
Role of Cell-Free DNA and Deoxyribonucleases in Tumor Progression


Abstract
1. Introduction
2. Brief Historical Insight
3. Composition of cfDNA
4. Participation of cfDNA of Tumor Origin in Malignant Transformation of Healthy Cells
5. Routes of Tumor-Derived cfDNA Penetration into Cells
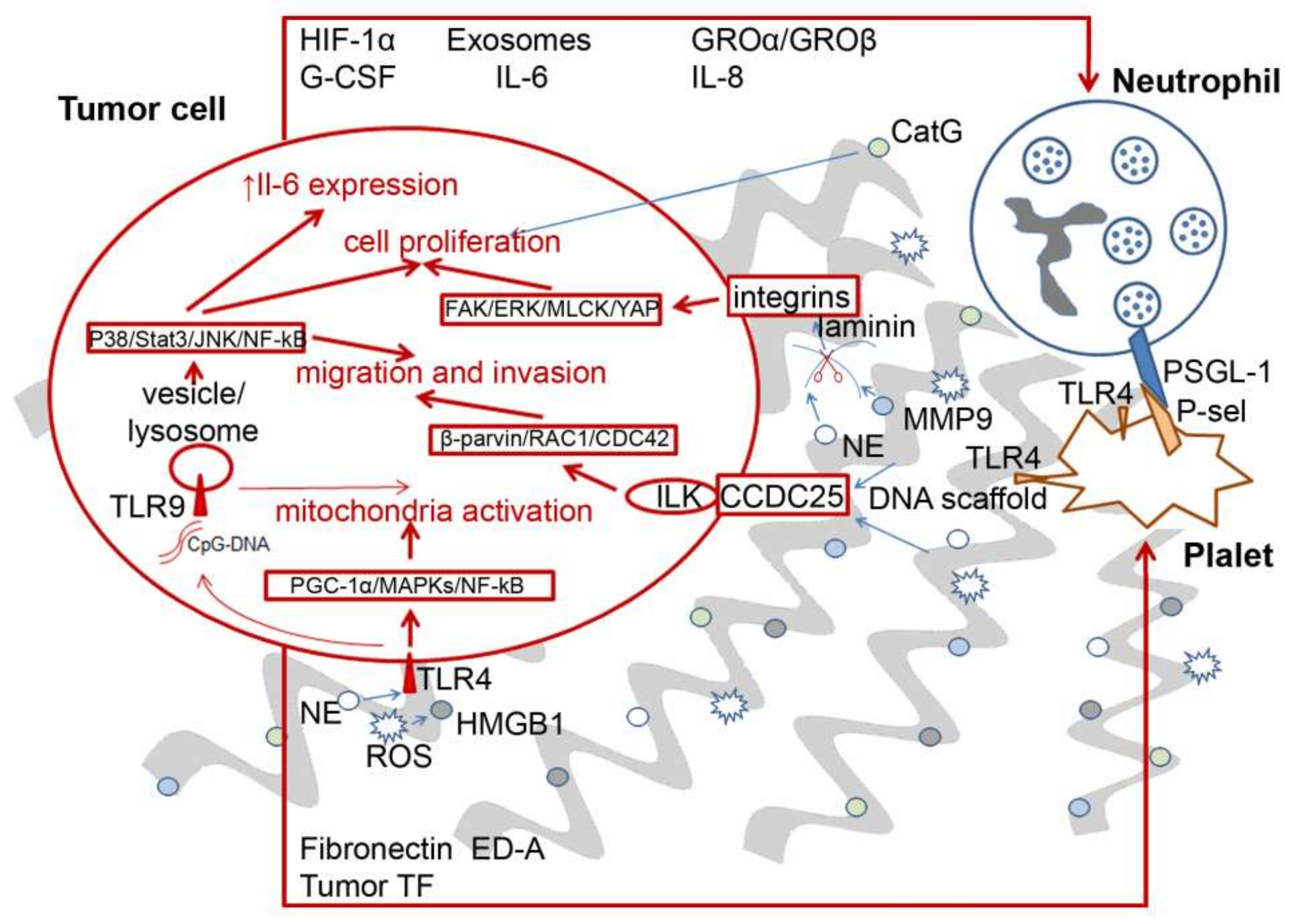
6. Pro-Tumor and Antitumor Effects of NETs
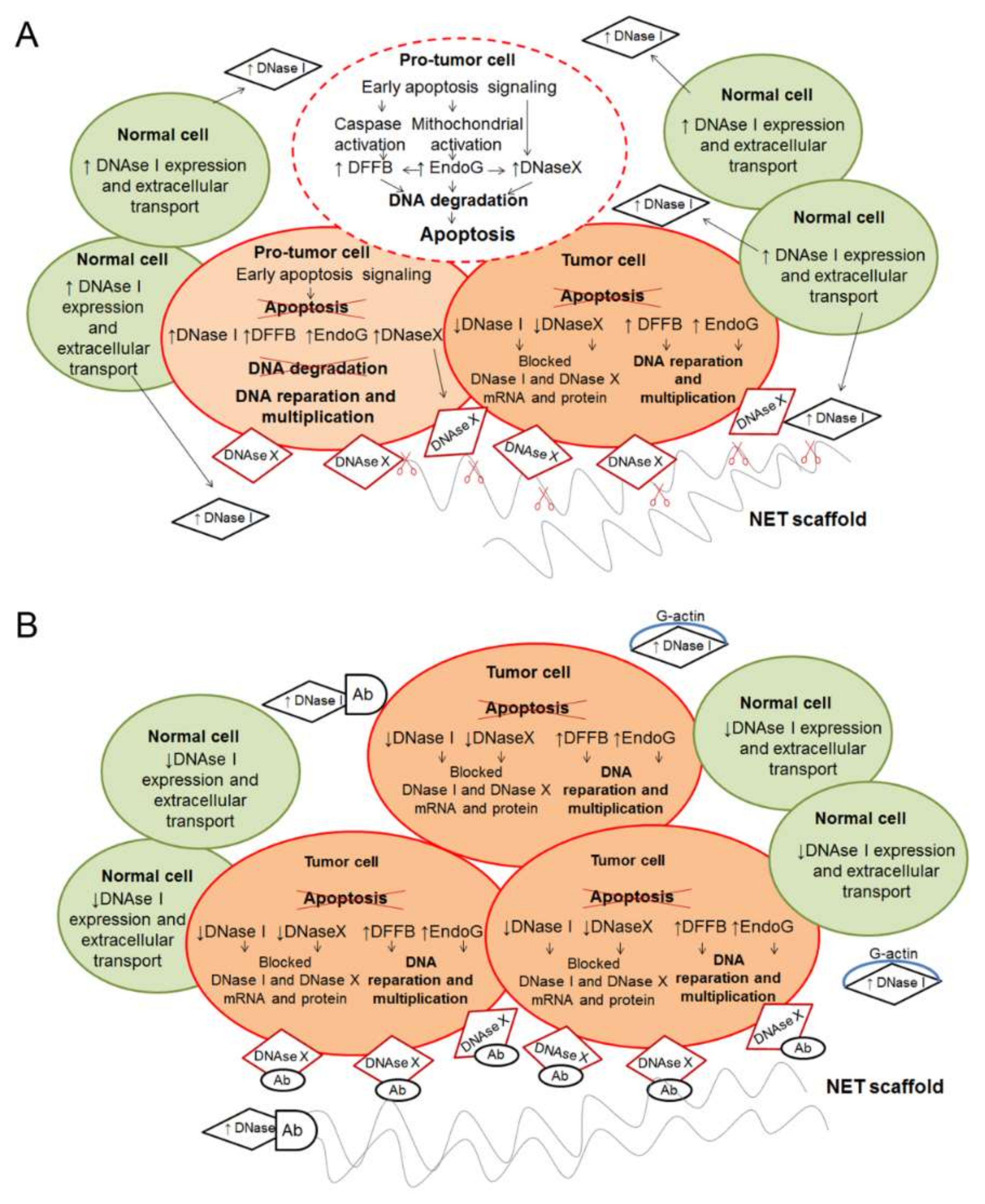
7. DNases and Their Role in the Maintenance of cfDNA Homeostasis under Pathological Conditions
7.1. Intracellular DNases
7.2. Extracellular Secreted DNases
7.3. DNase Activity of the Blood during Tumor Progression
8. Exogenous DNases as Antimetastatic and Antitumor Agents
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme. CR Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Stroun, M.; Anker, P.; Maurice, P.; Gahan, P.B. Circulating Nucleic Acids in Higher Organisms. Int. Rev. Cytol. 1977, 51, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Chiu, R.W. Sequencing of Circulating Cell-free DNA during Pregnancy. N. Engl. J. Med. 2018, 379, 464–473. [Google Scholar] [CrossRef]
- Dar, P.; Shani, H.; Evans, M.I. Cell-free DNA: Comparison of technologies. Clin. Lab. Med. 2016, 36, 199–211. [Google Scholar] [CrossRef]
- Lee, K.-H.; Shin, T.-J.; Kim, W.-H.; Lee, S.Y.; Cho, J.-Y. Methylation of LINE-1 in cell-free DNA serves as a liquid biopsy biomarker for human breast cancers and dog mammary tumors. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, B.C.; Paweletz, C.P. Cell-Free DNA in Oncology: Gearing up for Clinic. Ann. Lab. Med. 2018, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Oellerich, M.; Schütz, E.; Beck, J.; Kanzow, P.; Plowman, P.N.; Weiss, G.J.; Walson, P.D. Using circulating cell-free DNA to monitor personalized cancer therapy. Crit. Rev. Clin. Lab. Sci. 2017, 54, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Bedin, C.; Enzo, M.V.; Del Bianco, P.; Pucciarelli, S.; Nitti, D.; Agostini, M. Diagnostic and prognostic role of cell-free DNA testing for colorectal cancer patients. Int. J. Cancer 2016, 140, 1888–1898. [Google Scholar] [CrossRef]
- Tumor DNA circulating in the plasma might play a role in metastasis. The hypothesis of the genometastasis. Histol. Histopathol. 1999, 1159–1164. [CrossRef]
- García-Olmo, D.C.; Domínguez, C.; García-Arranz, M.; Anker, P.; Stroun, M.; García-Verdugo, J.M.; García-Olmo, D. Cell-Free Nucleic Acids Circulating in the Plasma of Colorectal Cancer Patients Induce the Oncogenic Transformation of Susceptible Cultured Cells. Cancer Res. 2010, 70, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Becerril, C.; Pérez-Cárdenas, E.; Taja-Chayeb, L.; Anker, P.; Herrera-Goepfert, R.; Medina-Velázquez, L.A.; Hidalgo-Miranda, A.; Montiel, M.D.P.; Chávez-Blanco, A.; Cruz-Velázquez, J.; et al. Cancer Progression Mediated by Horizontal Gene Transfer in an In Vivo Model. PLoS ONE 2012, 7, e52754. [Google Scholar] [CrossRef] [PubMed]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.-L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed]
- Gaiffe, E.; Prétet, J.-L.; Launay, S.; Jacquin, E.; Saunier, M.; Hetzel, G.; Oudet, P.; Mougin, C. Apoptotic HPV Positive Cancer Cells Exhibit Transforming Properties. PLoS ONE 2012, 7, e36766. [Google Scholar] [CrossRef]
- Beyer, C.; Pisetsky, D.S. The role of microparticles in the pathogenesis of rheumatic diseases. Nat. Rev. Rheumatol. 2009, 6, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Wartha, F.; Henriques-Normark, B. ETosis: A Novel Cell Death Pathway. Sci. Signal. 2008, 1, pe25. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, L.; Li, X.; Zhuo, W. Neutrophil Extracellular Traps in Tumor Metastasis: Pathological Functions and Clinical Applications. Cancers 2021, 13, 2832. [Google Scholar] [CrossRef]
- Lake, J.A.; Jain, R.; Rivera, M.C.; Sinelnikov, Y.D.; Chen, G.; Neuville, D.R.; Vaughan, M.T.; Liebermann, R.C. Mix and Match in the Tree of Life. Science 1999, 283, 2027–2028. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Stegemann, S.; Bock, R. Exchange of Genetic Material Between Cells in Plant Tissue Grafts. Science 2009, 324, 649–651. [Google Scholar] [CrossRef]
- Bhargava, P.M.; Shanmugam, G. Uptake of Nonviral Nucleic Acids by Mammalian Cells. Prog. Nucleic Acid Res. Mol. Biol. 1971, 11, 103–192. [Google Scholar] [CrossRef] [PubMed]
- Gahan, P.B.; Stroun, M. The biology of circulating nucleic acids in plasma and serum (CNAPS). In Extracellular Nucleic Acids: Nucleic Acids and Molecular Biology; Kikuchi, Y., Rykova, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 167–189. [Google Scholar]
- Karpfel, Z.; Šlotová, J.; Paleček, E. Chromosome aberrations produced by deoxyribonucleic acids in mice. Exp. Cell Res. 1963, 32, 147–148. [Google Scholar] [CrossRef]
- Szybalska, E.H.; Szybalski, W. Genetics of human cell lines, iv. dna-mediated heritable transformation of a biochemical trait. Proc. Natl. Acad. Sci. USA 1962, 48, 2026–2034. [Google Scholar] [CrossRef] [PubMed]
- Stroun, M.; Anker, P. Nucleic acids spontaneously released by living frog auricles. Biochem. J. 1972, 128, 100P–101P. [Google Scholar] [CrossRef]
- Stroun, M.; Anker, P. In vitro synthesis of DNA spontaneously released by bacteria or frog auricles. Biochimie 1972, 54, 1443–1452. [Google Scholar] [CrossRef]
- Gahan, P.B.; Stroun, M. The virtosome-a novel cytosolic informative entity and intercellular messenger. Cell Biochem. Funct. 2010, 28, 529–538. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Boddy, J.L.; Gal, S.; Malone, P.R.; Harris, A.; Wainscoat, J.S. Prospective Study of Quantitation of Plasma DNA Levels in the Diagnosis of Malignant versus Benign Prostate Disease. Clin. Cancer Res. 2005, 11, 1394–1399. [Google Scholar] [CrossRef]
- Wimberger, P.; Roth, C.; Pantel, K.; Kasimir-Bauer, S.; Kimmig, R.; Schwarzenbach, H. Impact of platinum-based chemotherapy on circulating nucleic acid levels, protease activities in blood and disseminated tumor cells in bone marrow of ovarian cancer patients. Int. J. Cancer 2010, 128, 2572–2580. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.A.; Bs, M.B.; Urbauer, D.L.; Dang, D.; Han, L.Y.; Godwin, A.K.; Karlan, B.Y.; Simpson, J.L.; Gershenson, D.M.; Coleman, R.L.; et al. Plasma cell-free DNA in ovarian cancer: An independent prognostic biomarker. Cancer 2010, 116, 1918–1925. [Google Scholar] [CrossRef]
- Sunami, E.; Vu, A.-T.; Nguyen, S.L.; Giuliano, A.E.; Hoon, D.S.B. Quantification of LINE1 in Circulating DNA as a Molecular Biomarker of Breast Cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 171–174. [Google Scholar] [CrossRef]
- Allen, D.; Butt, A.; Cahill, D.; Wheeler, M.; Popert, R.; Swaminathan, R. Role of Cell-Free Plasma DNA as a Diagnostic Marker for Prostate Cancer. Ann. N. Y. Acad. Sci. 2004, 1022, 76–80. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and Monitoring of Cell-Free DNA in Blood of Patients with Colorectal Cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Chun, F.K.-H.; Müller, I.; Lange, I.; Friedrich, M.G.; Erbersdobler, A.; Karakiewicz, P.I.; Graefen, M.; Pantel, K.; Huland, H.; Schwarzenbach, H. Circulating tumour-associated plasma DNA represents an independent and informative predictor of prostate cancer. BJU Int. 2006, 98, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Kisiel, J.; Roberts, L.R. Using cell-free DNA for HCC surveillance and prognosis. JHEP Rep. 2021, 3, 100304. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Incorvaia, L.; Del Re, M.; Malapelle, U.; Capoluongo, E.; Gristina, V.; Castiglia, M.; Danesi, R.; Fassan, M.; Giuffrè, G.; et al. The molecular profiling of solid tumors by liquid biopsy: A position paper of the AIOM–SIAPEC-IAP–SIBioC–SIC–SIF Italian Scientific Societies. ESMO Open 2021, 6, 100164. [Google Scholar] [CrossRef]
- Fleischhacker, M.; Schmidt, B. Circulating nucleic acids (CNAs) and cancer—A survey. Biochim. Biophys. Acta Rev. Cancer 2007, 1775, 181–232. [Google Scholar] [CrossRef] [PubMed]
- Jerónimo, C.; Nomoto, S.; Caballero, O.L.; Usadel, H.; Henrique, R.; Varzim, G.; Oliveira, J.; Lopes, C.; Fliss, M.S.; Sidransky, D. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 2001, 20, 5195–5198. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, S.; Yamashita, K.; Koshikawa, K.; Nakao, A.; Sidransky, D. Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin. Cancer Res. 2002, 8, 481–487. [Google Scholar]
- Belancio, V.P.; Engel, A.; Deininger, P.L. All y’all need to know ‘bout retroelements in cancer. Semin. Cancer Biol. 2010, 20, 200–210. [Google Scholar] [CrossRef]
- Carreira, P.; Richardson, S.; Faulkner, G.J. L1 retrotransposons, cancer stem cells and oncogenesis. FEBS J. 2013, 281, 63–73. [Google Scholar] [CrossRef]
- Kemp, J.R.; Longworth, M.S. Crossing the LINE Toward Genomic Instability: LINE-1 Retrotransposition in Cancer. Front. Chem. 2015, 3, 68. [Google Scholar] [CrossRef]
- Frickhofen, N.; Müller, E.; Sandherr, M.; Binder, T.; Bangerter, M.; Wiest, C.; Enz, M.; Heimpel, H. Rearranged Ig Heavy Chain DNA Is Detectable in Cell-Free Blood Samples of Patients With B-Cell Neoplasia. Blood 1997, 90, 4953–4960. [Google Scholar] [CrossRef]
- Cirmena, G.; Dameri, M.; Ravera, F.; Fregatti, P.; Ballestrero, A.; Zoppoli, G. Assessment of Circulating Nucleic Acids in Cancer: From Current Status to Future Perspectives and Potential Clinical Applications. Cancers 2021, 13, 3460. [Google Scholar] [CrossRef]
- Rhodes, C.H.; Honsinger, C.; Sorenson, G.D. Detection of Tumor-derived DNA in Cerebrospinal Fluid. J. Neuropathol. Exp. Neurol. 1994, 53, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Combaret, V.; Audoynaud, C.; Iacono, I.; Favrot, M.-C.; Schell, M.; Bergeron, C.; Puisieux, A. Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Res. 2002, 62, 3646–3648. [Google Scholar]
- Gotoh, T.; Hosoi, H.; Iehara, T.; Kuwahara, Y.; Osone, S.; Tsuchiya, K.; Ohira, M.; Nakagawara, A.; Kuroda, H.; Sugimoto, T. Prediction ofMYCNAmplification in Neuroblastoma Using Serum DNA and Real-Time Quantitative Polymerase Chain Reaction. J. Clin. Oncol. 2005, 23, 5205–5210. [Google Scholar] [CrossRef]
- Beck, J.; Urnovitz, H.B.; Riggert, J.; Clerici, M.; Schütz, E. Profile of the Circulating DNA in Apparently Healthy Individuals. Clin. Chem. 2009, 55, 730–738. [Google Scholar] [CrossRef] [PubMed]
- van der Vaart, M.; Semenov, D.V.; Kuligina, E.; Richter, V.A.; Pretorius, P.J. Characterisation of circulating DNA by parallel tagged sequencing on the 454 platform. Clin. Chim. Acta 2009, 409, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef]
- Akers, S.N.; Moysich, K.; Zhang, W.; Lai, G.C.; Miller, A.; Lele, S.; Odunsi, K.; Karpf, A.R. LINE1 and Alu repetitive element DNA methylation in tumors and white blood cells from epithelial ovarian cancer patients. Gynecol. Oncol. 2014, 132, 462–467. [Google Scholar] [CrossRef]
- Sikora, K.; Bedin, C.; Vicentini, C.; Malpeli, G.; D’Angelo, E.; Sperandio, N.; Lawlor, R.T.; Bassi, C.; Tortora, G.; Nitti, D.; et al. Evaluation of cell-free DNA as a biomarker for pancreatic malignancies. Int. J. Biol. Markers 2015, 30, 136–141. [Google Scholar] [CrossRef]
- Lehner, J.; Stötzer, O.J.; Fersching, D.; Nagel, D.; Holdenrieder, S. Circulating plasma DNA and DNA integrity in breast cancer patients undergoing neoadjuvant chemotherapy. Clin. Chim. Acta 2013, 425, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Iskow, R.C.; McCabe, M.T.; Mills, R.; Torene, S.; Pittard, W.S.; Neuwald, A.F.; Van Meir, E.G.; Vertino, P.M.; Devine, S.E. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell 2010, 141, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Duforestel, M.; Briand, J.; Bougras-Cartron, G.; Heymann, D.; Frenel, J.-S.; Vallette, F.M.; Cartron, P.-F. Cell-free circulating epimarks in cancer monitoring. Epigenomics 2020, 12, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Lee, K. Characteristics and Clinical Application of Extracellular Vesicle-Derived DNA. Cancers 2021, 13, 3827. [Google Scholar] [CrossRef]
- Helman, E.; Lawrence, M.S.; Stewart, C.; Sougnez, C.; Getz, G.; Meyerson, M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014, 24, 1053–1063. [Google Scholar] [CrossRef]
- Rodríguez-Martín, C.; Cidre, F.; Fernández-Teijeiro, A.; Gómez-Mariano, G.; De La Vega, L.; Ramos, P.; Zaballos, A.; Monzón, S.; Alonso, J. Familial retinoblastoma due to intronic LINE-1 insertion causes aberrant and noncanonical mRNA splicing of the RB1 gene. J. Hum. Genet. 2016, 61, 463–466. [Google Scholar] [CrossRef]
- Stacey, S.N.; Kehr, B.; Gudmundsson, J.; Zink, F.; Jonasdottir, A.; Gudjonsson, S.A.; Sigurdsson, A.; Halldorsson, B.V.; Agnarsson, B.A.; Benediktsdottir, K.R.; et al. Insertion of an SVA-E retrotransposon into theCASP8gene is associated with protection against prostate cancer. Hum. Mol. Genet. 2016, 25, 1008–1018. [Google Scholar] [CrossRef]
- Alekseeva, L.A.; Sen’Kova, A.V.; Zenkova, M.A.; Mironova, N.L. Targeting Circulating SINEs and LINEs with DNase I Provides Metastases Inhibition in Experimental Tumor Models. Mol. Ther.-Nucleic Acids 2020, 20, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Roshan, M.H.K.; Tambo, A.; Pace, N.P. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int. J. Inflamm. 2016, 2016, 1532832. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Richez, C.; Uccellini, M.; Richards, R.J.; Bonegio, R.; Akira, S.; Monestier, M.; Corley, R.; Viglianti, G.; Marshak-Rothstein, A.; et al. Requirement for DNA CpG Content in TLR9-Dependent Dendritic Cell Activation Induced by DNA-Containing Immune Complexes. J. Immunol. 2009, 183, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhao, E.; Wang, F.; Cui, H. CCDC25: Precise navigator for neutrophil extracellular traps on the prometastatic road. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Budker, V.; Godovikov, A.; Naumova, L.; Slepneva, I. Interaction of polynucleotides with natural and model membranes. Nucleic Acids Res. 1980, 8, 2499–2516. [Google Scholar] [CrossRef] [PubMed]
- Bryzgunova, O.; Laktionov, P. Generation of blood circulating DNA: The sources, peculiarities of circulation and structure. Biomeditsinskaya Khimiya 2015, 61, 409–426. [Google Scholar] [CrossRef]
- Kontouzov, S.; Cabrespines, A.; Amoura, Z.; Chabre, H.; Lotton, C.; Bach, J.-F. Binding of nucleosomes to a cell surface receptor: Redistribution and endocytosis in the presence of lupus antibodies. Eur. J. Immunol. 1996, 26, 472–486. [Google Scholar] [CrossRef]
- Hefeneider, S.H.; Cornell, K.A.; Brown, L.E.; Bakke, A.C.; McCoy, S.L.; Bennett, R.M. Nucleosomes and DNA bind to specific cell-surface molecules on murine cells and induce cytokine production. Clin. Immunol. Immunopathol. 1992, 63, 245–251. [Google Scholar] [CrossRef]
- Watson, K.; Gooderham, N.; Davies, D.S.; Edwards, R.J. Nucleosomes Bind to Cell Surface Proteoglycans. J. Biol. Chem. 1999, 274, 21707–21713. [Google Scholar] [CrossRef]
- Kubota, T.; Kanai, Y.; Miyasaka, N. Interpretation of the cross-reactivity of anti-DNA antibodies with cell surface proteins: The role of cell surface histones. Immunol. Lett. 1990, 23, 187–193. [Google Scholar] [CrossRef]
- Atluri, S.; Ragkousi, K.; Cortezzo, D.E.; Setlow, P. Cooperativity Between Different Nutrient Receptors in Germination of Spores of Bacillus subtilis and Reduction of This Cooperativity by Alterations in the GerB Receptor. J. Bacteriol. 2006, 188, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Marsman, G.; Zeerleder, S.; Luken, B.M. Extracellular histones, cell-free DNA, or nucleosomes: Differences in immunostimulation. Cell Death Dis. 2016, 7, e2518. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed]
- Msc, E.L.; Sanz-García, A.; Visakorpi, T.; Escobedo-Lucea, C.; Siljander, P.; Ayuso-Sacido, A.; Yliperttula, M. Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: Apoptotic bodies, microvesicles, and exosomes. Prostate 2014, 74, 1379–1390. [Google Scholar] [CrossRef]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef]
- Cai, J.; Wu, G.; Tan, X.; Han, Y.; Chen, C.; Li, C.; Wang, N.; Zou, X.; Chen, X.; Zhou, F.; et al. Transferred BCR/ABL DNA from K562 Extracellular Vesicles Causes Chronic Myeloid Leukemia in Immunodeficient Mice. PLoS ONE 2014, 9, e105200. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response toStaphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Braughton, K.R.; Whitney, A.R.; Voyich, J.M.; Schwan, T.G.; Musser, J.M.; DeLeo, F.R. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc. Natl. Acad. Sci. USA 2003, 100, 10948–10953. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, L. The pro-tumor effect and the anti-tumor effect of neutrophils extracellular traps. Biosci. Trends 2019, 13, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wagner, D.D. NETosis: A New Factor in Tumor Progression and Cancer-Associated Thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.D.; Houghton, A.M. Tumor-Associated Neutrophils: New Targets for Cancer Therapy: Figure 1. Cancer Res. 2011, 71, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Ho-Tin-Noé, B.; Carbo, C.; Demers, M.; Cifuni, S.M.; Goerge, T.; Wagner, D.D. Innate Immune Cells Induce Hemorrhage in Tumors during Thrombocytopenia. Am. J. Pathol. 2009, 175, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Berger-Achituv, S.; Brinkmann, V.; Abu Abed, U.; Kühn, L.I.; Ben-Ezra, J.; Elhasid, R.; Zychlinsky, A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front. Immunol. 2013, 4, 48. [Google Scholar] [CrossRef]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.A.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef]
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 2017, 140, 2321–2330. [Google Scholar] [CrossRef]
- Zhu, T.; Zou, X.; Yang, C.; Li, L.; Wang, B.; Li, R.; Li, H.; Xu, Z.; Huang, D.; Wu, Q. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelial-mesenchymal transition. Int. J. Mol. Med. 2021, 48, 1–13. [Google Scholar] [CrossRef]
- Rayes, R.F.; Mouhanna, J.G.; Nicolau, I.; Bourdeau, F.; Giannias, B.; Rousseau, S.; Quail, D.; Walsh, L.; Sangwan, V.; Bertos, N.; et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis-promoting effects. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Papagoras, C.; Miltiades, P.; Angelidou, I.; Mitsios, A.; Kotsianidis, I.; Skendros, P.; Sivridis, E.; et al. Gradient Infiltration of Neutrophil Extracellular Traps in Colon Cancer and Evidence for Their Involvement in Tumour Growth. PLoS ONE 2016, 11, e0154484. [Google Scholar] [CrossRef]
- Schedel, F.; Mayer-Hain, S.; Pappelbaum, K.I.; Metze, D.; Stock, M.; Goerge, T.; Loser, K.; Sunderkötter, C.; Luger, T.A.; Weishaupt, C. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment. Cell Melanoma Res. 2019, 33, 63–73. [Google Scholar] [CrossRef]
- Breitbach, C.J.; De Silva, N.S.; Falls, T.; Aladl, U.; Evgin, L.; Paterson, J.; Sun, Y.Y.; Roy, D.; Rintoul, J.L.; Daneshmand, M.; et al. Targeting Tumor Vasculature with an Oncolytic Virus. Mol. Ther. 2011, 19, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef]
- Jin, W.; Xu, H.-X.; Zhang, S.-R.; Li, H.; Wang, W.-Q.; Gao, H.-L.; Wu, C.-T.; Xu, J.-Z.; Qi, Z.-H.; Li, S.; et al. Tumor-Infiltrating NETs Predict Postsurgical Survival in Patients with Pancreatic Ductal Adenocarcinoma. Ann. Surg. Oncol. 2018, 26, 635–643. [Google Scholar] [CrossRef]
- McInturff, A.M.; Cody, M.J.; Elliott, E.A.; Glenn, J.W.; Rowley, J.W.; Rondina, M.T.; Yost, C.C. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 2012, 120, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.; Weitz, J.I.; Liaw, P.C. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arter. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A.; et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2013, 73, 1854–1863. [Google Scholar] [CrossRef]
- Kambas, K.; Mitroulis, I.; Apostolidou, E.; Girod, A.; Chrysanthopoulou, A.; Pneumatikos, I.; Skendros, P.; Kourtzelis, I.; Koffa, M.; Kotsianidis, I.; et al. Autophagy Mediates the Delivery of Thrombogenic Tissue Factor to Neutrophil Extracellular Traps in Human Sepsis. PLoS ONE 2012, 7, e45427. [Google Scholar] [CrossRef]
- Cedervall, J.; Zhang, Y.; Huang, H.; Zhang, L.; Femel, J.; Dimberg, A.; Olsson, A.-K. Neutrophil Extracellular Traps Accumulate in Peripheral Blood Vessels and Compromise Organ Function in Tumor-Bearing Animals. Cancer Res. 2015, 75, 2653–2662. [Google Scholar] [CrossRef]
- Janus, N.; Launay-Vacher, V.; Byloos, E.; Machiels, J.P.; Duck, L.; Kerger, J.; Wynendaele, W.; Canon, J.-L.; Lybaert, W.; Nortier, J.; et al. Cancer and renal insufficiency results of the BIRMA study. Br. J. Cancer 2010, 103, 1815–1821. [Google Scholar] [CrossRef]
- Launay-Vacher, V. Epidemiology of Chronic Kidney Disease in Cancer Patients: Lessons from the IRMA Study Group. Semin. Nephrol. 2010, 30, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Launay-Vacher, V.; Oudard, S.; Janus, N.; Gligorov, J.; Pourrat, X.; Rixe, O.; Morere, J.-F.; Beuzeboc, P.; Deray, G. On behalf of the Renal Insufficiency and Cancer Medications (IRMA) Study Group. Prevalence of Renal Insufficiency in cancer patients and implications for anticancer drug management: The renal insufficiency and anticancer medications (IRMA) study. Cancer 2007, 110, 1376–1384. [Google Scholar] [CrossRef]
- Thålin, C.; Demers, M.; Blomgren, B.; Wong, S.L.; von Arbin, M.; von Heijne, A.; Laska, A.C.; Wallén, H.; Wagner, D.D.; Aspberg, S. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb. Res. 2016, 139, 56–64. [Google Scholar] [CrossRef]
- Yu, S.C.; Lee, S.W.; Jiang, P.; Leung, T.Y.; Chan, K.A.; Chiu, R.W.; Lo, Y.D. High-Resolution Profiling of Fetal DNA Clearance from Maternal Plasma by Massively Parallel Sequencing. Clin. Chem. 2013, 59, 1228–1237. [Google Scholar] [CrossRef]
- Han, D.S.; Lo, Y.D. The Nexus of cfDNA and Nuclease Biology. Trends Genet. 2021, 37, 758–770. [Google Scholar] [CrossRef]
- Coy, J.F.; Velhagen, I.; Himmele, R.; Delius, H.; Poustka, A.; Zentgraf, H. Isolation, differential splicing and protein expression of a DNase on the human X chromosome. Cell Death Differ. 1996, 3, 199–206. [Google Scholar] [PubMed]
- Shiokawa, D.; Matsushita, T.; Shika, Y.; Shimizu, M.; Maeda, M.; Tanuma, S.-I. DNase X Is a Glycosylphosphatidylinositol-anchored Membrane Enzyme That Provides a Barrier to Endocytosis-mediated Transfer of a Foreign Gene. J. Biol. Chem. 2007, 282, 17132–17140. [Google Scholar] [CrossRef] [PubMed]
- Shiokawa, D.; Tanuma, S.-I. Characterization of Human DNase I Family Endonucleases and Activation of DNase γ during Apoptosis. Biochemistry 2000, 40, 143–152. [Google Scholar] [CrossRef]
- Pal, K.; Zhao, Y.; Wang, Y.; Wang, X. Ubiquitous membrane-bound DNase activity in podosomes and invadopodia. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Grimm, M.; Cetindis, M.; Lehmann, M.; Biegner, T.; Munz, A.; Teriete, P.; Reinert, S. Apoptosis resistance-related ABCB5 and DNaseX (Apo10) expression in oral carcinogenesis. Acta Odontol. Scand. 2014, 73, 336–342. [Google Scholar] [CrossRef]
- Taper, H.S. Altered deoxyribonuclease activity in cancer cells and its role in non toxic adjuvant cancer therapy with mixed vitamins C and K3. Anticancer. Res. 2008, 28, 2727–2732. [Google Scholar] [PubMed]
- Grimm, M.; Schmitt, S.; Teriete, P.; Biegner, T.; Stenzl, A.; Hennenlotter, J.; Muhs, H.-J.; Munz, A.; Nadtotschi, T.; König, K.; et al. A biomarker based detection and characterization of carcinomas exploiting two fundamental biophysical mechanisms in mammalian cells. BMC Cancer 2013, 13, 569. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Sarkar, A.; Wang, X. Optical sensor revealed abnormal nuclease spatial activity on cancer cell membrane. J. Biophotonics 2018, 12, e201800351. [Google Scholar] [CrossRef] [PubMed]
- Coy, J.F. EDIM-TKTL1/Apo10 Blood Test: An Innate Immune System Based Liquid Biopsy for the Early Detection, Characterization and Targeted Treatment of Cancer. Int. J. Mol. Sci. 2017, 18, 878. [Google Scholar] [CrossRef] [PubMed]
- Woo, E.-J.; Kim, Y.-G.; Kim, M.-S.; Han, W.-D.; Shin, S.; Robinson, H.; Park, S.-Y.; Oh, B.-H. Structural Mechanism for Inactivation and Activation of CAD/DFF40 in the Apoptotic Pathway. Mol. Cell 2004, 14, 531–539. [Google Scholar] [CrossRef]
- Los, M.; Neubüser, D.; Coy, J.F.; Mozoluk, M.; Poustka, A.; Schulze-Osthoff, K. Functional Characterization of DNase X, a Novel Endonuclease Expressed in Muscle Cells. Biochemistry 2000, 39, 7365–7373. [Google Scholar] [CrossRef]
- Widlak, P.; Li, L.Y.; Wang, X.; Garrard, W.T. Action of Recombinant Human Apoptotic Endonuclease G on Naked DNA and Chromatin Substrates. J. Biol. Chem. 2001, 276, 48404–48409. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Sally, B.; D’Agati, V.; Martinez-Ortiz, W.; Özçakar, Z.; David, J.; Rashidfarrokhi, A.; Yeste, A.; Panea, C.; Chida, A.S.; et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166, 88–101. [Google Scholar] [CrossRef]
- Miles, M.A.; Harris, M.A.; Hawkins, C.J. Proteasome inhibitors trigger mutations via activation of caspases and CAD, but mutagenesis provoked by the HDAC inhibitors vorinostat and romidepsin is caspase/CAD-independent. Apoptosis 2019, 24, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, X.; Wang, T.; Zhang, Y.; Wang, Q.; Hu, Y. HNRNPCL1, PRAMEF1, CFAP74, and DFFB: Common Potential Biomarkers for Sporadic and Suspected Lynch Syndrome Endometrial Cancer. Cancer Manag. Res. 2020, 12, 11231–11241. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, D.D.; Fahmi, T.; Wang, X.; Apostolov, E.O.; Sokolov, N.N.; Javadov, S.; Basnakian, A.G. Regulation of Apoptotic Endonucleases by EndoG. DNA Cell Biol. 2015, 34, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, D.D.; Gladilina, Y.A.; Pokrovsky, V.S.; Grishin, D.V.; Grachev, V.; Orlova, V.; Pokrovskaya, M.V.; Alexandrova, S.S.; Plyasova, A.A.; Sokolov, N.N. Endonuclease G modulates the alternative splicing of deoxyribonuclease 1 mRNA in human CD4+ T lymphocytes and prevents the progression of apoptosis. Biochimie 2018, 157, 158–176. [Google Scholar] [CrossRef]
- Barés, G.; Beà, A.; Hernández, L.; Navaridas, R.; Felip, I.; Megino, C.; Blasco, N.; Nadeu, F.; Campo, E.; Llovera, M.; et al. ENDOG Impacts on Tumor Cell Proliferation and Tumor Prognosis in the Context of PI3K/PTEN Pathway Status. Cancers 2021, 13, 3803. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Y.; Zhao, Y.; He, S.; Zhao, R.; Song, Y.; Cheng, J.; Gong, Y.; Xie, J.; Wang, Y.; et al. Caspase-3 knockout attenuates radiation-induced tumor repopulation via impairing the ATM/p53/Cox-2/PGE2 pathway in non-small cell lung cancer. Aging 2020, 12, 21758–21776. [Google Scholar] [CrossRef] [PubMed]
- Hawes, M.C.; Curlango-Rivera, G.; Wen, F.; White, G.J.; VanEtten, H.D.; Xiong, Z. Extracellular DNA: The tip of root defenses? Plant Sci. 2011, 180, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Han, D.S.; Ni, M.; Chan, R.W.; Chan, V.W.; Lui, K.; Chiu, R.W.; Lo, Y.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020, 106, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Kishi, K.; Yasuda, T.; Takeshita, H. DNase I: Structure, function, and use in medicine and forensic science. Leg. Med. 2001, 3, 69–83. [Google Scholar] [CrossRef]
- Kochanek, S.; Renz, D.; Doerfler, W. Differences in the accessibility of methylated and unmethylated DNA to DNase I. Nucleic Acids Res. 1993, 21, 5843–5845. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eulitz, D.; Mannherz, H.G. Inhibition of deoxyribonuclease I by actin is to protect cells from premature cell death. Apoptosis 2007, 12, 1511–1521. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Rodina, D.; Nomuraa, H.; Morton, C.C.; Weremowiczb, S.; Schneider, M.C. Identification, Localization, and Expression of Two Novel Human Genes Similar to Deoxyribonuclease I. Genomics 1997, 42, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Wilber, A.; Lu, M.; Schneider, M.C. Deoxyribonuclease I-like III Is an Inducible Macrophage Barrier to Liposomal Transfection. Mol. Ther. 2002, 6, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Napirei, M.; Wulf, S.; Eulitz, D.; Mannherz, H.G.; Kloeckl, T. Comparative characterization of rat deoxyribonuclease 1 (Dnase1) and murine deoxyribonuclease 1-like 3 (Dnase1l3). Biochem. J. 2005, 389, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, R.; Mizuta, M.; Araki, S.; Shiokawa, D.; Tanuma, S.-I.; Kitamura, D. Action of apoptotic endonuclease DNase γ on naked DNA and chromatin substrates. Biochem. Biophys. Res. Commun. 2006, 345, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Economidou-Karaoglou, A.; Lans, M.; Taper, H.S.; Michaux, J.L.; Roberfroid, M. variations in serum alkaline dnase activity. A new means for therapeutic monitoring of malignant lymphomas. Cancer 1988, 61, 1838–1843. [Google Scholar] [CrossRef]
- Tamkovich, S.; Cherepanova, A.V.; Kolesnikova, E.V.; Rykova, E.Y.; Pyshnyi, D.V.; Vlassov, V.V.; Laktionov, P.P. Circulating DNA and DNase Activity in Human Blood. Ann. N. Y. Acad. Sci. 2006, 1075, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, A.V.; Tamkovich, S.N.; Vlassov, V.V.; Laktionov, P.P. Blood deoxyribonuclease activity in health and diseases. Biochem. Suppl. Ser. B: Biomed. Chem. 2007, 1, 299–304. [Google Scholar] [CrossRef]
- Golonka, R.M.; Yeoh, B.S.; Petrick, J.L.; Weinstein, S.J.; Albanes, D.; Gewirtz, A.T.; McGlynn, K.A.; Vijay-Kumar, M. Deoxyribonuclease I Activity, Cell-Free DNA, and Risk of Liver Cancer in a Prospective Cohort. JNCI Cancer Spectr. 2018, 2, pky083. [Google Scholar] [CrossRef]
- Liu, J.; Yi, J.; Zhang, Z.; Cao, D.; Li, L.; Yao, Y. Deoxyribonuclease 1-like 3 may be a potential prognostic biomarker associated with immune infiltration in colon cancer. Aging 2021, 13, 16513–16526. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Dinsdale, R.J.; Naumann, D.N.; Acharjee, A.; Bishop, J.R.B.; Lord, J.M.; Harrison, P. Traumatic injury is associated with reduced deoxyribonuclease activity and dysregulation of the actin scavenging system. Burn. Trauma 2021, 9, tkab001. [Google Scholar] [CrossRef]
- Yasuda, T.; Kawai, Y.; Ueki, M.; Kishi, K. Clinical applications of DNase I, a genetic marker already used for forensic identification. Leg. Med. 2005, 7, 274–277. [Google Scholar] [CrossRef] [PubMed]
- De Lamirande, G. Action of Deoxyribonuclease and Ribonuclease on the Growth of Ehrlich Ascites Carcinoma in Mice. Nature 1961, 192, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Belyaeva, M.I.; Kyune, M.F.; Nuxhina, A.M. Effect of bacterial deoxyribonuclease on Ehrlich’s ascities carcinoma cells in vitro. Fed. Proc. Transl. Suppl. 1964, 23, 345–348. [Google Scholar] [PubMed]
- Salganik, R.I.; Martynova, R.P.; Matienko, N.A.; Ronichevskaya, G.M. Effect of Deoxyribonuclease on the Course of Lymphatic Leukaemia in AKR Mice. Nature 1967, 214, 100–102. [Google Scholar] [CrossRef]
- Sugihara, S.; Yamamoto, T.; Tanaka, H.; Kambara, T.; Hiraoka, T.; Miyauchi, Y. Deoxyribonuclease treatment prevents blood-borne liver metastasis of cutaneously transplanted tumour cells in mice. Br. J. Cancer 1993, 67, 66–70. [Google Scholar] [CrossRef]
- Alcázar-Leyva, S.; Cerón, E.; Masso, F.; Montaño, L.F.; Gorocica, P.; Alvarado-Vásquez, N. Incubation with DNase I inhibits tumor cell proliferation. Med Sci. Monit. 2009, 15, 55. [Google Scholar]
- Patutina, O.; Mironova, N.; Ryabchikova, E.; Popova, N.; Nikolin, V.; Kaledin, V.; Vlassov, V.; Zenkova, M. Inhibition of metastasis development by daily administration of ultralow doses of RNase A and DNase I. Biochimie 2011, 93, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Shen, A.; Choi, A.; Gerner, E.W.; Shi, J. Extracellular DNA in Pancreatic Cancer Promotes Cell Invasion and Metastasis. Cancer Res. 2013, 73, 4256–4266. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Chen, X.; Xiao, J.; Zhou, D.B.; Lu, X.X.; Li, W.; Xie, B.; Kuang, X.; Chen, Q. Neutrophil extracellular traps promote lipopolysaccharide-induced airway inflammation and mucus hypersecretion in mice. Oncotarget 2018, 9, 13276–13286. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, T.; Fisher, J.; Bakochi, A.; Neumann, A.; Cardoso, J.F.P.; Karlsson, C.A.Q.; Pavan, C.; Lundgaard, I.; Nilson, B.; Reinstrup, P.; et al. Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nat. Commun. 2019, 10, 1667. [Google Scholar] [CrossRef] [PubMed]
- Demkow, U. Neutrophil Extracellular Traps (NETs) in Cancer Invasion, Evasion and Metastasis. Cancers 2021, 13, 4495. [Google Scholar] [CrossRef]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Ferri, L.E. Neutrophils Promote Liver Metastasis via Mac-1–Mediated Interactions with Circulating Tumor Cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef]
- Hisada, Y.; Grover, S.P.; Maqsood, A.; Houston, R.; Ay, C.; Noubouossie, D.F.; Cooley, B.C.; Wallén, H.; Key, N.S.; Thålin, C.; et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica 2019, 105, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, L.A.; Mironova, N.; Brenner, E.V.; Kurilshikov, A.; Patutina, O.A.; Zenkova, M.A. Alteration of the exDNA profile in blood serum of LLC-bearing mice under the decrease of tumour invasion potential by bovine pancreatic DNase I treatment. PLoS ONE 2017, 12, e0171988. [Google Scholar] [CrossRef]
- Rosner, K. DNase1: A new personalized therapy for cancer? Expert Rev. Anticancer. Ther. 2011, 11, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Haig, J.D.M. Eradication of Human Ovarian Cancer Cells by Transgenic Expression of Recombinant DNASE1, DNASE1L3, DNASE2, and DFFB Controlled by EGFR Promoter: Novel Strategy for Targeted Therapy of Cancer. J. Genet. Syndr. Gene Ther. 2013, 4, 152. [Google Scholar] [CrossRef]
- Xia, Y.; He, J.; Zhang, H.; Wang, H.; Tetz, G.; Maguire, C.A.; Wang, Y.; Onuma, A.; Genkin, D.; Tetz, V.; et al. AAV-mediated gene transfer of DNase I in the liver of mice with colorectal cancer reduces liver metastasis and restores local innate and adaptive immune response. Mol. Oncol. 2020, 14, 2920–2935. [Google Scholar] [CrossRef]
- Yang, L.-Y.; Luo, Q.; Lu, L.; Zhu, W.-W.; Sun, H.-T.; Wei, R.; Lin, Z.-F.; Wang, X.-Y.; Wang, C.-Q.; Lu, M.; et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J. Hematol. Oncol. 2020, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.-H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef]
- Silverman, R.A.; Foley, F.; Dalipi, R.; Kline, M.; Lesser, M. The use of rhDNAse in severely ill, non-intubated adult asthmatics refractory to bronchodilators: A pilot study. Respir. Med. 2012, 106, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Mittal, B.B.; Wang, E.; Sejpal, S.; Agulnik, M.; Mittal, A.; Harris, J. Effect of Recombinant Human Deoxyribonuclease on Oropharyngeal Secretions in Patients with Head-and-Neck Cancers Treated with Radiochemotherapy. Int. J. Radiat. Oncol. 2013, 87, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Assallum, H.; Song, T.Y.; DeLorenzo, L.; Harris, K. Bronchoscopic instillation of DNase to manage refractory lobar atelectasis in a lung cancer patient. Ann. Transl. Med. 2019, 7, 363. [Google Scholar] [CrossRef] [PubMed]
- Várady, C.B.; Oliveira, A.C.; Monteiro, R.Q.; Gomes, T. Recombinant human DNase I for the treatment of cancer-associated thrombosis: A pre-clinical study. Thromb. Res. 2021, 203, 131–137. [Google Scholar] [CrossRef]
- Genkin, D.; Tets, V.; Tets, G. Method for Treating Oncological Diseases. U.S. Patent WO-2005004903-A1, 20 January 2005. [Google Scholar]
- Genkin, D.; Tets, G.; Tets, V. Method to Improve Safety and Efficacy of Anti-Cancer Therapy 2015. U.S. Patent 2020/0188493, 20 February 2020. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Name | Source 1 | Location | Specificity 2 | Cleavage Products 3 | Ion Dependency | Optimal pH | Structure | Details |
|---|---|---|---|---|---|---|---|---|
| Intracellular catabolism of DNA | ||||||||
| DNase IL1 (DNase X) | All cells | Endoplasmic reticulum, extracellular membrane | Chromatin | 3′OH, 5′P oligonucleosomes with ds-breaks [127] | Ca2+, Mg2+ | 6–8 | Capable of homodimerization [128] | C-terminal signal peptide, an N-linked glycosylation site and C-terminal hydrophobic domain; inactivated with Zn2+ and Apo10 |
| DNA fragmentation factor B (DFFB, DFF40 or CAD) | All cells | Endoplasmic reticulum and nucleus | 5′A(G)→3′X >> 5′C(T)→3′X chromatin and naked DNA [127] | 3′OH, 5′P oligonucleosomes with ds-breaks [127] | Mg2+ | 6–8 | Heterodimer | Usually bound with inhibitor DFFA; activated by cleavage of inhibitor with caspase-3 |
| Endonuclease G (EndoG) | All cells | Mitochondria, migrates to the nucleus under apoptosis | poly(dG), poly(dC) >> others; ssDNA and dsDNA in chromatin; DNA/RNA heteroduplexes [129] | 3′OH, 5′P oligonucleosomes with ds-breaks and internal ss- nicks [129] | Mg2+/Mn2+ | Biphasic pH optima: 9 and 7 [129] | Homodimer | ββα-Me-finger; normally bound by Hsp70 and CHIP; inactivated with Fe2+ and Zn2+ [129] |
| Extracellular catabolism of cfDNA | ||||||||
| DNase I | Predominantly expressed in exocrine cells in the gastrointestinal tract, salivary glands, and kidneys; endothelial cells | Extracellular space | 5′-T > C >> A,G→3′X; naked dsDNA >> ssDNA; DNA in DNA/RNA heteroduplexes; slight efficacy to chromatin [127] | 3′OH,5′P mononucleosomes and tetranucleotides [127] | Ca2+, Mg2+ | 6–8 | Monomer | Inactivated with Zn2+ and G-actin |
| DNase IL3 (DNAse γ) | Predominantly expressed in the liver and spleen; endothelial cells; macrophages and dendritic cells | Endoplasmic reticulum, nucleus and extracellular space | 5′ C > T >> A, G→3′X; chromatin DNA in lipid–membrane particles [130] | 3′OH, 5′P mononucleosomes [127] | Ca2+, Mg2+ | 6–8 | Inactivated with Zn2+ and heparin; it has a positively charged C-terminal sequence allowing transfer to the nucleus and encapsulation in MVs | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alekseeva, L.; Mironova, N. Role of Cell-Free DNA and Deoxyribonucleases in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12246. https://doi.org/10.3390/ijms222212246
Alekseeva L, Mironova N. Role of Cell-Free DNA and Deoxyribonucleases in Tumor Progression. International Journal of Molecular Sciences. 2021; 22(22):12246. https://doi.org/10.3390/ijms222212246
Chicago/Turabian StyleAlekseeva, Ludmila, and Nadezhda Mironova. 2021. "Role of Cell-Free DNA and Deoxyribonucleases in Tumor Progression" International Journal of Molecular Sciences 22, no. 22: 12246. https://doi.org/10.3390/ijms222212246
APA StyleAlekseeva, L., & Mironova, N. (2021). Role of Cell-Free DNA and Deoxyribonucleases in Tumor Progression. International Journal of Molecular Sciences, 22(22), 12246. https://doi.org/10.3390/ijms222212246