
Multiscale Modeling of Amyloid Fibrils Formed by Aggregating Peptides Derived from the Amyloidogenic Fragment of the A-Chain of Insulin


Abstract
:1. Introduction
2. Materials and Methods
2.1. Modeling Peptide Aggregation Using CABS-Dock
2.2. Test Prediction of Known Protofilament Structures
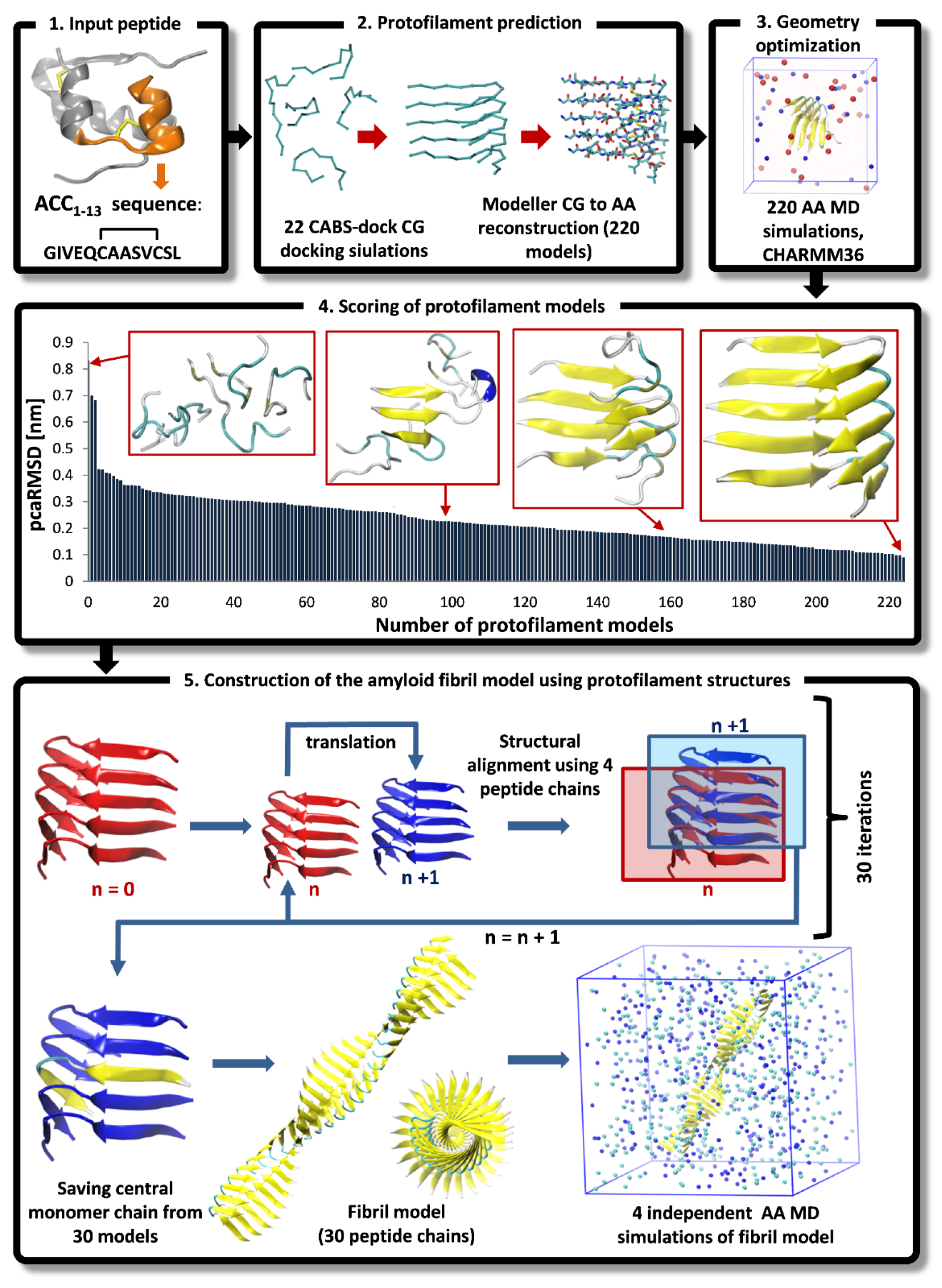
2.3. Multiscale Modeling Procedure for the Structure Prediction of Amyloid Fibrils
- 1
- Input peptide. The amino acid sequence of ACC1-13 consisting of 13 residues was extracted from the N-terminal part of bovine insulin’s A-chain (PDB ID: 2A3G) [30]. The Cys residue at the 7th position (which is engaged in intramolecular disulfide bonds in the native insulin) was substituted with an Ala residue.
- 2
- Protofilament prediction. The CABS-dock program [13] was used for predicting a large number of peptide assemblies formed by 5 interacting monomers (details of docking simulation are described in Section 2.1. During CG docking simulations, additional distance restraints were imposed on pairs of C-alpha atoms of appropriate cysteine residues forming the disulfide bridge in the A-chain of insulin (Cys6-Cys11). The length of these restraints in all peptide monomers was set to 6.5 Å (the average distance observed during the AA MD simulation of a single peptide monomer in explicit solvent). We conducted a total of 22 CG docking simulations, resulting in 220 top-scored protofilament models, each composed of 5 interacting monomers in C-alpha representations. The resulting CG models were converted into their AA representations using the Modeller version 9.25 program [31].
- 3
- Geometry optimization. Each protofilament model was inserted into simulation boxes and solvated with 6000 water molecules, 23 Na+ ions, and 28 Cl− ions. The system was modeled at a low pH of 1.9; therefore, the side chains of Glu residues were protonated in all peptide chains. Furthermore, the N-termini of all peptide monomers started with a positively charged protonated amino group (-NH3+), while the C-termini were capped by a neutral protonated carboxyl group (COOH). System equilibration included 20,000 steps of the steepest descent algorithm, followed by 10 ns MD simulations using weak position restraints imposed on the backbone atoms of peptide monomers. This retained the initial conformation of the peptide monomers, allowing for the correct orientation of the surrounding water molecules. Eventually, the production run included unrestrained MD simulations lasting 100 ns. The PME method [32] was used for the treatment of long-range electrostatic interactions. Hydrogen bonds were restrained using the LINC algorithm [33]. The simulation step was 2 fs. The Charmm36 force field [34] parameter set was applied and the TIP3P water model [35] was used for solvent molecules. All simulations were conducted by applying periodic boundary conditions (PBC) at 1013 hPa. The temperature was set to 310 K using the velocity rescale (V-rescale) thermostat. A total of 220 protofilament models extracted from the final frames of the MD trajectories were saved for further analysis.
- 4
- Scoring of protofilament models. A straightforward scoring method has been proposed to evaluate and select the most reliable structure of protofilaments for the further construction of amyloid fibril models. Since amyloid fibrils consist of a large number of identical peptide chains forming long stacks of β-sheets with a quasi-translational symmetry along the long fibril axis [23], all peptide monomers in the predicted protofilament model should have very similar chain conformations. We used the symmetry criteria to select the best models of protofilaments. Therefore, for each model, we defined the peptide-chain-average-RMSD parameter (pcaRMSD). The value of pcaRMSD was calculated for each protofilament model using the set of RMSD values obtained during the mutual comparison of all 5 monomers. The RMSD value for each peptide pair was calculated after the structural fitting of two monomers. In this way, the conformations of 5 peptides were compared with each other in a given protofilament model. Low pcaRMSD values correlate with a high level of translational symmetry of monomers within the protofilament structure. The pcaRMSD values were calculated for all 220 models using the following formula:where M is the number of peptide chains in the protofilament model, N is the number of C-alpha atoms, Xk is the coordinate vector for the target C-alpha atom k, and Yk is the coordinate vector for the reference C-alpha atom k, whereas i and j indicate indexes of a particular pair of compared peptide chains in the protofilament model.
- 5
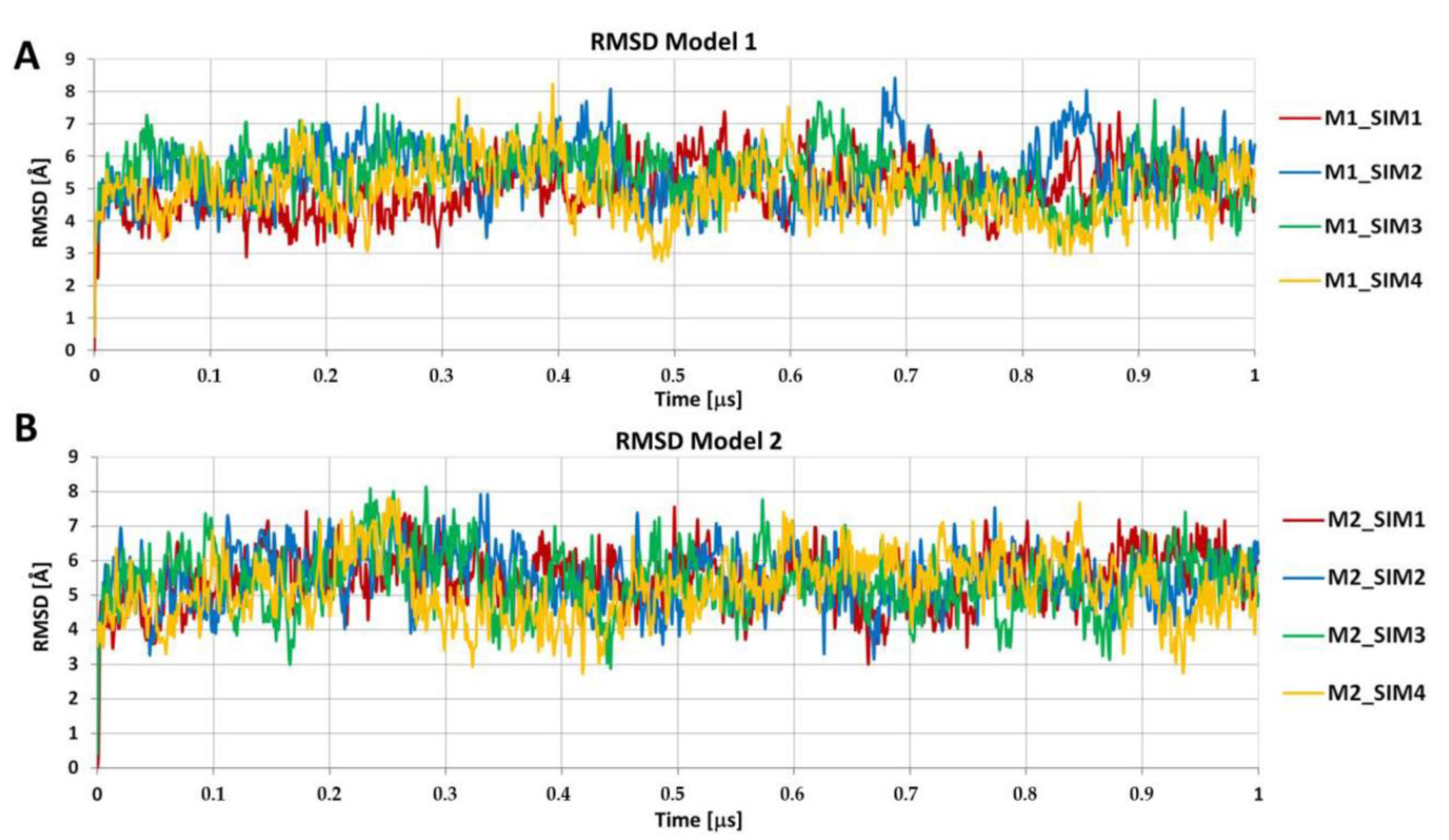
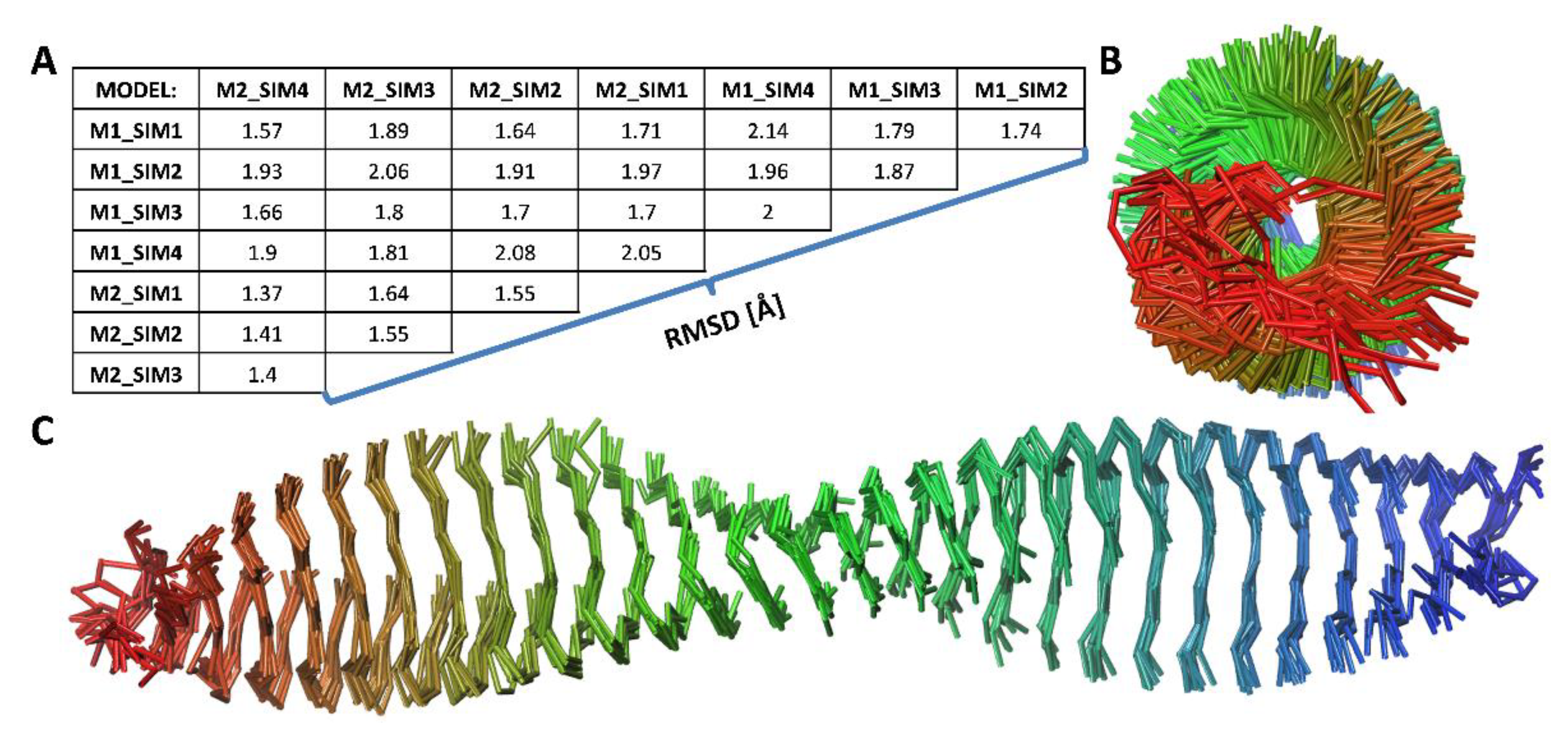
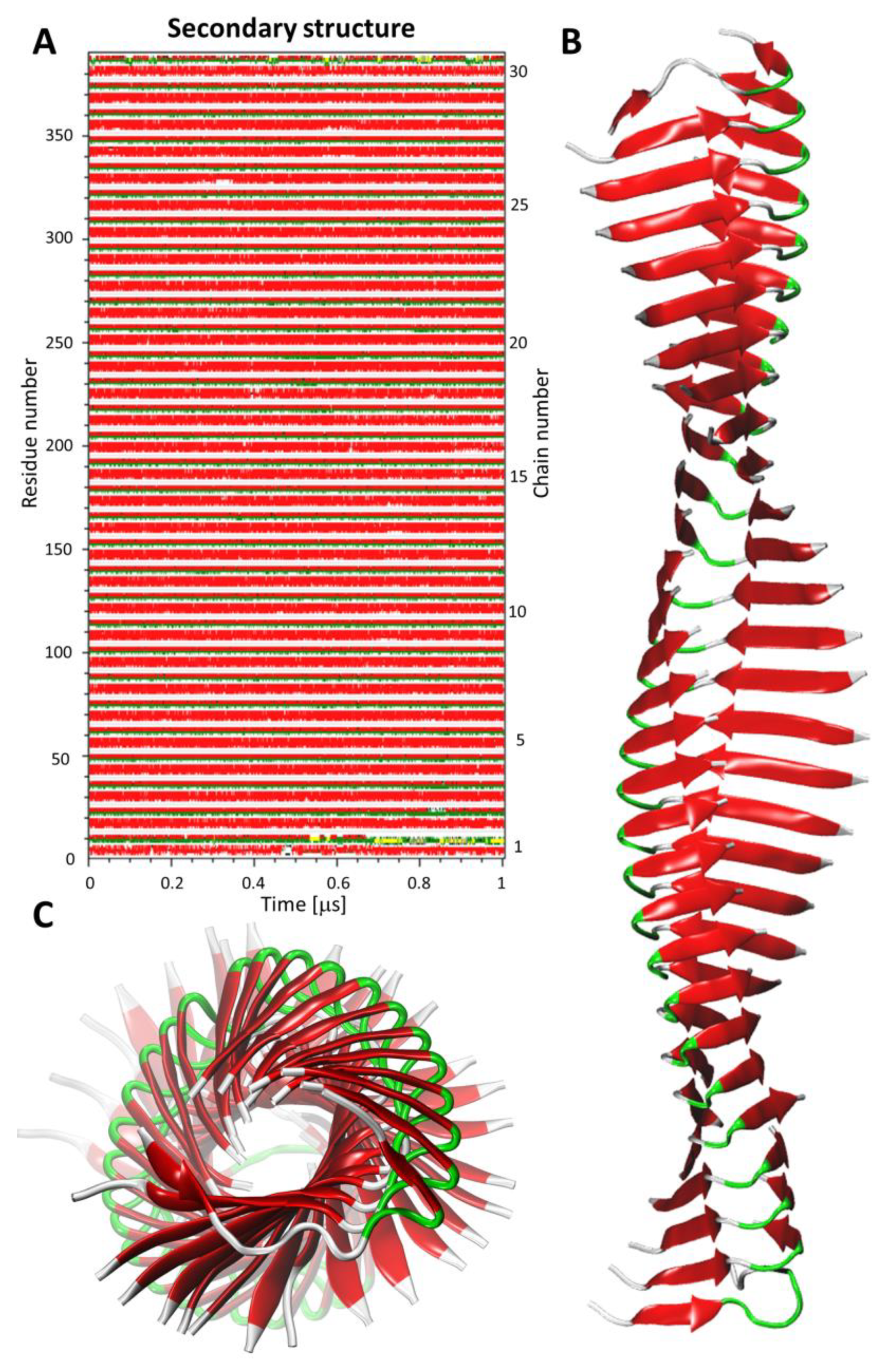
- Construction of the amyloid fibril model using protofilament structures. The amyloid fibril model was constructed using the top-scored protofilament structures as building blocks. The fibril assembly procedure included a series of translations. First, two copies of single protofilament models were superimposed using structural alignment based on only four monomer chains in each protofilament model. More specifically, peptide chains from the second to the fifth (monomers 2–5) of the starting protofilament model (Figure 1, protofilament shown in red color) were used for structural fitting of the next identical protofilament model (Figure 1, protofilament shown in blue color), using peptide chains from the first to the fourth (monomers 1–4). During the structural fitting, the coordinates of the starting protofilament model were kept fixed while the second protofilament was translated along the long axis of the formed fibril structure. The thus obtained “translated protofilament” structure was marked with index n + 1 (n being the number of translations/procedure iterations), saved after the first cycle of the procedure and used as a starting protofilament structure for the next iteration of the fibril assembly. The procedure was repeated 30 times, resulting in 30 protofilament structures that were consequently translated along the long axis of the predicted fibril. The central peptide chain (chain number 3) was extracted from each of the 30 translated protofilaments and saved. Finally, all extracted central peptides were combined, forming a plausible amyloid fibril model consisting of 30 well-matching peptide chains. The whole modeling procedure was repeated twice (each with 30 iterations) with the two top-scored protofilaments (showing the lowest value of the pcaRMSD parameter) used as a building block, therefore leading to two distinct amyloid fibril models. Both thus obtained fibril models were then subjected to AA MD simulation in explicit solvent. Each system consisted of the fibril model (made of 30 peptide chains), 106,176 water molecules, 420 Cl- ions, and 390 Na+ ions. The Simulation box dimensions were 148.1 Å × 148.1 Å × 148.1 Å and in total, the system included 324,618 atoms). At first, each system was equilibrated during the MD simulation with position restraints imposed on the backbone atoms of all peptide chains for the first 100 ns. The production run included unrestrained MD simulations lasting 1 μs and trajectory frames were recorded every 10 ps. For each fibril model, four independent MD simulations were conducted using random initial velocities. Simulation conditions were identical to those used at the earlier step of protofilament optimization (see step 3 Geometry optimization of the multi-scale modeling procedure). All AA MD simulations were conducted using the GROMACS version 5.14 program [36]. VMD version 1.9.3 [37] software was used for data analysis and visualization.
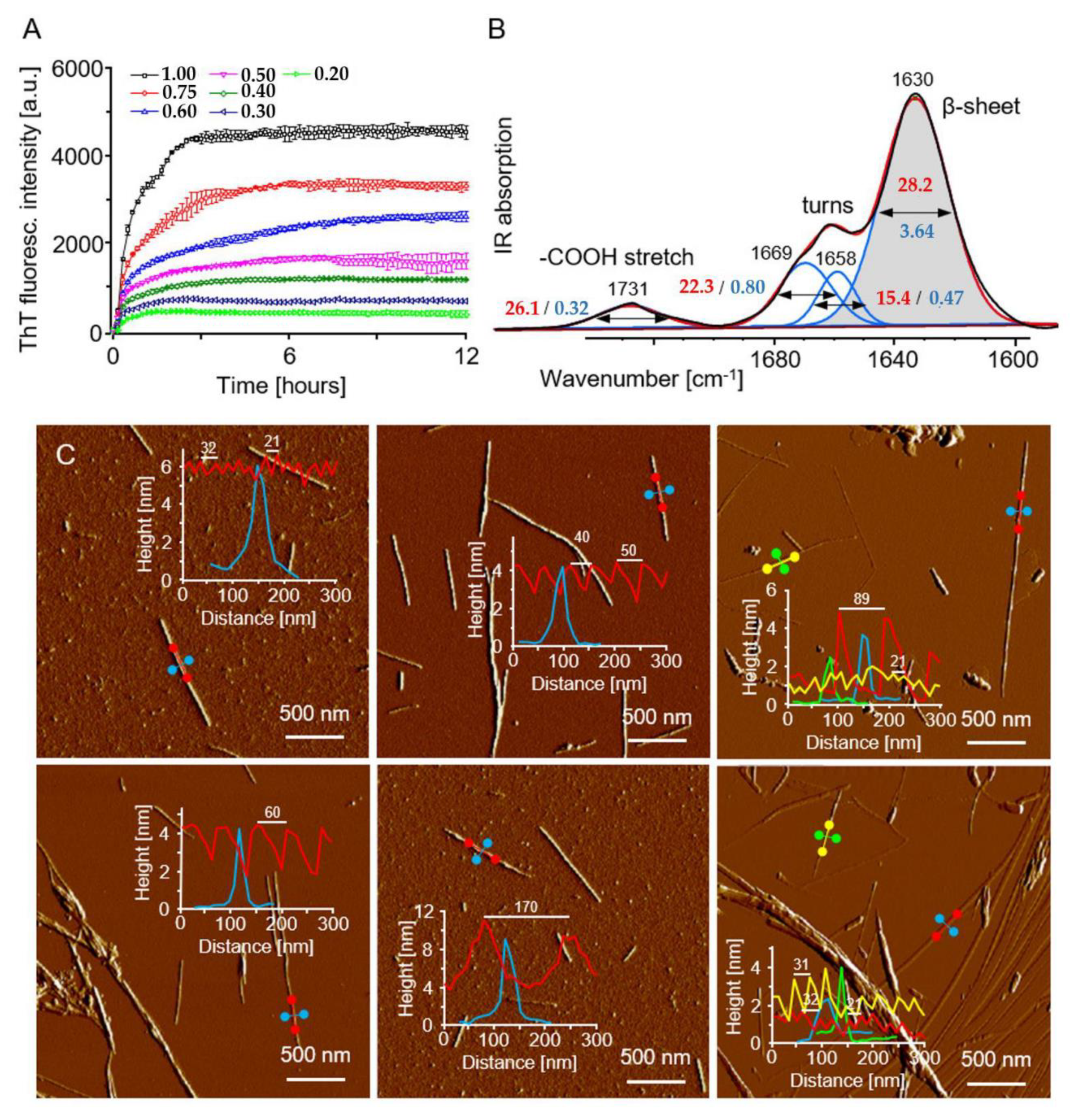
2.4. Experimental: Evaluation of the β-Sheet Content and Morphology of ACC1-13 Amyloid Fibrils
3. Results and Discussion
3.1. Test Prediction of Fibrils with Known Structures
3.2. Amyloid Fibril Models Predicted for Insulin-Derived ACC1-13 Peptide
3.3. β-Sheet Content in Predicted Fibril Models—Comparison to Experimental Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. The “correctly folded” state of proteins: Is it a metastable state? Angew. Chem. Int. Ed. 2002, 41, 257–259. [Google Scholar] [CrossRef]
- Knowles, T.P.; Buehler, M.J. Nanomechanics of functional and pathological amyloid materials. Nat. Nanotechnol. 2011, 6, 469–479. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef]
- Epstein, E.A.; Chapman, M.R. Polymerizing the fibre between bacteria and host cells: The biogenesis of functional amyloid fibres. Cell. Microbiol. 2008, 10, 1413–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [Green Version]
- Petkova, A.; Buntkowsky, G.; Dyda, F.; Leapman, R.; Yau, W.-M.; Tycko, R. Solid state NMR reveals a pH-dependent antiparallel β-sheet registry in fibrils formed by a β-amyloid peptide. J. Mol. Biol. 2004, 335, 247–260. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Buchete, N.-V.; Tycko, R.; Hummer, G. Molecular dynamics simulations of Alzheimer’s β-amyloid protofilaments. J. Mol. Biol. 2005, 353, 804–821. [Google Scholar] [CrossRef]
- Strodel, B. Amyloid aggregation simulations: Challenges, advances and perspectives. Curr. Opin. Struct. Biol. 2021, 67, 145–152. [Google Scholar] [CrossRef]
- Ilie, I.M.; Caflisch, A. Simulation studies of amyloidogenic polypeptides and their aggregates. Chem. Rev. 2019, 119, 6956–6993. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurcinski, M.; Pawel Ciemny, M.; Oleniecki, T.; Kuriata, A.; Badaczewska-Dawid, A.E.; Kolinski, A.; Kmiecik, S. CABS-dock standalone: A toolbox for flexible protein–peptide docking. Bioinformatics 2019, 35, 4170–4172. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Badaczewska-Dawid, A.; Kolinski, M.; Kolinski, A.; Kmiecik, S. Flexible docking of peptides to proteins using CABS-dock. Protein Sci. 2020, 29, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dec, R.; Koliński, M.; Dzwolak, W. Beyond amino acid sequence: Disulfide bonds and the origins of the extreme amyloidogenic properties of insulin’s H-fragment. FEBS J. 2019, 286, 3194–3205. [Google Scholar] [CrossRef]
- Dec, R.; Dzwolak, W. Extremely Amyloidogenic Single-Chain Analogues of Insulin’s H-Fragment: Structural Adaptability of an Amyloid Stretch. Langmuir 2020, 36, 12150–12159. [Google Scholar] [CrossRef]
- Dec, R.; Dzwolak, W. A tale of two tails: Self-assembling properties of A- and B-chain parts of insulin’s highly amyloidogenic H-fragment. Int. J. Biol. Macromol. 2021, 186, 510–518. [Google Scholar] [CrossRef]
- Piejko, M.; Dec, R.; Babenko, V.; Hoang, A.; Szewczyk, M.; Mak, P.; Dzwolak, W. Highly Amyloidogenic Two-chain Peptide Fragments Are Released upon Partial Digestion of Insulin with Pepsin. J. Biol. Chem. 2015, 290, 5947–5958. [Google Scholar] [CrossRef] [Green Version]
- Koliński, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [CrossRef] [Green Version]
- Pulawski, W.; Jamroz, M.; Kolinski, M.; Kolinski, A.; Kmiecik, S. Coarse-grained simulations of membrane insertion and folding of small helical proteins using the CABS model. J. Chem. Inf. Modeling 2016, 56, 2207–2215. [Google Scholar] [CrossRef]
- Kmiecik, S.; Jamroz, M.; Kolinski, M. Structure prediction of the second extracellular loop in G-protein-coupled receptors. Biophys. J. 2014, 106, 2408–2416. [Google Scholar] [CrossRef] [Green Version]
- Kolinski, M.; Filipek, S. Study of a structurally similar kappa opioid receptor agonist and antagonist pair by molecular dynamics simulations. J. Mol. Modeling 2010, 16, 1567–1576. [Google Scholar] [CrossRef] [Green Version]
- Badaczewska-Dawid, A.E.; Kmiecik, S.; Koliński, M. Docking of peptides to GPCRs using a combination of CABS-dock with FlexPepDock refinement. Brief. Bioinform. 2021, 22, bbaa109. [Google Scholar] [CrossRef]
- Koliński, M.; Kmiecik, S.; Dec, R.; Piejko, M.; Mak, P.; Dzwolak, W. Docking interactions determine early cleavage events in insulin proteolysis by pepsin: Experiment and simulation. Int. J. Biol. Macromol. 2020, 149, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Kmiecik, S.; Zalewski, M.; Kolinski, A. Protein–Protein Docking with Large-Scale Backbone Flexibility Using Coarse-Grained Monte-Carlo Simulations. Int. J. Mol. Sci. 2021, 22, 7341. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krotee, P.; Griner, S.L.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Shi, D.; Philipp, S.; Murray, K.; Saelices, L.; Lee, J.; et al. Common fibrillar spines of amyloid-beta and human islet amyloid polypeptide revealed by microelectron diffraction and structure-based inhibitors. J. Biol. Chem. 2018, 293, 2888–2902. [Google Scholar] [CrossRef] [Green Version]
- Iwata, K.; Fujiwara, T.; Matsuki, Y.; Akutsu, H.; Takahashi, S.; Naiki, H.; Goto, Y. 3D structure of amyloid protofilaments of β2-microglobulin fragment probed by solid-state NMR. Proc. Natl. Acad. Sci. USA 2006, 103, 18119–18124. [Google Scholar] [CrossRef] [Green Version]
- Sgourakis, N.G.; Yau, W.-M.; Qiang, W. Modeling an in-register, parallel “iowa” aβ fibril structure using solid-state NMR data from labeled samples with rosetta. Structure 2015, 23, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.D.; Pangborn, W.A.; Blessing, R.H. The structure of T6 bovine insulin. Acta Crystallographica. Sect. D Biol. Crystallogr. 2005, 61, 1476–1482. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, M.W.; Jorgensen, W.L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 2000, 112, 8910–8922. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Raussens, V.; Ruysschaert, J.M. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta 1999, 1422, 105–185. [Google Scholar] [CrossRef]
- de Jongh, H.H.; Goormaghtigh, E.; Ruysschaert, J.M. The different molar absorptivities of the secondary structure types in the amide I region: An attenuated total reflection infrared study on globular proteins. Anal. Biochem. 1996, 242, 95–103. [Google Scholar] [CrossRef]
- Zalewski, M.; Kmiecik, S.; Koliński, M. Molecular Dynamics Scoring of Protein–Peptide Models Derived from Coarse-Grained Docking. Molecules 2021, 26, 3293. [Google Scholar] [CrossRef]
- Dec, R.; Kolinski, M.; Kouza, M.; Dzwolak, W. Rapid self-association of highly amyloidogenic H-fragments of insulin: Experiment and molecular dynamics simulations. Int. J. Biol. Macromol. 2020, 150, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.P.; Debinski, A.; Paczkowska, M.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. Protein-peptide molecular docking with large-scale conformational changes: The p53-MDM2 interaction. Sci. Rep. 2016, 6, 1–8. [Google Scholar]
- Ciemny, M.P.; Badaczewska-Dawid, A.E.; Pikuzinska, M.; Kolinski, A.; Kmiecik, S. Modeling of disordered protein structures using monte carlo simulations and knowledge-based statistical force fields. Int. J. Mol. Sci. 2019, 20, 606. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolym. Orig. Res. Biomol. 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandomeneghi, G.; Krebs, M.R.; McCammon, M.G.; Fändrich, M. FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 2004, 13, 3314–3321. [Google Scholar] [CrossRef] [Green Version]
- Aggeli, A.; Nyrkova, I.A.; Bell, M.; Harding, R.; Carrick, L.; McLeish, T.C.; Semenov, A.N.; Boden, N. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide β-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. USA 2001, 98, 11857–11862. [Google Scholar] [CrossRef] [Green Version]
- Dzwolak, W. Chirality and chiroptical properties of amyloid fibrils. Chirality 2014, 26, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Gruziel, M.; Dzwolak, W.; Szymczak, P. Chirality inversions in self-assembly of fibrillar superstructures: A computational study. Soft Matter 2013, 9, 8005–8013. [Google Scholar] [CrossRef]
- Maslova, M.; Gerasimova, L.; Forsling, W. Surface properties of cleaved mica. Colloid J. 2004, 66, 322–328. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | d [Å] a | α [Deg] b | % Res. with H-Bonds c | No. Res. with H-Bonds d |
|---|---|---|---|---|
| M1_SIM1 | 134.13 ± 2.33 | 9.15 ± 0.61 | 49.92 ± 1.21 | 194.7 ± 4.72 |
| M1_SIM2 | 133.32 ± 2.21 | 9.06 ± 0.72 | 50.23 ± 1.22 | 195.9 ± 4.78 |
| M1_SIM3 | 133.94 ± 2.27 | 9.02 ± 0.68 | 49.85 ± 2 | 194.42 ± 7.81 |
| M1_SIM4 | 134.14 ± 2.34 | 9.16 ± 0.72 | 49.18 ± 1.4 | 191.82 ± 5.46 |
| M2_SIM1 | 133.76 ± 2.25 | 9.03 ± 0.68 | 49.92 ± 1.17 | 194.69 ± 4.6 |
| M2_SIM2 | 134.4 ± 2.37 | 8.9 ± 0.67 | 49.73 ± 1.96 | 193.98 ± 7.67 |
| M2_SIM3 | 134.3 ± 2.32 | 9.16 ± 0.69 | 49.58 ± 1.22 | 193.37 ± 4.77 |
| M2_SIM4 | 134.23 ± 2.34 | 9.16 ± 0.68 | 49.48 ± 1.24 | 192.99 ± 4.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koliński, M.; Dec, R.; Dzwolak, W. Multiscale Modeling of Amyloid Fibrils Formed by Aggregating Peptides Derived from the Amyloidogenic Fragment of the A-Chain of Insulin. Int. J. Mol. Sci. 2021, 22, 12325. https://doi.org/10.3390/ijms222212325
Koliński M, Dec R, Dzwolak W. Multiscale Modeling of Amyloid Fibrils Formed by Aggregating Peptides Derived from the Amyloidogenic Fragment of the A-Chain of Insulin. International Journal of Molecular Sciences. 2021; 22(22):12325. https://doi.org/10.3390/ijms222212325
Chicago/Turabian StyleKoliński, Michał, Robert Dec, and Wojciech Dzwolak. 2021. "Multiscale Modeling of Amyloid Fibrils Formed by Aggregating Peptides Derived from the Amyloidogenic Fragment of the A-Chain of Insulin" International Journal of Molecular Sciences 22, no. 22: 12325. https://doi.org/10.3390/ijms222212325