokine Hubs. N. Engl. J. Med. 2021, 385, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Yin, Y.; Li, X.; Shan, G.; Wu, X.; Liang, D.; Li, Y.; Zhang, X. The status of rheumatoid factor and anti-cyclic citrullinated peptide antibody are not associated with the effect of anti-TNFalpha agent treatment in patients with rheumatoid arthritis: A meta-analysis. PLoS ONE 2014, 9, e89442. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, M.; Shipa, M.; Yeoh, S.-A.; Roussou, E.; Mukerjee, D.; Ehrenstein, M.R. Methotrexate reduces withdrawal rates of TNF inhibitors due to ineffectiveness in rheumatoid arthritis but only in patients who are seropositive. Ann. Rheum. Dis. 2020, 79, 1516–1517. [Google Scholar] [CrossRef]
- Witte, T. Methotrexate as combination partner of TNF inhibitors and tocilizumab. What is reasonable from an immunological viewpoint? Clin. Rheumatol. 2015, 34, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.; Drutskaya, M.; Schlienz, D.; Gorshkova, E.; Kurz, K.; Morawietz, L.; Nedospasov, S. Contrasting contributions of TNF from distinct cellular sources in arthritis. Ann. Rheum. Dis. 2020, 79, 1453–1459. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Holweg, C.T.; Kummerfeld, S.K.; Choy, D.F.; Setiadi, A.F.; Hackney, J.A.; Haverty, P.M.; Gilbert, H.; Lin, W.Y.; Diehl, L.; et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res. Ther. 2014, 16, R90. [Google Scholar] [CrossRef]
- Humby, F.; Durez, P.; Buch, M.H.; Lewis, M.J.; Rizvi, H.; Rivellese, F.; Nerviani, A.; Giorli, G.; Mahto, A.; Montecucco, C.; et al. Rituximab versus tocilizumab in anti-TNF inadequate responder patients with rheumatoid arthritis (R4RA): 16-week outcomes of a stratified, biopsy-driven, multicentre, open-label, phase 4 randomised controlled trial. Lancet 2021, 397, 305–317. [Google Scholar] [CrossRef]
- Szekanecz, Z.; McInnes, I.B.; Schett, G.; Szamosi, S.; Benkő, S.; Szűcs, G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 2021, 17, 585–595. [Google Scholar] [CrossRef]
- Cohen, S.; Hurd, E.; Cush, J.; Schiff, M.; Weinblatt, M.E.; Moreland, L.; Kremer, J.; Bear, M.B.; Rich, W.J.; McCabe, D. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: Results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 614–624. [Google Scholar] [CrossRef]
- Patel, D.D.; Lee, D.M.; Kolbinger, F.; Antoni, C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann. Rheum. Dis. 2013, 72 (Suppl. 2), iii116–iii123. [Google Scholar] [CrossRef]
- Orr, C.; Vieira-Sousa, E.; Boyle, D.L.; Buch, M.H.; Buckley, C.D.; Cañete, J.D.; Catrina, A.I.; Choy, E.H.S.; Emery, P.; Fearon, U.; et al. Synovial tissue research: A state-of-the-art review. Nat. Rev. Rheumatol. 2017, 13, 463–475. [Google Scholar] [CrossRef]
- Manzo, A.; Bugatti, S.; Caporali, R.F.; Montecucco, C. Histopathology of the synovial tissue: Perspectives for biomarker development in chronic inflammatory arthritides. Reumatismo 2018, 70, 121–132. [Google Scholar] [CrossRef]
- Manzo, A.; Bugatti, S.; Rossi, S. Clinical Applications of Synovial Biopsy. Front. Med. 2019, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Bozzalla Cassione, E.; De Stefano, L.; Manzo, A. Established rheumatoid arthritis. The pathogenic aspects. Best Pract. Res. Clin. Rheumatol. 2019, 33, 101478. [Google Scholar] [CrossRef]
- Bugatti, S.; Manzo, A.; Bombardieri, M.; Vitolo, B.; Humby, F.; Kelly, S.; Montecucco, C.; Pitzalis, C. Synovial Tissue Heterogeneity and Peripheral Blood Biomarkers. Curr. Rheumatol. Rep. 2011, 13, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, M.G.; Baeten, D.L. Immunopathology of synovitis: From histology to molecular pathways. Rheumatology 2016, 55, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Humby, F.; Lewis, M.; Ramamoorthi, N.; Hackney, J.; Barnes, M.R.; Bombardieri, M.; Setiadi, A.F.; Kelly, S.; Bene, F.; DiCicco, M.; et al. Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients. Ann. Rheum. Dis. 2019, 78, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Manzo, A.; Vitolo, B.; Benaglio, F.; Binda, E.; Scarabelli, M.; Humby, F.; Caporali, R.; Pitzalis, C.; Montecucco, C. High expression levels of the B cell chemoattractant CXCL13 in rheumatoid synovium are a marker of severe disease. Rheumatology 2014, 53, 1886–1895. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Manzo, A.; Benaglio, F.; Klersy, C.; Vitolo, B.; Todoerti, M.; Sakellariou, G.; Montecucco, C.; Caporali, R. Serum levels of CXCL13 are associated with ultrasonographic synovitis and predict power Doppler persistence in early rheumatoid arthritis treated with non-biological disease-modifying anti-rheumatic drugs. Arthritis Res. Ther. 2012, 14, R34. [Google Scholar] [CrossRef] [PubMed]
- Orr, C.; Najm, A.; Biniecka, M.; McGarry, T.; Ng, C.T.; Young, F.; Fearon, U.; Veale, D.J. Synovial Immunophenotype and Anti-Citrullinated Peptide Antibodies in Rheumatoid Arthritis Patients: Relationship to Treatment Response and Radiologic Prognosis. Arthritis Rheumatol. 2017, 69, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Lliso-Ribera, G.; Humby, F.; Lewis, M.; Nerviani, A.; Mauro, D.; Rivellese, F.; Kelly, S.; Hands, R.; Bene, F.; Ramamoorthi, N.; et al. Synovial tissue signatures enhance clinical classification and prognostic/treatment response algorithms in early inflammatory arthritis and predict requirement for subsequent biological therapy: Results from the pathobiology of early arthritis cohort (PEAC). Ann. Rheum. Dis. 2019, 78, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, F.; Humby, F.; Bugatti, S.; Fossati-Jimack, L.; Rizvi, H.; Lucchesi, D.; Lliso-Ribera, G.; Nerviani, A.; Hands, R.E.; Giorli, G.; et al. B Cell Synovitis and Clinical Phenotypes in Rheumatoid Arthritis: Relationship to Disease Stages and Drug Exposure. Arthritis Rheumatol. 2020, 72, 714–725. [Google Scholar] [CrossRef]
- Bugatti, S.; Manzo, A.; Caporali, R.F.; Montecucco, C. Assessment of synovitis to predict bone erosions in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2012, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, M.; Bajema, I.; Levarht, E.W.; Toes, R.E.; Huizinga, T.W.; van Laar, J.M. Differences in synovial tissue infiltrates between anti-cyclic citrullinated peptide-positive rheumatoid arthritis and anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheum. 2008, 58, 53–60. [Google Scholar] [CrossRef]
- Floudas, A.; Canavan, M.; McGarry, T.; Mullan, R.; Nagpal, S.; Veale, D.; Fearon, U. ACPA Status Correlates with Differential Immune Profile in Patients with Rheumatoid Arthritis. Cells 2021, 10, 647. [Google Scholar] [CrossRef]
- Wu, X.; Liu, Y.; Jin, S.; Wang, M.; Jiao, Y.; Yang, B.; Lu, X.; Ji, X.; Fei, Y.; Yang, H.; et al. Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis. Nat. Commun. 2021, 12, 4977. [Google Scholar] [CrossRef]
- Cañete, J.D.; Santiago, B.; Cantaert, T.; Sanmartí, R.; Palacin, A.; Celis, R.; Graell, E.; Gil-Torregrosa, B.; Baeten, D.; Pablos, J.L. Ectopic lymphoid neogenesis in psoriatic arthritis. Ann. Rheum. Dis. 2007, 66, 720–726. [Google Scholar] [CrossRef]
- Alivernini, S.; Bruno, D.; Tolusso, B.; Bui, L.; Petricca, L.; Gigante, M.R.; Birra, D.; Fedele, A.L.; Peluso, G.; Federico, F.; et al. Differential synovial tissue biomarkers among psoriatic arthritis and rheumatoid factor/anti-citrulline antibody-negative rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 116. [Google Scholar] [CrossRef]
- Taylor, P.; Atzeni, F.; Balsa, A.; Gossec, L.; Müller-Ladner, U.; Pope, J. The Key Comorbidities in Patients with Rheumatoid Arthritis: A Narrative Review. J. Clin. Med. 2021, 10, 509. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.E.; Choy, E.H. C-reactive protein and implications in rheumatoid arthritis and associated comorbidities. Semin. Arthritis Rheum. 2021, 51, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Goodson, N.J.; Wiles, N.J.; Lunt, M.; Barrett, E.M.; Silman, A.J.; Symmons, D.P.M. Mortality in early inflammatory polyarthritis: Cardiovascular mortality is increased in seropositive patients. Arthritis Rheum. 2002, 46, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Mohning, M.P.; Amigues, I.; Demoruelle, M.K.; Pérez, E.R.F.; Huie, T.J.; Keith, R.K.; Olson, A.L.; Yunt, Z.X.; Chung, J.H.; Hobbs, S.; et al. Duration of rheumatoid arthritis and the risk of developing interstitial lung disease. ERJ Open Res. 2021, 7, 00633–2020. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Bogliolo, L.; Vitolo, B.; Manzo, A.; Montecucco, C.; Caporali, R. Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 226. [Google Scholar] [CrossRef]
- Dong, H.; Julien, P.J.; Demoruelle, M.K.; Deane, K.D.; Weisman, M.H. Interstitial lung abnormalities in patients with early rheumatoid arthritis: A pilot study evaluating prevalence and progression. Eur. J. Rheumatol. 2018, 6, 193–198. [Google Scholar] [CrossRef]
- Bugatti, S.; Bogliolo, L.; Manzo, A.; De Stefano, L.; Delvino, P.; Motta, F.; Montecucco, C. Impact of Anti-Citrullinated Protein Antibodies on Progressive Systemic Bone Mineral Density Loss in Patients with Early Rheumatoid Arthritis After Two Years of Treat-to-Target. Front. Immunol. 2021, 12, 701922. [Google Scholar] [CrossRef]
- Barra, L.J.; Pope, J.; Hitchon, C.; Boire, G.; Schieir, O.; Lin, D.; Thorne, C.J.; Tin, D.; Keystone, E.C.; Haraoui, B.; et al. The effect of rheumatoid arthritis–associated autoantibodies on the incidence of cardiovascular events in a large inception cohort of early inflammatory arthritis. Rheumatology 2017, 56, 768–776. [Google Scholar] [CrossRef][Green Version]
- Kastbom, A.; Ärlestig, L.; Rantapää-Dahlqvist, S. Genetic Variants of the NLRP3 Inflammasome Are Associated with Stroke in Patients with Rheumatoid Arthritis. J. Rheumatol. 2015, 42, 1740–1745. [Google Scholar] [CrossRef]
- Lwin, M.N.; Serhal, L.; Holroyd, C.; Edwards, C.J. Rheumatoid Arthritis: The Impact of Mental Health on Disease: A Narrative Review. Rheumatol. Ther. 2020, 7, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Süß, P.; Rothe, T.; Hoffmann, A.; Schlachetzki, J.C.M.; Winkler, J. The Joint-Brain Axis: Insights from Rheumatoid Arthritis on the Crosstalk Between Chronic Peripheral Inflammation and the Brain. Front. Immunol. 2020, 11, 612104. [Google Scholar] [CrossRef] [PubMed]


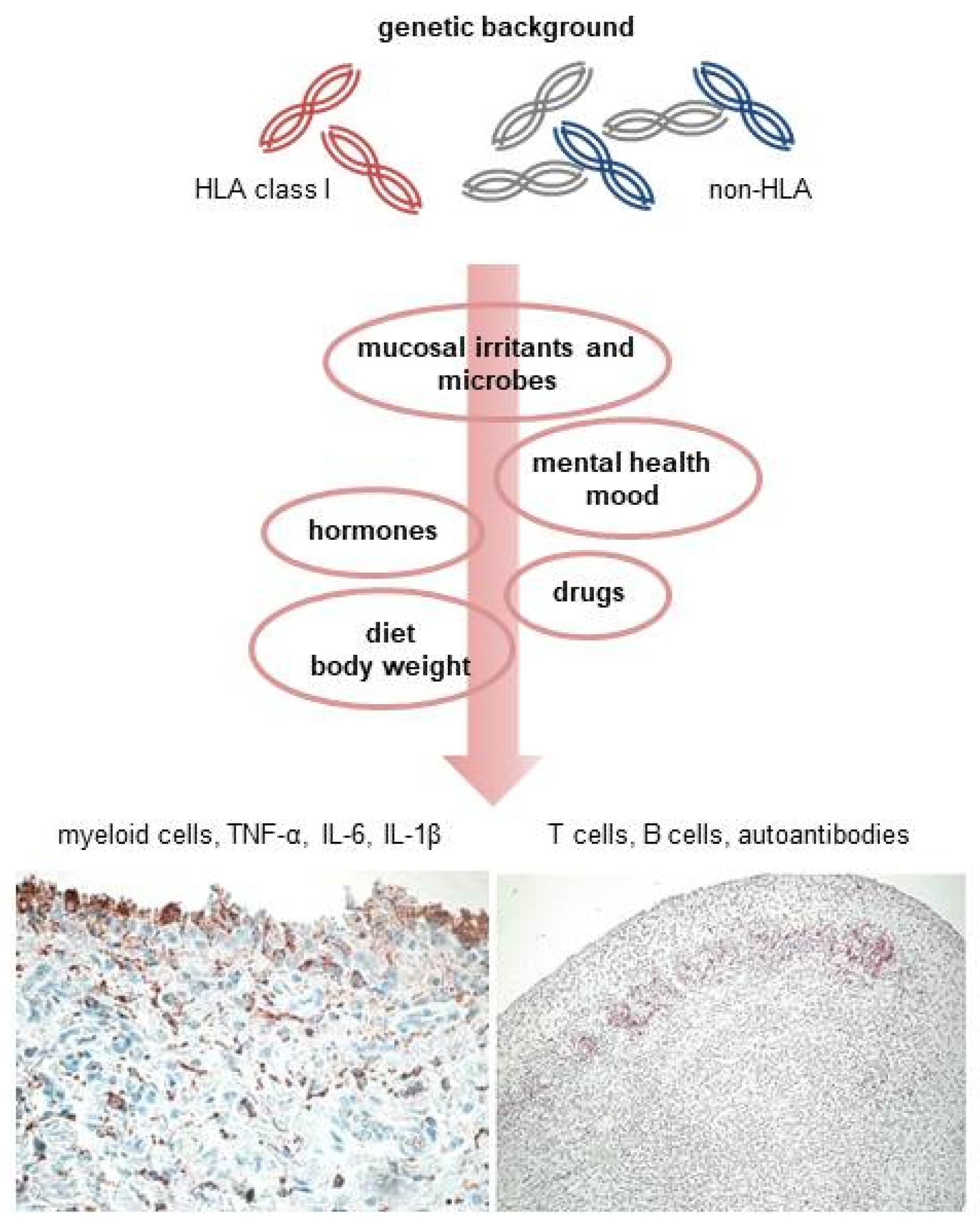{kind=link}
| Autoantibody-Positive | Autoantibody-Negative | |
|---|---|---|
| Genetic associations | shared epitope-containing HLA-DRB1 alleles | HLA-B*08 with aspartate at position 9 and DRB1*03 with serine at position 11 non-HLA genes |
| Environmental and endogenous factors | Smoking silica air pollution organic dusts asbestos pulmonary inflammation dysbiosis early menopause | air pollution organic dusts asbestos pulmonary inflammation dysbiosis excess body weight postpartum early menopause depression post-traumatic stress immune checkpoint inhibitors |
| Autoantibodies Anti-CarP Anti-acetylated Anti-PAD | 15–65% 40–60% 2–18% | 10–15% 10–25% 3–19% |
| Cytokines | TNF-α (lymphoid) IL-6 | TNF-α (myeloid) IL-6 IL-1β |
| Synovial pathology | mostly lympho-myeloid pattern higher levels of CD19+ B cells, CD3+ T cells, lymphoid aggregates, germinal centers | lympho-myeloid, diffuse-myeloid and pauci-immune pattern lower antigen processing and presentation activity in B cells lower cytotoxic and exhausted gene expression in T cells higher proinflammatory cytokine expression in macrophages |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Stefano, L.; D’Onofrio, B.; Manzo, A.; Montecucco, C.; Bugatti, S. The Genetic, Environmental, and Immunopathological Complexity of Autoantibody-Negative Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 12386. https://doi.org/10.3390/ijms222212386
De Stefano L, D’Onofrio B, Manzo A, Montecucco C, Bugatti S. The Genetic, Environmental, and Immunopathological Complexity of Autoantibody-Negative Rheumatoid Arthritis. International Journal of Molecular Sciences. 2021; 22(22):12386. https://doi.org/10.3390/ijms222212386
Chicago/Turabian StyleDe Stefano, Ludovico, Bernardo D’Onofrio, Antonio Manzo, Carlomaurizio Montecucco, and Serena Bugatti. 2021. "The Genetic, Environmental, and Immunopathological Complexity of Autoantibody-Negative Rheumatoid Arthritis" International Journal of Molecular Sciences 22, no. 22: 12386. https://doi.org/10.3390/ijms222212386
APA StyleDe Stefano, L., D’Onofrio, B., Manzo, A., Montecucco, C., & Bugatti, S. (2021). The Genetic, Environmental, and Immunopathological Complexity of Autoantibody-Negative Rheumatoid Arthritis. International Journal of Molecular Sciences, 22(22), 12386. https://doi.org/10.3390/ijms222212386