
Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect



Abstract
:1. Introduction
2. Ex Vivo-Generated DC Vaccines
2.1. Maturation Protocols
2.2. Extensive-vs. Short-Culture Period
2.3. Tumor Antigens and DCs Loading
2.4. Clinical Trials
3. Immunotherapy in Combination with the Standards of Care: May It Help?
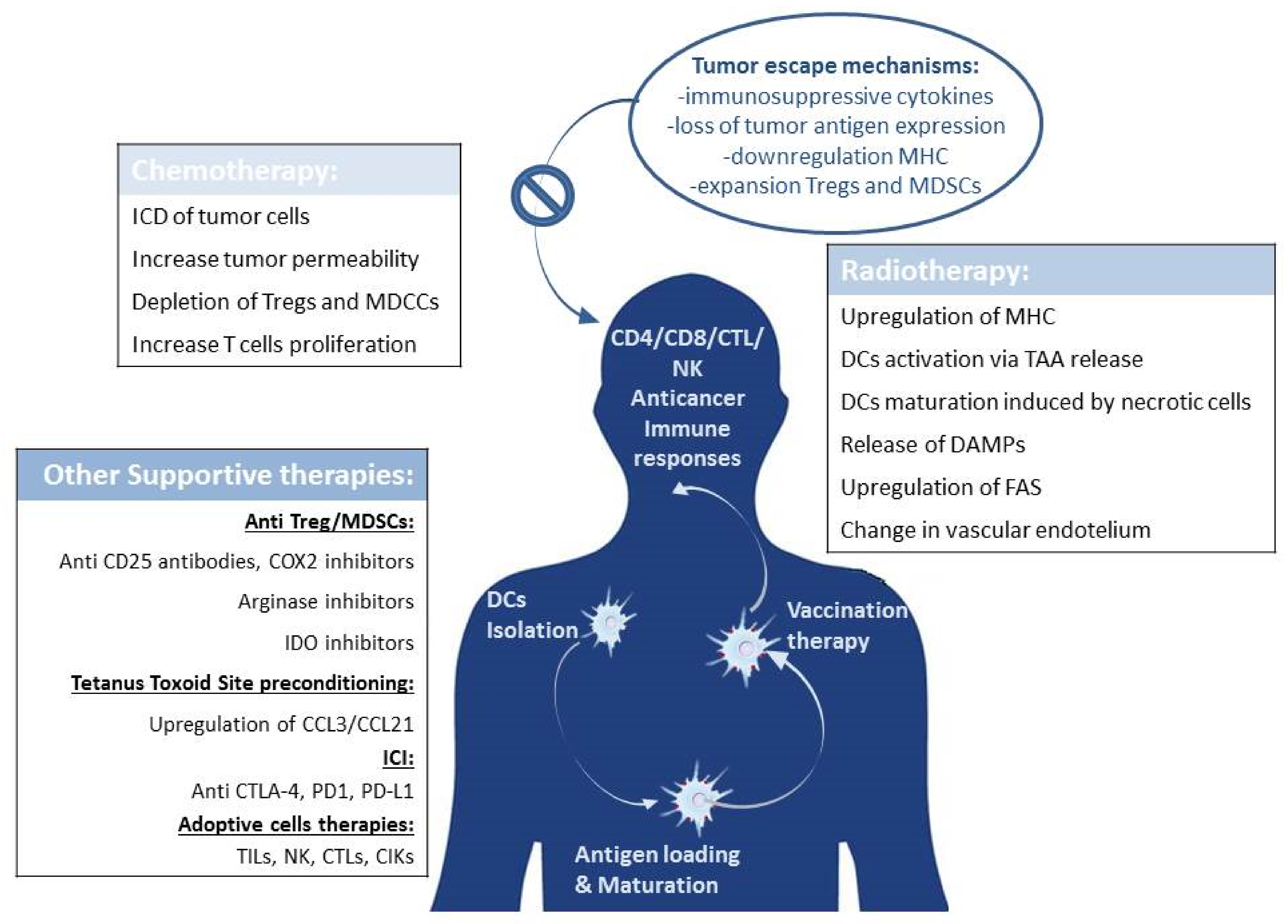
3.1. DCs Immunotherapy Combined with Chemotherapy
3.2. DCs Therapy Combined with Radiotherapy
3.3. Cryoablation
4. Combating Tumor Immune Evasion
4.1. Dendritic Cells Combined with Immune Checkpoint Inhibition
4.2. Dendritic Cells Vaccination Combined with Adoptive Cell Therapies
5. How to Further Improve the DCs Vaccine Efficacy: Lessons Learned from Site Preconditioning in Glioblastoma
6. Rationale for DC Vaccination in the Adjuvant Treatment of Cancer
7. Conclusions and Future Prospects
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MHC | Major Histocompatibility Complex |
| TAA | tumor associated antigens |
| DAMPS | damage-associated molecular patterns |
| MDSCs | myeloid-derived suppressor cells |
| COX2 | cyclooxygenase-2 |
| IDO | indoleamine 2,3-dioxygenase |
| CCL3 | C-C motif chemokine ligand 3 |
| CCL21 | C-C motif chemokine ligand 21 |
| TILs | tumor infiltrating lymphocytes |
| CTL | cytotoxic T lymphocytes |
| NK | natural killer cells |
| ICI | immune checkpoint inhibitors |
| CIK | cytokines induced killer cells |
References
- Bol, K.F.; Chreibelt, G.; Gerritsen, W.R.; de Vries, J.M.; Figdor, C.G. Dendritic Cell–Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, S.; Yang, J.; Ronchese, F. Monocyte-Derived Dendritic Cells Are Essential for CD8(+) T Cell Activation and Antitumor Responses After Local Immunotherapy. Front. Immunol. 2015, 6, 584. [Google Scholar] [CrossRef] [Green Version]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Draube, A.; Klein-Gonzalez, N.; Mattheus, S.; Brillant, C.; Hellmich, M.; Engert, A.; von Bergwelt-Baildon, M. Dendritic cell based tumor vaccination in prostate and renal cell cancer: A systematic review and meta-analysis. PLoS ONE 2011, 6, e18801. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Ugurel, S.; Schuler-Thurner, B.; Nestle, F.O.; Enk, A.; Brocker, E.B.; Grabbe, S.; Rittgen, W.; Edler, L.; Sucker, A.; et al. Dacarbazine (DTIC) versus vaccination with autologous peptidepulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: A randomized phase III trial of the DC study group of the DeCOG. Ann. Oncol. 2006, 17, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Bernstein, G.T.; Corman, J.M.; Glode, L.M.; Hall, S.J.; Poll, W.L.; Schellhammer, P.F.; Jones, L.A.; Xu, Y.; Kylstra, J.W.; et al. Randomized trial of autologous cellular immunotherapy with sipuleucel-T in androgen-dependent prostate cancer. Clin. Cancer Res. 2011, 17, 4558–4567. [Google Scholar] [CrossRef] [Green Version]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. for the IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Enomoto, Y.; Kume, H.; Nakagawa, T.; Fukuhara, H.; Suzuki, M.; Fujimura, T.; Homma, Y.; Kakimi, K. A pilot study of autologous tumor lysate-loaded dendritic cell vaccination combined with sunitinib for metastatic renal cell carcinoma. J. Immunother. Cancer 2014, 2, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, Y.; Shen, J.; He, F.; Zhang, D.; Chen, Z.; Duan, Y.; Sun, J. Feasibility study of DCs/CIKs combined with thoracic radiotherapy for patients with locally advanced or metastatic non-small-cell lung cancer. Radiat. Oncol. 2016, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, R.; Koizumi, T.; Koya, T.; Sano, K.; Koido, S.; Nagai, K.; Kobayashi, M.; Okamoto, M.; Sugiyama, H.; Shimodaira, S. WT1-pulsed Dendritic Cell Vaccine Combined with Chemotherapy for Resected Pancreatic Cancer in a Phase I Study. Anticancer Res. 2018, 38, 2217–2225. [Google Scholar]
- Laurell, A.; Lönnemark, M.; Brekkan, E.; Magnusson, A.; Tolf, A.; Wallgren, A.C.; Andersson, B.; Adamson, L.; Kiessling, R.; Karlsson-Parra, A. Intratumorally injected pro-inflammatory allogeneic dendritic cells as immune enhancers: A first-in-human study in unfavourable risk patients with metastatic renal cell carcinoma. J. Immunother. Cancer 2017, 5, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J. Immunother. 2015, 38, 71–76. [Google Scholar] [CrossRef]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.; Duiveman-de Boer, T.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [Green Version]
- Van Eck van der Sluijs, J.; van Ens, D.; Thordardottir, S.; Vodegel, D.; Hermens, I.; van der Waart, A.B.; Falkenburg, J.H.F.; Kester, M.G.D.; de Rink, I.; Heemskerk, M.H.M.; et al. Clinically applicable CD34+-derived blood dendritic cell subsets exhibit key subset-specific features and potently boost anti-tumor T and NK cell responses. Cancer Immunol. Immunother. 2021, 70, 3167–3181. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, I.J.; Eggert, A.A.; Scharenborg, N.M.; Vissers, J.L.; Lesterhuis, W.J.; Boerman, O.C.; Punt, C.J.; Adema, G.J.; Figdor, C.G. Phenotypical and functional characterization of clinical grade dendritic cells. J. Immunother. 2002, 25, 429–438. [Google Scholar] [CrossRef] [Green Version]
- McIlroy, D.; Gregoire, M. Optimizing dendritic cell-based anticancer immunotherapy: Maturation state does have clinical impact. Cancer Immunol. Immunother. 2003, 52, 583–591. [Google Scholar] [CrossRef] [PubMed]
- De Vries, I.J.; Krooshoop, D.J.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.; Van Muijen, G.N.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.; et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar] [PubMed]
- Dhodapkar, M.V.; Steinman, R.M.; Sapp, M.; Desai, H.; Fossella, C.; Krasovsky, J.; Donahoe, S.M.; Dunbar, P.R.; Cerundolo, V.; Nixon, D.F.; et al. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J. Clin. Investig. 1999, 104, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonuleit, H.; Kühn, U.; Müller, G.; Steinbrink, K.; Paragnik, L.; Schmitt, E.; Knop, J.; Enk, A.H. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J. Immunol. 1997, 27, 3135–3142. [Google Scholar] [CrossRef]
- Bol, K.F.; Aarntzen, E.H.; Hout, F.E.; Schreibelt, G.; Creemers, J.H.; Lesterhuis, W.J.; Gerritsen, W.R.; Grunhagen, D.J.; Verhoef, C.; Punt, C.J.; et al. Favorable overall survival in stage III melanoma patients after adjuvant dendritic cell vaccination. Oncoimmunology 2015, 5, e1057673. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Fields, R.C.; Giedlin, M.; Mulé, J.J. Systemic administration of interleukin 2 enhances the therapeutic efficacy of dendritic cell-based tumor vaccines. Proc. Natl. Acad. Sci. USA 1999, 96, 2268–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bol, K.F.; Aarntzen, E.H.; Pots, J.M.; Olde Nordkamp, M.A.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; van Oorschot, T.G.; Croockewit, S.A.; Blokx, W.A.; et al. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol. Immunother. 2016, 65, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Lövgren, T.; Sarhan, D.; Truxová, I.; Choudhary, B.; Maas, R.; Melief, J.; Nyström, M.; Edbäck, U.; Vermeij, R.; Scurti, G.; et al. Enhanced stimulation of human tumor-specific T cells by dendritic cells matured in the presence of interferon-γ and multiple toll-like receptor agonists. Cancer Immunol. Immunother. 2017, 66, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Boullart, A.C.; Aarntzen, E.H.; Verdijk, P.; Jacobs, J.F.; Schuurhuis, D.H.; Benitez-Ribas, D.; Schreibelt, G.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.; et al. Maturation of monocyte-derived dendritic cells with Toll-like receptor 3 and 7/8 ligands combined with prostaglandin E2 results in high interleukin-12 production and cell migration. Cancer Immunol. Immunother. 2008, 57, 1589–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, H.O.; Kim, H.J.; Lee, K.; Kim, H.S. Generation of functionally mature dendritic cells from elutriated monocytes using polyinosinic: Polycytidylic acid and soluble CD40 ligand for clinical application. Clin. Exp. Immunol. 2008, 154, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, S.I.M.; Ju, X.; Horvath, L.G.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef]
- Bonehill, A.; Van Nuffel, A.M.; Corthals, J.; Tuyaerts, S.; Heirman, C.; François, V.; Colau, D.; van der Bruggen, P.; Neyns, B.; Thielemans, K. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef] [Green Version]
- Pen, J.J.; De Keersmaecker, B.; Maenhout, S.K.; Van Nuffel, A.M.; Heirman, C.; Corthals, J.; Escors, D.; Bonehill, A.; Thielemans, K.; Breckpot, K.; et al. Modulation of regulatory T cell function by monocyte-derived dendritic cells matured through electroporation with mRNA encoding CD40 ligand, constitutively active TLR4, and CD70. J. Immunol. 2013, 191, 1976–1983. [Google Scholar] [CrossRef] [Green Version]
- Nava, S.; Lisini, D.; Frigerio, S.; Pogliani, S.; Pellegatta, S.; Gatti, L.; Finocchiaro, G.; Bersano, A.; Parati, E.A. PGE2 Is Crucial for the Generation of FAST Whole-Tumor-Antigens Loaded Dendritic Cells Suitable for Immunotherapy in Glioblastoma. Pharmaceutics 2020, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Bürdek, M.; Spranger, S.; Wilde, S.; Frankenberger, B.; Schendel, D.J.; Geiger, C. Three-day dendritic cells for vaccine development: Antigen uptake, processing and presentation. J. Transl. Med. 2010, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekete, N.; Rojewski, M.T.; Fürst, D.; Kreja, L.; Ignatius, A.; Dausend, J.; Schrezenmeier, H. GMP-compliant isolation and large-scale expansion of bone marrow-derived MSC. PLoS ONE 2012, 7, e43255. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef]
- Strome, S.E.; Voss, S.; Wilcox, R.; Wakefield, T.L.; Tamada, K.; Flies, D.; Chapoval, A.; Lu, J.; Kasperbauer, J.L.; Padley, D.; et al. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res. 2002, 62, 1884–1889. [Google Scholar]
- Nava, S.; Dossena, M.; Pogliani, S.; Pellegatta, S.; Antozzi, C.; Baggi, F.; Gellera, C.; Pollo, B.; Parati, E.A.; Finocchiaro, G.; et al. An optimized method for manufacturing a clinical scale dendritic cell-based vaccine for the treatment of glioblastoma. PLoS ONE 2012, 7, e52301. [Google Scholar] [CrossRef]
- Boudousquié, C.; Boand, V.; Lingre, E.; Dutoit, L.; Balint, K.; Danilo, M.; Harari, A.; Gannon, P.O.; Kandalaft, L.E. Development and Optimization of a GMP-Compliant Manufacturing Process for a Personalized Tumor Lysate Dendritic Cell Vaccine. Vaccines 2020, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Bosch, M.L.; Salgaller, M.L. Current methods for loading dendritic cells with tumor antigen for the induction of antitumor immunity. J. Immunother. 2002, 25, 289–303. [Google Scholar] [CrossRef]
- Chiang, C.L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [Green Version]
- Martin-Lluesma, S.; Graciotti, M.; Grimm, A.J.; Boudousquié, C.; Chiang, C.L.; Kandalaft, L.E. Are dendritic cells the most appropriate therapeutic vaccine for patients with ovarian cancer? Curr. Opin. Biotechnol. 2020, 65, 190–196. [Google Scholar] [CrossRef]
- Matera, L. The choice of the antigen in the dendritic cell-based vaccine therapy for prostate cancer. Cancer Treat. Rev. 2010, 36, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Celluzzi, C.M.; Falo, L.D., Jr. Physical interaction between dendritic cells and tumor cells results in an immunogen that induces protective and therapeutic tumor rejection. J. Immunol. 1998, 160, 3081–3085. [Google Scholar] [PubMed]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, P.F.; Chodak, G.; Whitmore, J.B.; Sims, R.; Frohlich, M.W.; Kantoff, P.W. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-T in the Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT) trial. Urology 2013, 81, 1297–1302. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Lawson, D.; Richards, J.; Conry, R.M.; Miller, D.; Triesman, J.; Gailani, F.; Riley, L.B.; Vena, D.; Hwu, P. A phase III multi-institutional randomized study of immunization with the gp100:209–217 (210M) peptide followed by high-dose IL-2 compared with high-dose IL-2 alone in patients with metastatic melanoma. J. Clin. Oncol. Abstr. 2009, 27, CRA9011. [Google Scholar] [CrossRef]
- Schuster, S.J.; Neelapu, S.S.; Gause, B.L.; Muggia, F.M.; Gockerman, J.P.; Sotomayor, J.E.M.; Winter, N.; Flowers, C.R.; Stergiou, A.M.; Kwak, L.W. Idiotype vaccine therapy (BiovaxID) in follicular lymphoma in first complete remission: Phase III clinical trial results. J. Clin. Oncol. Abstr. 2009, 27, 2. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Glode, L.M.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Schmitt, N.; Klechevsky, E.; Pedroza-Gonzalez, A.; Matsui, T.; Zurawski, G.; Oh, S.; Fay, J.; Pascual, V.; Banchereau, J.; et al. Harnessing human dendritic cell subsets for medicine. Immunol. Rev. 2010, 234, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, K.; Ueno, H.; Roberts, L.; Fay, J.; Banchereau, J. Dendritic cells: Are they clinically relevant? Cancer J. 2010, 16, 318–324. [Google Scholar] [CrossRef]
- Gilboa, E. The makings of a tumor rejection antigen. Immunity 1999, 11, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Parmiani, G.; De Filippo, A.; Novellino, L.; Castelli, C. Unique human tumor antigens: Immunobiology and use in clinical trials. J. Immunol. 2007, 178, 1975–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, T.; Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P. Human T cell responses against melanoma. Annu. Rev. Immunol. 2006, 24, 175–208. [Google Scholar] [CrossRef]
- Finn, O.J. Cancer immunology. N. Engl. J. Med. 2008, 358, 2704–2715. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Douek, D.C.; Price, D.A. CD8+ T cell efficacy in vaccination and disease. Nat. Med. 2008, 14, 623–628. [Google Scholar] [CrossRef]
- Finn, O.J. Cancer vaccines: Between the idea and the reality. Nat. Rev. Immunol. 2003, 3, 630–641. [Google Scholar] [CrossRef]
- Harari, A.; Graciotti, M.; Bassani-Sternberg, M.; Kandalaft, L.E. Antitumour dendritic cell vaccination in a priming and boosting approach. Nat. Rev. Drug Discov. 2020, 19, 635–652. [Google Scholar] [CrossRef]
- Prins, R.M.; Odesa, S.K.; Liau, L.M. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res. 2003, 63, 8487–8491. [Google Scholar]
- Harada, M.; Li, Y.F.; El-Gamil, M.; Ohnmacht, G.A.; Rosenberg, S.A.; Robbins, P.F. Melanoma-Reactive CD8+ T cells recognize a novel tumor antigen expressed in a wide variety of tumor types. J. Immunother. 2001, 24, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gulijk, M.; Dammeijer, F.; Aerts, J.G.J.V.; Vroman, H. Combination Strategies to Optimize Efficacy of Dendritic Cell-Based Immunotherapy. Front. Immunol. 2018, 9, 2759. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, M.V.; June, C.H. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin. Cancer Res. 2016, 22, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlom, J. Therapeutic cancer vaccines: Current status moving forward. J. Natl. Cancer Inst. 2012, 104, 599–613. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, C.J.; Das, A.; Liu, G.; Yu, J.S.; Black, K.L. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin. Cancer Res. 2004, 10, 5316–5326. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; André, F.; Tesniere, A.; Kroemer, G. The anticancer immune response: Indispensable for therapeutic success? J. Clin. Investig. 2008, 118, 1991–2001. [Google Scholar] [CrossRef] [Green Version]
- Sadeghzadeh, M.; Bornehdeli, S.; Mohahammadrezakhani, H.; Abolghasemi, M.; Poursaei, E.; Asadi, M.; Zafari, V.; Aghebati-Maleki, L.; Shanehbandi, D. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020, 254, 117580. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Sun, W.; Hussain, S.F.; Dey, M.; Crutcher, L.; Aldape, K.; Gilbert, M.; Hassenbusch, S.J.; Sawaya, R.; Schmittling, B.; et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: Case study. Neuro Oncol. 2008, 10, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Fritzell, S.; Sandén, E.; Eberstål, S.; Visse, E.; Darabi, A.; Siesjö, P. Intratumoral temozolomide synergizes with immunotherapy in a T cell-dependent fashion. Cancer Immunol. Immunother. 2013, 62, 1463–1474. [Google Scholar] [CrossRef]
- Litterman, A.J.; Zellmer, D.M.; Grinnen, K.L.; Hunt, M.A.; Dudek, A.Z.; Salazar, A.M.; Ohlfest, J.R. Profound impairment of adaptive immune responses by alkylating chemotherapy. J. Immunol. 2013, 190, 6259–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litterman, A.J.; Dudek, A.Z.; Largaespada, D.A. Alkylating chemotherapy may exert a uniquely deleterious effect upon neo-antigen-targeting anticancer vaccination. Oncoimmunology 2013, 2, e26294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegatta, S.; Eoli, M.; Cuccarini, V.; Anghileri, E.; Pollo, B.; Pessina, S.; Frigerio, S.; Servida, M.; Cuppini, L.; Antozzi, C.; et al. Survival gain in glioblastoma patients treated with dendritic cell immunotherapy is associated with increased NK but not CD8+ T cell activation in the presence of adjuvant temozolomide. Oncoimmunology 2018, 7, e1412901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eoli, M.; Corbetta, C.; Anghileri, E.; Di Ianni, N.; Milani, M.; Cuccarini, V.; Musio, S.; Paterra, R.; Frigerio, S.; Nava, S.; et al. Expansion of effector and memory T cells is associated with increased survival in recurrent glioblastomas treated with dendritic cell immunotherapy. Neuro Oncol. Adv. 2019, 1, vdz022. [Google Scholar] [CrossRef] [Green Version]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.A.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro Oncol. 2019, 21, 730–741. [Google Scholar] [CrossRef]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011, 3, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Lamph, W.W. Cross-coupling of AP-1 and intracellular hormone receptors. Cancer Cells 1991, 3, 183–185. [Google Scholar]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Kimura, H.; Matsui, Y.; Ishikawa, A.; Nakajima, T.; Yoshino, M.; Sakairi, Y. Randomized controlled phase III trial of adjuvant chemo-immunotherapy with activated killer T cells and dendritic cells in patients with resected primary lung cancer. Cancer Immunol. Immunother. 2015, 64, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellebaek, E.; Engell-Noerregaard, L.; Iversen, T.Z.; Froesig, T.M.; Munir, S.; Hadrup, S.R.; Andersen, M.H.; Svane, I.M. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: Results from a phase II trial. Cancer Immunol. Immunother. 2012, 61, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, H.; Li, L.; Zhang, Y. Effects of a gemcitabine plus platinum regimen combined with a dendritic cell-cytokine induced killer immunotherapy on recurrence and survival rate of non-small cell lung cancer patients. Exp. Ther. Med. 2014, 7, 1403–1407. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.A.; Kim, J.E.; Ruzevick, J.; Li, G.; Lim, M. The future of glioblastoma therapy: Synergism of standard of care and immunotherapy. Cancers 2014, 6, 1953–1985. [Google Scholar] [CrossRef] [Green Version]
- Gough, M.J.; Crittenden, M.R. Combination approaches to immunotherapy: The radiotherapy example. Immunotherapy 2009, 1, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.M.; Fan, S.; Rockwell, S.; Goldberg, I.D. The molecular and cellular basis of radiosensitivity: Implications for understanding how normal tissues and tumors respond to therapeutic radiation. Cancer Investig. 1999, 17, 56–72. [Google Scholar] [CrossRef]
- Stone, H.B.; Peters, L.J.; Milas, L. Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J. Natl. Cancer Inst. 1979, 63, 1229–1235. [Google Scholar]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef]
- Gerber, S.A.; Sedlacek, A.L.; Cron, K.R.; Murphy, S.P.; Frelinger, J.G.; Lord, E.M. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am. J. Pathol. 2013, 182, 2345–2354. [Google Scholar] [CrossRef] [Green Version]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar] [PubMed]
- Sharabi, A.B.; Nirschl, C.J.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Velarde, E.; Deweese, T.L.; Drake, C.G. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol. Res. 2015, 3, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Garnett, C.T.; Palena, C.; Chakraborty, M.; Tsang, K.Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, M.; Abrams, S.I.; Coleman, C.N.; Camphausen, K.; Schlom, J.; Hodge, J.W. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004, 64, 4328–4337. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef] [PubMed]
- Wiendl, H.; Mitsdoerffer, M.; Hofmeister, V.; Wischhusen, J.; Bornemann, A.; Meyermann, R.; Weiss, E.H.; Melms, A.; Weller, M. A functional role of HLA-G expression in human gliomas: An alternative strategy of immune escape. J. Immunol. 2002, 168, 4772–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef]
- Newcomb, E.W.; Demaria, S.; Lukyanov, Y.; Shao, Y.; Schnee, T.; Kawashima, N.; Lan, L.; Dewyngaert, J.K.; Zagzag, D.; McBride, W.H.; et al. The combination of ionizing radiation and peripheral vaccination produces long-term survival of mice bearing established invasive GL261 gliomas. Clin. Cancer Res. 2006, 12, 4730–4737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharabi, A.B.; Lim, M.; DeWeese, T.L.; Drake, C.G. Radiation and checkpoint blockade immunotherapy: Radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015, 16, e498–e509. [Google Scholar] [CrossRef]
- Hoffmann, T.K.; Meidenbauer, N.; Dworacki, G.; Kanaya, H.; Whiteside, T.L. Generation of tumor-specific T-lymphocytes by cross-priming with human dendritic cells ingesting apoptotic tumor cells. Cancer Res. 2000, 60, 3542–3549. [Google Scholar] [PubMed]
- Sauter, B.; Albert, M.L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000, 191, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, T.; Purushothaman, K.; Puthiyottil, S.S.; Bhattacharjee, A.; Muttah, G. Immunological interactions in radiotherapy-opening a new window of opportunity. Ann. Transl. Med. 2016, 4, 51. [Google Scholar]
- Brix, N.; Tiefenthaller, A.; Anders, H.; Belka, C.; Lauber, K. Abscopal, immunological effects of radiotherapy: Narrowing the gap between clinical and preclinical experiences. Immunol. Rev. 2017, 280, 249–279. [Google Scholar] [CrossRef]
- De la Cruz-Merino, L.; Illescas-Vacas, A.; Grueso-López, A.; Barco-Sánchez, A.; Míguez-Sánchez, C. Cancer Immunotherapies Spanish Group (GETICA). Radiation for Awakening the Dormant Immune System, a Promising Challenge to be Explored. Front. Immunol. 2014, 5, 102. [Google Scholar] [CrossRef] [Green Version]
- Teitz-Tennenbaum, S.; Li, Q.; Okuyama, R.; Davis, M.A.; Sun, R.; Whitfield, J.; Knibbs, R.N.; Stoolman, L.M.; Chang, A.E. Mechanisms involved in radiation enhancement of intratumoral dendritic cell therapy. J. Immunother. 2008, 31, 345–358. [Google Scholar] [CrossRef]
- Guo, C.; Yi, H.; Yu, X.; Zuo, D.; Qian, J.; Yang, G.; Foster, B.A.; Subjeck, J.R.; Sun, X.; Mikkelsen, R.B.; et al. In situ vaccination with CD204 gene-silenced dendritic cell, not unmodified dendritic cell, enhances radiation therapy of prostate cancer. Mol. Cancer Ther. 2012, 11, 2331–2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Weng, D.; Song, B.; Koido, S.; Calderwood, S.K.; Gong, J. Immunotherapy of radioresistant mammary tumors with early metastasis using molecular chaperone vaccines combined with ionizing radiation. J. Immunol. 2013, 191, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akutsu, Y.; Matsubara, H.; Urashima, T.; Komatsu, A.; Sakata, H.; Nishimori, T.; Yoneyama, Y.; Hoshino, I.; Murakami, K.; Usui, A.; et al. Combination of direct intratumoral administration of dendritic cells and irradiation induces strong systemic antitumor effect mediated by GRP94/gp96 against squamous cell carcinoma in mice. Int. J. Oncol. 2007, 31, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Tatsuta, K.; Tanaka, S.; Tajiri, T.; Shibata, S.; Komaru, A.; Ueda, Y.; Inoue, M.; Hasegawa, M.; Suita, S.; Sueishi, K.; et al. Complete elimination of established neuroblastoma by synergistic action of gamma-irradiation and DCs treated with rSeV expressing interferon-beta gene. Gene Ther. 2009, 16, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Teitz-Tennenbaum, S.; Li, Q.; Davis, M.A.; Wilder-Romans, K.; Hoff, J.; Li, M.; Chang, A.E. Radiotherapy combined with intratumoral dendritic cell vaccination enhances the therapeutic efficacy of adoptive T-cell transfer. J. Immunother. 2009, 32, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Teitz-Tennenbaum, S.; Li, Q.; Rynkiewicz, S.; Ito, F.; Davis, M.A.; McGinn, C.J.; Chang, A.E. Radiotherapy potentiates the therapeutic efficacy of intratumoral dendritic cell administration. Cancer Res. 2003, 63, 8466–8475. [Google Scholar]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 2003, 170, 6338–6347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, M.; Takahashi, K.; Inanami, O. Induction of apoptosis through the activation of SAPK/JNK followed by the expression of death receptor Fas in X-irradiated cells. J. Radiat. Res. 2003, 44, 203–209. [Google Scholar] [CrossRef]
- Hallahan, D.; Kuchibhotla, J.; Wyble, C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996, 56, 5150–5155. [Google Scholar] [PubMed]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Ariyoshi, K.; Takabatake, T.; Shinagawa, M.; Kadono, K.; Daino, K.; Imaoka, T.; Kakinuma, S.; Nishimura, M.; Shimada, Y. Age dependence of hematopoietic progenitor survival and chemokine family gene induction after gamma irradiation in bone marrow tissue in C3H/He mice. Radiat. Res. 2014, 181, 302–313. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Camphausen, K.; Moses, M.A.; Ménard, C.; Sproull, M.; Beecken, W.D.; Folkman, J.; O’Reilly, M.S. Radiation abscopal antitumor effect is mediated through p53. Cancer Res. 2003, 63, 1990–1993. [Google Scholar]
- Cao, Z.A.; Daniel, D.; Hanahan, D. Sub-lethal radiation enhances anti-tumor immunotherapy in a transgenic mouse model of pancreatic cancer. BMC Cancer 2002, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Aarts, B.M.; Klompenhouwer, E.G.; Rice, S.L.; Imani, F.; Baetens, T.; Bex, A.; Horenblas, S.; Kok, M.; Haanen, J.B.A.G.; Beets-Tan, R.G.H.; et al. Cryoablation and immunotherapy: An overview of evidence on its synergy. Insights Imaging. 2019, 10, 53. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Grauer, O.M.; Sutmuller, R.P.; van Maren, W.; Jacobs, J.F.; Bennink, E.; Toonen, L.W.; Nierkens, S.; Adema, G.J. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int. J. Cancer 2008, 122, 1794–1802. [Google Scholar] [CrossRef]
- Jacobs, J.F.; Punt, C.J.; Lesterhuis, W.J.; Sutmuller, R.P.; Brouwer, H.M.; Scharenborg, N.M.; Klasen, I.S.; Hilbrands, L.B.; Figdor, C.G.; de Vries, I.J.; et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: A phase I/II study in metastatic melanoma patients. Clin. Cancer Res. 2010, 16, 5067–5078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Hobeika, A.C.; Osada, T.; Serra, D.; Niedzwiecki, D.; Lyerly, H.K.; Clay, T.M. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood 2008, 112, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Baur, A.S.; Lutz, M.B.; Schierer, S.; Beltrame, L.; Theiner, G.; Zinser, E.; Ostalecki, C.; Heidkamp, G.; Haendle, I.; Erdmann, M.; et al. Denileukin diftitox (ONTAK) induces a tolerogenic phenotype in dendritic cells and stimulates survival of resting Treg. Blood 2013, 122, 2185–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesolowski, R.; Markowitz, J.; Carson, W.E., 3rd. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunother. Cancer 2013, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.C.; Lapenta, C.; Santini, S.M. Advances and perspectives of dendritic cell-based active immunotherapies in follicular lymphoma. Cancer Immunol. Immunother. 2020, 69, 913–925. [Google Scholar] [CrossRef]
- Bachy, E.; Seymour, J.F.; Feugier, P.; Offner, F.; López-Guillermo, A.; Belada, D.; Xerri, L.; Catalano, J.V.; Brice, P.; Lemonnier, F.; et al. Sustained Progression-Free Survival Benefit of Rituximab Maintenance in Patients with Follicular Lymphoma: Long-Term Results of the PRIMA Study. J. Clin. Oncol. 2019, 37, 2815–2824. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, F.; Schreibelt, G.; Sköld, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm Shift in Dendritic Cell-Based Immunotherapy: From in vitro Generated Monocyte-Derived DCs to Naturally Circulating DC Subsets. Front. Immunol. 2014, 5, 165. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.M.; Lapenta, C.; Logozzi, M.; Parlato, S.; Spada, M.; Di Pucchio, T.; Belardelli, F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 2000, 191, 1777–1788. [Google Scholar] [CrossRef]
- Parlato, S.; Santini, S.M.; Lapenta, C.; Di Pucchio, T.; Logozzi, M.; Spada, M.; Giammarioli, A.M.; Malorni, W.; Fais, S.; Belardelli, F. Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: Importance for the rapid acquisition of potent migratory and functional activities. Blood 2001, 98, 3022–3029. [Google Scholar] [CrossRef] [Green Version]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Ock, C.Y.; Keam, B.; Kim, S.; Lee, J.S.; Kim, M.; Kim, T.M.; Jeon, Y.K.; Kim, D.W.; Chung, D.H.; Heo, D.S. Pan-Cancer Immunogenomic Perspective on the Tumor Microenvironment Based on PD-L1 and CD8 T-Cell Infiltration. Clin. Cancer Res. 2016, 22, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Carroll, P.; Weinberg, V.; Chan, S.; Lewis, J.; Corman, J.; Amling, C.L.; Stephenson, R.A.; Simko, J.; Sheikh, N.A.; et al. Activated lymphocyte recruitment into the tumor microenvironment following preoperative sipuleucel-T for localized prostate cancer. J. Natl. Cancer Inst. 2014, 106, dju268. [Google Scholar] [CrossRef]
- Ribas, A.; Comin-Anduix, B.; Chmielowski, B.; Jalil, J.; de la Rocha, P.; McCannel, T.A.; Ochoa, M.T.; Seja, E.; Villanueva, A.; Oseguera, D.K.; et al. Dendritic cell vaccination combined with CTLA4 blockade in patients with metastatic melanoma. Clin. Cancer Res. 2009, 15, 6267–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilgenhof, S.; Corthals, J.; Van Nuffel, A.M.; Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Neyns, B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol. Immunother. 2015, 64, 381–388. [Google Scholar] [CrossRef]
- Van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef]
- Wada, S.; Jackson, C.M.; Yoshimura, K.; Yen, H.R.; Getnet, D.; Harris, T.J.; Goldberg, M.V.; Bruno, T.C.; Grosso, J.F.; Durham, N.; et al. Sequencing CTLA-4 blockade with cell-based immunotherapy for prostate cancer. J. Transl. Med. 2013, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, Z. Efficacy and safety of dendritic cells co-cultured with cytokine-induced killer cells immunotherapy for non-small-cell lung cancer. Int. Immunopharmacol. 2015, 28, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Deng, X.; Wu, H.; Peng, P.; Wen, B.; Li, F. Combined immunotherapy with dendritic cells and cytokine-induced killer cells for malignant tumors: A systematic review and meta-analysis. Int. Immunopharmacol. 2014, 22, 451–464. [Google Scholar] [CrossRef]
- Olioso, P.; Giancola, R.; Di Riti, M.; Contento, A.; Accorsi, P.; Iacone, A. Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: A pilot clinical trial. Hematol. Oncol. 2009, 27, 130–139. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, W.; Qi, X.; Li, H.; Yu, J.; Wei, S.; Hao, X.; Ren, X. Randomized study of autologous cytokine-induced killer cell immunotherapy in metastatic renal carcinoma. Clin. Cancer Res. 2012, 18, 1751–1759. [Google Scholar] [CrossRef] [Green Version]
- Leemhuis, T.; Wells, S.; Scheffold, C.; Edinger, M.; Negrin, R.S. A phase I trial of autologous cytokine-induced killer cells for the treatment of relapsed Hodgkin disease and non-Hodgkin lymphoma. Biol. Blood Marrow Transplant. 2005, 11, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, K.M.; Cirignano, B.; Chamian, F.; Zagzag, D.; Miller, D.C.; Finlay, J.L.; Steinman, R.M. Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell-mediated expansion. Int. J. Cancer 2004, 109, 893–899. [Google Scholar] [CrossRef]
- Liu, H.; Chen, L.; Liu, J.; Meng, H.; Zhang, R.; Ma, L.; Wu, L.; Yu, S.; Shi, F.; Li, Y.; et al. Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett. 2017, 411, 182–190. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef]
- Pantel, K.; Riethmüller, G. Micrometastasis detection and treatment with monoclonal antibodies. Curr. Top. MicroBiol. Immunol. 1996, 213, 1–18. [Google Scholar] [PubMed]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L. TGFbeta, a potent regulator of tumor microenvironment and host immune response, implication for therapy. Curr. Mol. Med. 2010, 10, 374–380. [Google Scholar] [CrossRef]
- Johnson, B.F.; Clay, T.M.; Hobeika, A.C.; Lyerly, H.K.; Morse, M.A. Vascular endothelial growth factor and immunosuppression in cancer: Current knowledge and potential for new therapy. Expert Opin. Biol. Ther. 2007, 7, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Gulley, J.L.; Madan, R.A.; Schlom, J. Impact of tumour volume on the potential efficacy of therapeutic vaccines. Curr. Oncol. 2011, 18, e150–e157. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Boudewijns, S.; Koornstra, R.H.; Westdorp, H.; Schreibelt, G.; van den Eertwegh, A.J.; Geukes Foppen, M.H.; Haanen, J.B.; de Vries, I.J.; Figdor, C.G.; Bol, K.F.; et al. Ipilimumab administered to metastatic melanoma patients who progressed after dendritic cell vaccination. Oncoimmunology 2016, 5, e1201625. [Google Scholar] [CrossRef]
- Ardon, H.; Van Gool, S.W.; Verschuere, T.; Maes, W.; Fieuws, S.; Sciot, R.; Wilms, G.; Demaerel, P.; Goffin, J.; Van Calenbergh, F.; et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: Results of the HGG-2006 phase I/II trial. Cancer Immunol. Immunother. 2012, 61, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas: With a report of ten original cases. Am. J. Med. Sci. 1893, 105, 487–511. [Google Scholar] [CrossRef]

{kind=link}
| Description | N. of Trials | Disease(s) | Status | Phase |
|---|---|---|---|---|
| DCs based adjuvant therapy versus standard of care (radio/chemotherapy) | 79 | 24 Brain tumors; 13 Melanoma; 12 Breast Cancer; 6 Liver/colo-rectal cancer; 4 pancreatic cancer; 20 other. | 15 Recruiting; 19 Not (yet) recruiting; 38 completed/terminated; 7 Unknown/withdrawn | 3 in phase 3; other phase 1, 2 |
| DCs based adjuvant therapy plus ICI | 42 | 11 Melanoma; 3 brain tumor; 4 Gastric cancer; 4 lung cancer; 3 pancreatic cancers; 5 Myeloma/Lymphoma; 12 other | 15 Recruiting; 9 Not (yet) recruiting; 12 completed/terminated; 6 Unknown/withdrawn | 5 in phase 3/4; other phase 1, 2 |
| DCs based adjuvant therapy plus other adoptive cells therapy | 29 | 6 Brain tumors; 3 Melanoma; 1 lung; 7 epato/gastrict tract cancer; 4 Myeloma/Lymphoma; 8 other | 8 Recruiting; 4 Not (yet) recruiting; 5 completed/terminated; 12 Unknown/withdrawn | 2 in phase 3; other phase 1, 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nava, S.; Lisini, D.; Frigerio, S.; Bersano, A. Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect. Int. J. Mol. Sci. 2021, 22, 12339. https://doi.org/10.3390/ijms222212339
Nava S, Lisini D, Frigerio S, Bersano A. Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect. International Journal of Molecular Sciences. 2021; 22(22):12339. https://doi.org/10.3390/ijms222212339
Chicago/Turabian StyleNava, Sara, Daniela Lisini, Simona Frigerio, and Anna Bersano. 2021. "Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect" International Journal of Molecular Sciences 22, no. 22: 12339. https://doi.org/10.3390/ijms222212339
APA StyleNava, S., Lisini, D., Frigerio, S., & Bersano, A. (2021). Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect. International Journal of Molecular Sciences, 22(22), 12339. https://doi.org/10.3390/ijms222212339