
A Yeast-Based Repurposing Approach for the Treatment of Mitochondrial DNA Depletion Syndromes Led to the Identification of Molecules Able to Modulate the dNTP Pool

 , , and
, , and
Abstract
:1. Introduction
2. Results
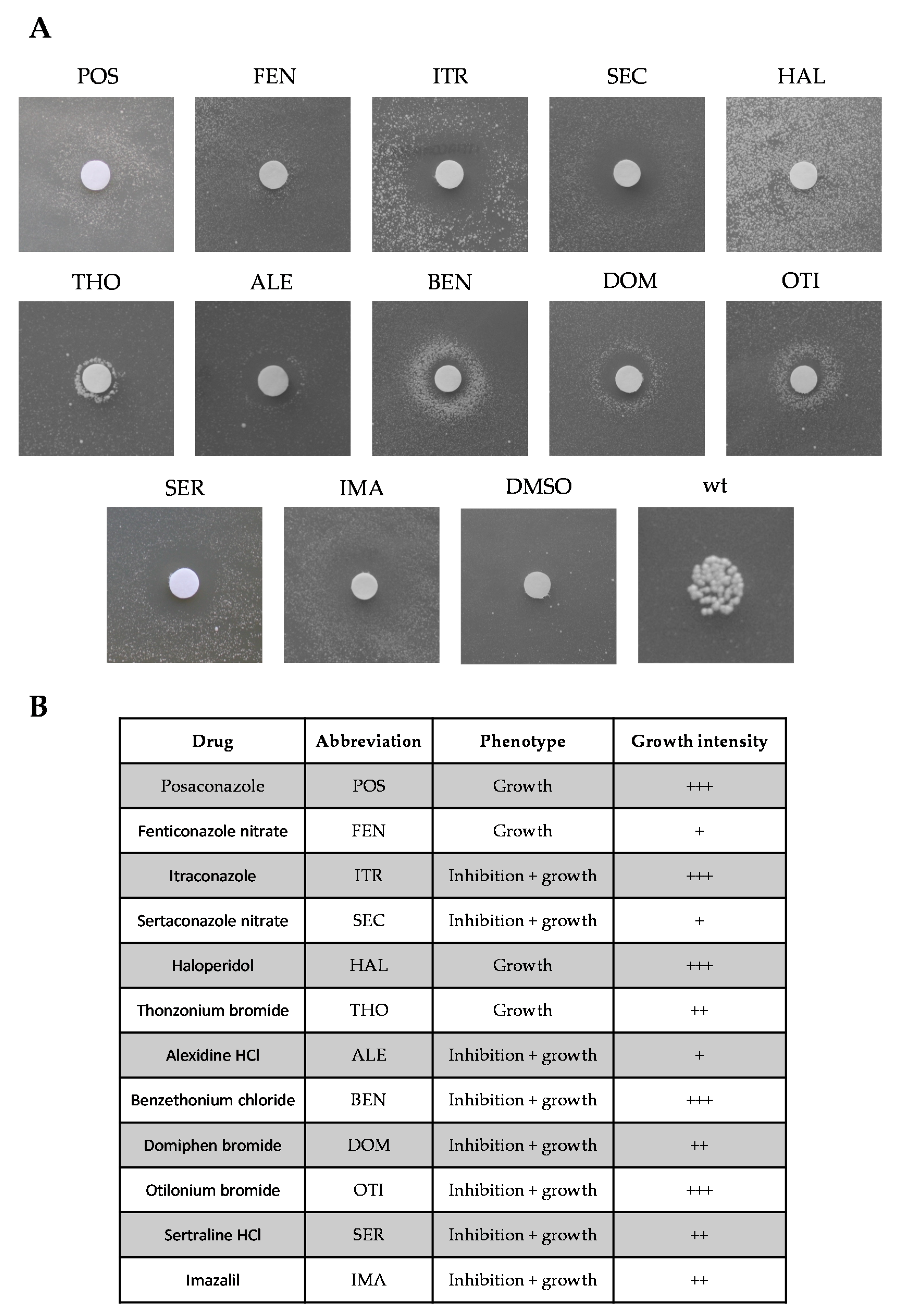
2.1. Identification of Molecules Preventing the Oxidative Phosphorylation Defect Associated with the Sym1 Mutation
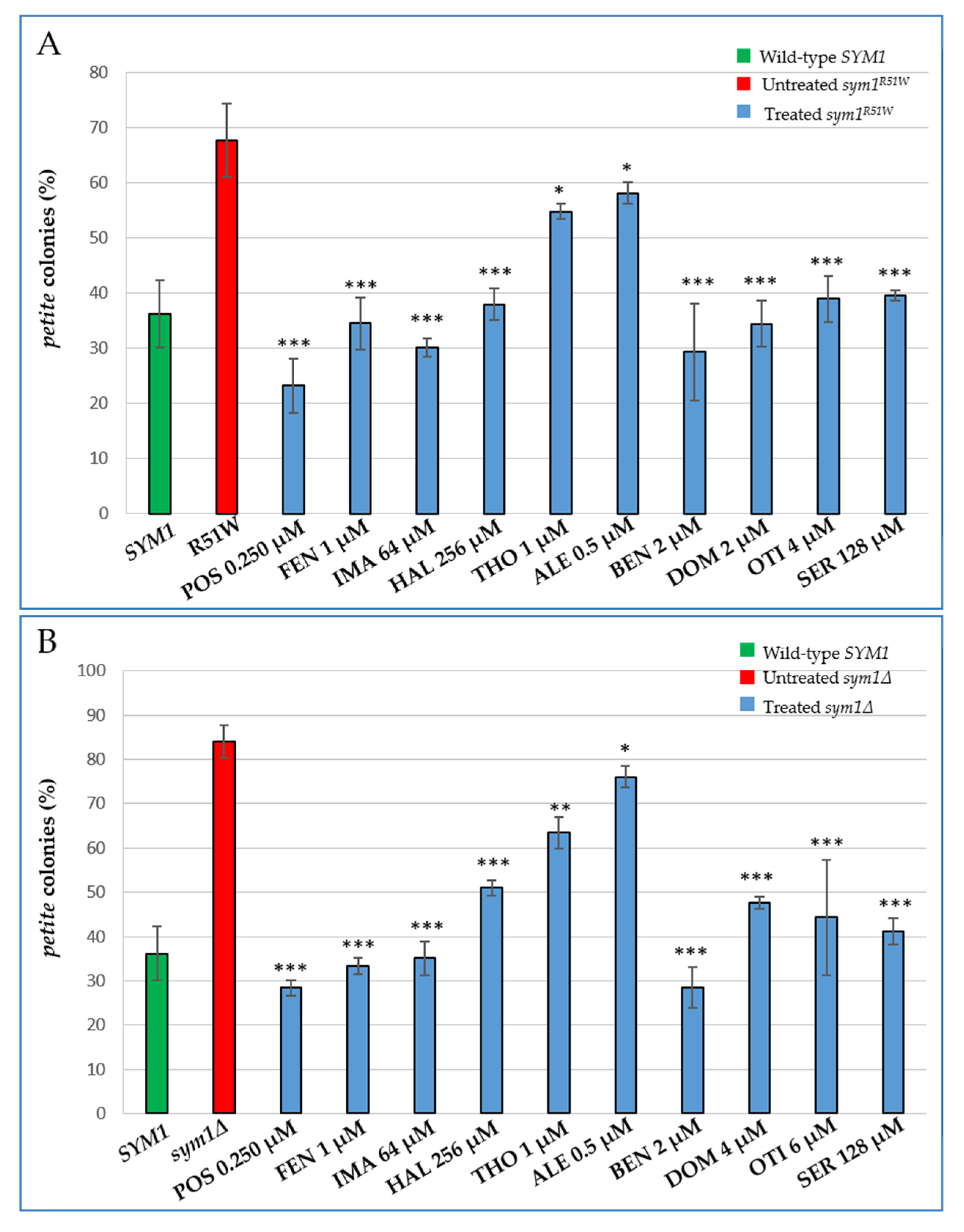
2.2. Characterization of the Effects of the Identified Molecules on mtDNA Maintenance
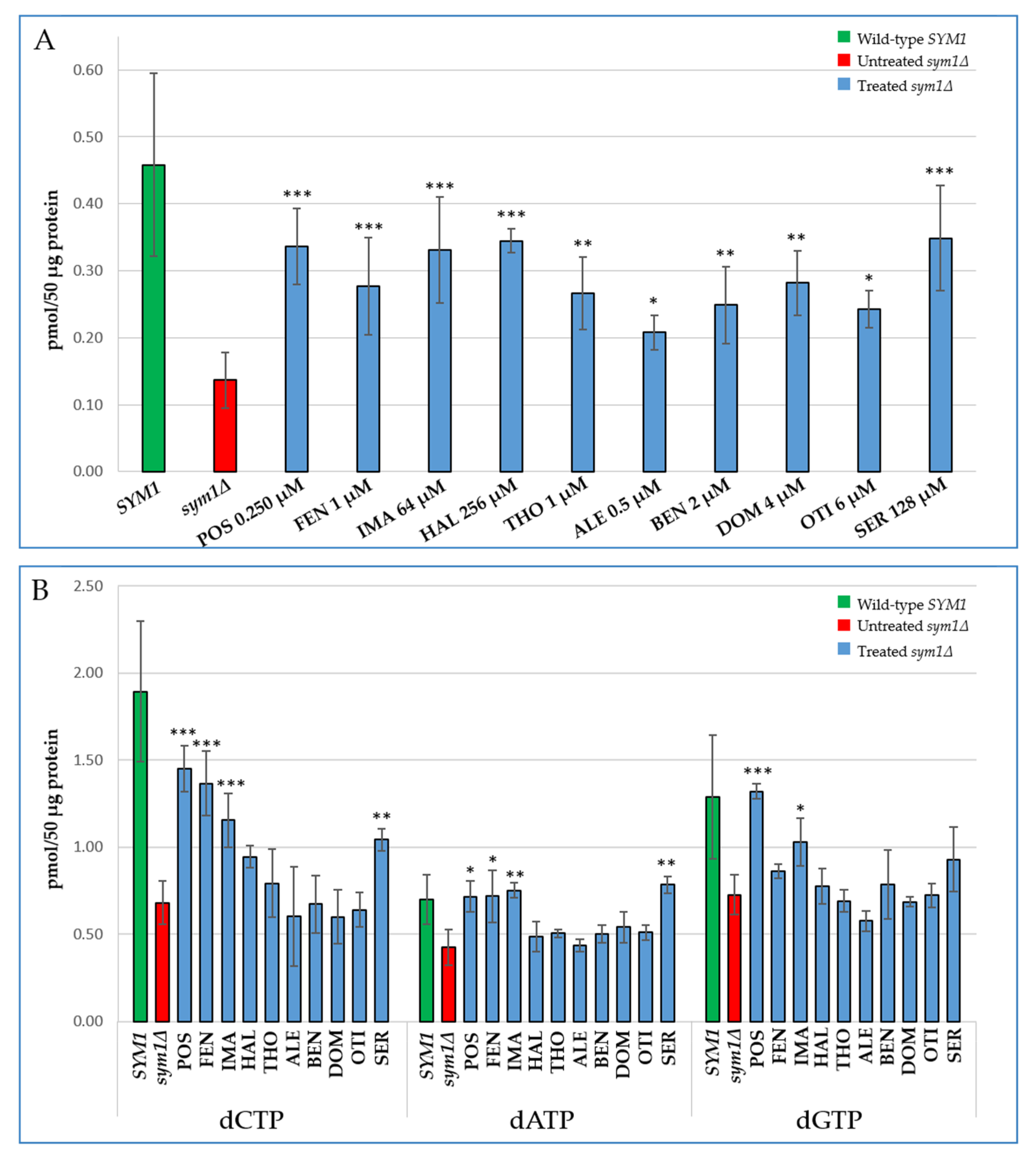
2.3. Characterization of the Effects of the Identified Molecules on the Mitochondrial dNTP Pool
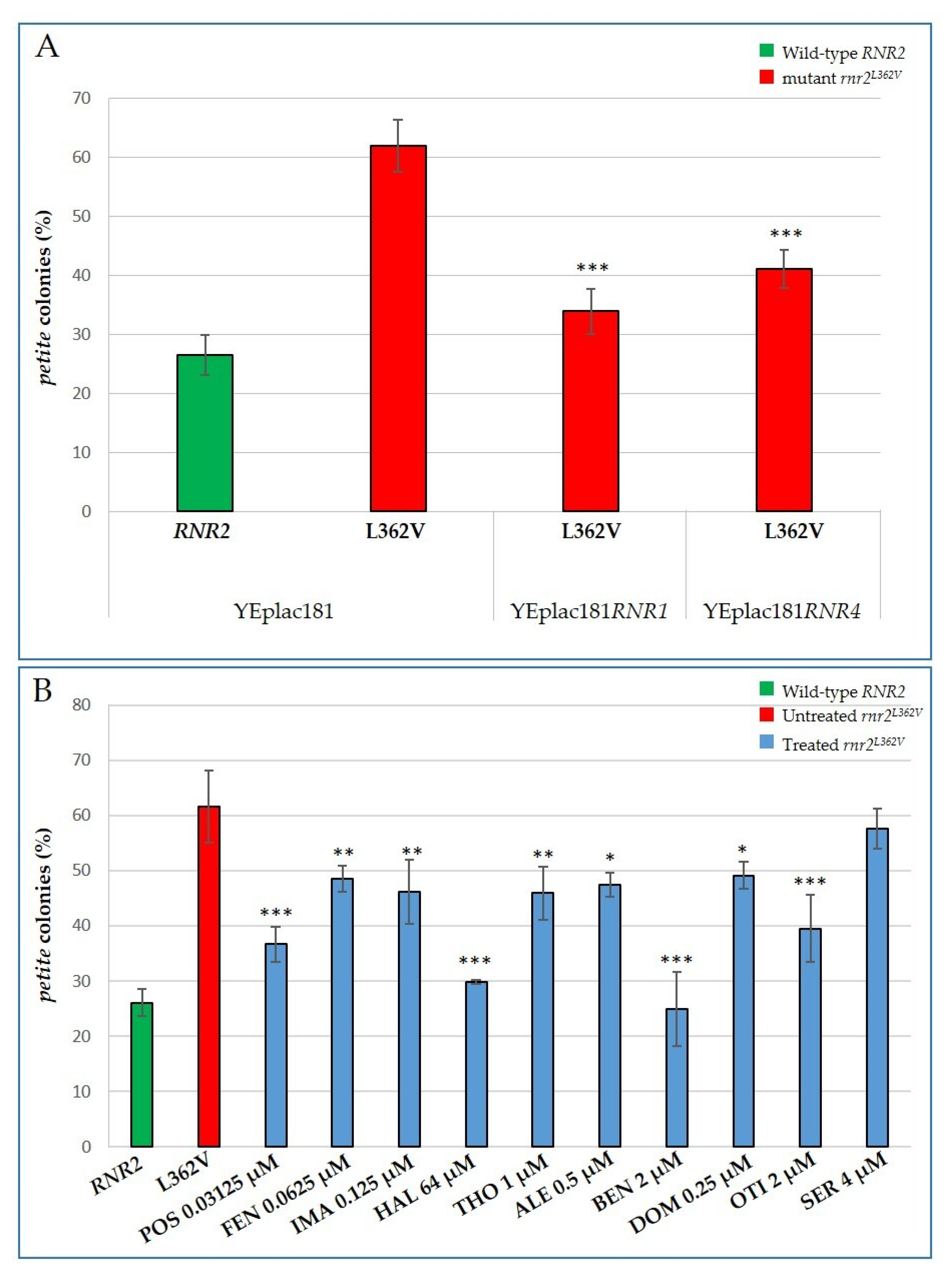
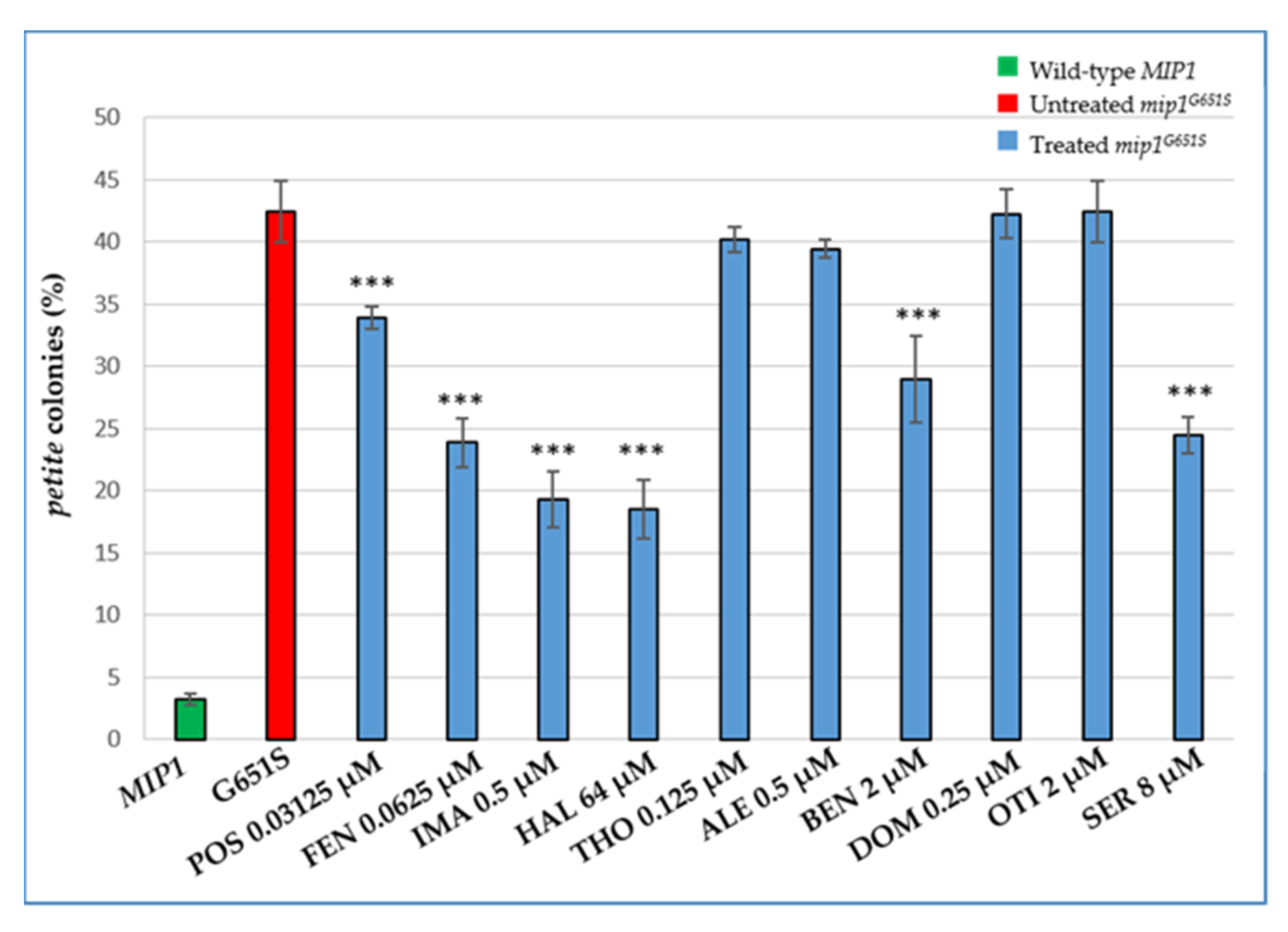
2.4. Characterization of the Effects of the Identified Molecules on other MDS Yeast Models: The rnr2 and mip1 Mutants
3. Discussion
4. Materials and Methods
4.1. Yeast Strains and Culture Media
4.2. High-Throughput Screening: Drug Drop Test
4.3. Petite Frequency and Fitness Test
4.4. Whole Cell Dexoxyribonucleoside Triphosphate Extraction
4.5. Mitochondrial Extraction and Separation of Mitochondrial and Cytosolic dNTPs
4.6. DNA Polymerase Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suomalainen, A.; Isohanni, P. Mitochondrial DNA Depletion Syndromes--Many Genes, Common Mechanisms. Neuromuscul. Disord. NMD 2010, 20, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Dornas, W.; Schuppan, D. Mitochondrial Oxidative Injury: A Key Player in Nonalcoholic Fatty Liver Disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Karasawa, M.; Zwacka, R.M.; Reuter, A.; Fink, T.; Hsieh, C.L.; Lichter, P.; Francke, U.; Weiher, H. The Human Homolog of the Glomerulosclerosis Gene Mpv17: Structure and Genomic Organization. Hum. Mol. Genet. 1993, 2, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 Encodes an Inner Mitochondrial Membrane Protein and Is Mutated in Infantile Hepatic Mitochondrial DNA Depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef]
- Karadimas, C.L.; Vu, T.H.; Holve, S.A.; Chronopoulou, P.; Quinzii, C.; Johnsen, S.D.; Kurth, J.; Eggers, E.; Palenzuela, L.; Tanji, K.; et al. Navajo Neurohepatopathy Is Caused by a Mutation in the MPV17 Gene. Am. J. Hum. Genet. 2006, 79, 544–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löllgen, S.; Weiher, H. The Role of the Mpv17 Protein Mutations of Which Cause Mitochondrial DNA Depletion Syndrome (MDDS): Lessons from Homologs in Different Species. Biol. Chem. 2015, 396, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Dallabona, C.; Marsano, R.M.; Arzuffi, P.; Ghezzi, D.; Mancini, P.; Zeviani, M.; Ferrero, I.; Donnini, C. Sym1, the Yeast Ortholog of the MPV17 Human Disease Protein, Is a Stress-Induced Bioenergetic and Morphogenetic Mitochondrial Modulator. Hum. Mol. Genet. 2010, 19, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Bottani, E.; Giordano, C.; Civiletto, G.; Di Meo, I.; Auricchio, A.; Ciusani, E.; Marchet, S.; Lamperti, C.; d’Amati, G.; Viscomi, C.; et al. AAV-Mediated Liver-Specific MPV17 Expression Restores MtDNA Levels and Prevents Diet-Induced Liver Failure. Mol. Ther. 2014, 22, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, R.; Krüger, V.; Meinecke, M.; Schulz, C.; Schmidt, B.; Grunau, S.D.; Guiard, B.; Wiedemann, N.; van der Laan, M.; Wagner, R.; et al. The Channel-Forming Sym1 Protein Is Transported by the TIM23 Complex in a Presequence-Independent Manner. Mol. Cell. Biol. 2012, 32, 5009–5021. [Google Scholar] [CrossRef] [Green Version]
- Antonenkov, V.D.; Isomursu, A.; Mennerich, D.; Vapola, M.H.; Weiher, H.; Kietzmann, T.; Hiltunen, J.K. The Human Mitochondrial DNA Depletion Syndrome Gene MPV17 Encodes a Non-Selective Channel That Modulates Membrane Potential. J. Biol. Chem. 2015, 290, 13840–13861. [Google Scholar] [CrossRef] [Green Version]
- Luna-Sanchez, M.; Benincá, C.; Cerutti, R.; Brea-Calvo, G.; Yeates, A.; Scorrano, L.; Zeviani, M.; Viscomi, C. Opa1 Overexpression Protects from Early-Onset Mpv17−/−-Related Mouse Kidney Disease. Mol. Ther. 2020, 28, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Dalla Rosa, I.; Cámara, Y.; Durigon, R.; Moss, C.F.; Vidoni, S.; Akman, G.; Hunt, L.; Johnson, M.A.; Grocott, S.; Wang, L.; et al. MPV17 Loss Causes Deoxynucleotide Insufficiency and Slow DNA Replication in Mitochondria. PLoS Genet. 2016, 12, e1005779. [Google Scholar] [CrossRef]
- Krauss, J.; Astrinidis, P.; Astrinides, P.; Frohnhöfer, H.G.; Walderich, B.; Nüsslein-Volhard, C. Transparent, a Gene Affecting Stripe Formation in Zebrafish, Encodes the Mitochondrial Protein Mpv17 That Is Required for Iridophore Survival. Biol. Open 2013, 2, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorano, L.; Peron, M.; Laquatra, C.; Lidron, E.; Facchinello, N.; Meneghetti, G.; Tiso, N.; Rasola, A.; Ghezzi, D.; Argenton, F. The Zebrafish Orthologue of the Human Hepatocerebral Disease Gene MPV17 Plays Pleiotropic Roles in Mitochondria. Dis. Model. Mech. 2019, 12, dmm037226. [Google Scholar] [CrossRef] [Green Version]
- Viscomi, C.; Zeviani, M. Strategies for Fighting Mitochondrial Diseases. J. Intern. Med. 2020, 287, 665–684. [Google Scholar] [CrossRef]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics 2020, 12, 1083. [Google Scholar] [CrossRef]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 Overexpression Ameliorates the Phenotype of Two Mitochondrial Disease Mouse Models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for Mitochondrial Diseases and Current Clinical Trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Rinaldi, T.; Dallabona, C.; Ferrero, I.; Frontali, L.; Bolotin-Fukuhara, M. Mitochondrial Diseases and the Role of the Yeast Models. FEMS Yeast Res. 2010, 10, 1006–1022. [Google Scholar] [CrossRef] [PubMed]
- Lasserre, J.-P.; Dautant, A.; Aiyar, R.S.; Kucharczyk, R.; Glatigny, A.; Tribouillard-Tanvier, D.; Rytka, J.; Blondel, M.; Skoczen, N.; Reynier, P.; et al. Yeast as a System for Modeling Mitochondrial Disease Mechanisms and Discovering Therapies. Dis. Model. Mech. 2015, 8, 509–526. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.V.R.; Bremner, L.; Barros, M.H. Human Mitochondrial Pathologies of the Respiratory Chain and ATP Synthase: Contributions from Studies of Saccharomyces cerevisiae. Life 2020, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Figuccia, S.; Degiorgi, A.; Ceccatelli Berti, C.; Baruffini, E.; Dallabona, C.; Goffrini, P. Mitochondrial Aminoacyl-TRNA Synthetase and Disease: The Yeast Contribution for Functional Analysis of Novel Variants. Int. J. Mol. Sci. 2021, 22, 4524. [Google Scholar] [CrossRef] [PubMed]
- Ceccatelli Berti, C.; di Punzio, G.; Dallabona, C.; Baruffini, E.; Goffrini, P.; Lodi, T.; Donnini, C. The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases. Genes 2021, 12, 300. [Google Scholar] [CrossRef] [PubMed]
- Couplan, E.; Aiyar, R.S.; Kucharczyk, R.; Kabala, A.; Ezkurdia, N.; Gagneur, J.; St Onge, R.P.; Salin, B.; Soubigou, F.; Le Cann, M.; et al. A Yeast-Based Assay Identifies Drugs Active against Human Mitochondrial Disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 11989–11994. [Google Scholar] [CrossRef] [Green Version]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol. Cell 2017, 67, 711–723.e7. [Google Scholar] [CrossRef]
- Aiyar, R.S.; Bohnert, M.; Duvezin-Caubet, S.; Voisset, C.; Gagneur, J.; Fritsch, E.S.; Couplan, E.; von der Malsburg, K.; Funaya, C.; Soubigou, F.; et al. Mitochondrial Protein Sorting as a Therapeutic Target for ATP Synthase Disorders. Nat. Commun. 2014, 5, 5585. [Google Scholar] [CrossRef] [Green Version]
- Pitayu, L.; Baruffini, E.; Rodier, C.; Rötig, A.; Lodi, T.; Delahodde, A. Combined Use of Saccharomyces cerevisiae, Caenorhabditis elegans and Patient Fibroblasts Leads to the Identification of Clofilium Tosylate as a Potential Therapeutic Chemical against POLG-Related Diseases. Hum. Mol. Genet. 2016, 25, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Panozzo, C.; Laleve, A.; Tribouillard-Tanvier, D.; Ostojić, J.; Sellem, C.H.; Friocourt, G.; Bourand-Plantefol, A.; Burg, A.; Delahodde, A.; Blondel, M.; et al. Chemicals or Mutations That Target Mitochondrial Translation Can Rescue the Respiratory Deficiency of Yeast Bcs1 Mutants. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2297–2307. [Google Scholar] [CrossRef]
- Delerue, T.; Tribouillard-Tanvier, D.; Daloyau, M.; Khosrobakhsh, F.; Emorine, L.J.; Friocourt, G.; Belenguer, P.; Blondel, M.; Arnauné-Pelloquin, L. A Yeast-Based Screening Assay Identifies Repurposed Drugs That Suppress Mitochondrial Fusion and MtDNA Maintenance Defects. Dis. Model. Mech. 2019, 12, dmm036558. [Google Scholar] [CrossRef] [Green Version]
- Di Punzio, G.; Di Noia, M.A.; Delahodde, A.; Sellem, C.; Donnini, C.; Palmieri, L.; Lodi, T.; Dallabona, C. A Yeast-Based Screening Unravels Potential Therapeutic Molecules for Mitochondrial Diseases Associated with Dominant ANT1 Mutations. Int. J. Mol. Sci. 2021, 22, 4461. [Google Scholar] [CrossRef]
- Aleo, S.J.; Del Dotto, V.; Fogazza, M.; Maresca, A.; Lodi, T.; Goffrini, P.; Ghelli, A.; Rugolo, M.; Carelli, V.; Baruffini, E.; et al. Drug Repositioning as a Therapeutic Strategy for Neurodegenerations Associated with OPA1 Mutations. Hum. Mol. Genet. 2021, 29, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Aubé, J. Drug Repurposing and the Medicinal Chemist. ACS Med. Chem. Lett. 2012, 3, 442–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilberti, M.; Baruffini, E.; Donnini, C.; Dallabona, C. Pathological Alleles of MPV17 Modeled in the Yeast Saccharomyces cerevisiae Orthologous Gene SYM1 Reveal Their Inability to Take Part in a High Molecular Weight Complex. PLoS ONE 2018, 13, e0205014. [Google Scholar] [CrossRef]
- Dujon, B. Mitochondrial genetics and functions. In The Molecular Biology of the Yeast Saccharomyces. Life Cycle and Inheritance; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1981; pp. 505–635. [Google Scholar]
- Pontarin, G.; Ferraro, P.; Bee, L.; Reichard, P.; Bianchi, V. Mammalian Ribonucleotide Reductase Subunit P53R2 Is Required for Mitochondrial DNA Replication and DNA Repair in Quiescent Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13302–13307. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, B.; Area, E.; Flanigan, K.M.; Ganesh, J.; Jayakar, P.; Swoboda, K.J.; Coku, J.; Naini, A.; Shanske, S.; Tanji, K.; et al. Mitochondrial DNA Depletion Syndrome Due to Mutations in the RRM2B Gene. Neuromuscul. Disord. NMD 2008, 18, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Copeland, W.C. POLG-Related Disorders and Their Neurological Manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef]
- Kasiviswanathan, R.; Longley, M.J.; Chan, S.S.L.; Copeland, W.C. Disease Mutations in the Human Mitochondrial DNA Polymerase Thumb Subdomain Impart Severe Defects in Mitochondrial DNA Replication. J. Biol. Chem. 2009, 284, 19501–19510. [Google Scholar] [CrossRef] [Green Version]
- Baruffini, E.; Lodi, T.; Dallabona, C.; Puglisi, A.; Zeviani, M.; Ferrero, I. Genetic and Chemical Rescue of the Saccharomyces cerevisiae Phenotype Induced by Mitochondrial DNA Polymerase Mutations Associated with Progressive External Ophthalmoplegia in Humans. Hum. Mol. Genet. 2006, 15, 2846–2855. [Google Scholar] [CrossRef] [Green Version]
- Mandel, H.; Szargel, R.; Labay, V.; Elpeleg, O.; Saada, A.; Shalata, A.; Anbinder, Y.; Berkowitz, D.; Hartman, C.; Barak, M.; et al. The Deoxyguanosine Kinase Gene Is Mutated in Individuals with Depleted Hepatocerebral Mitochondrial DNA. Nat. Genet. 2001, 29, 337–341. [Google Scholar] [CrossRef]
- Moss, C.F.; Dalla Rosa, I.; Hunt, L.E.; Yasukawa, T.; Young, R.; Jones, A.W.E.; Reddy, K.; Desai, R.; Virtue, S.; Elgar, G.; et al. Aberrant Ribonucleotide Incorporation and Multiple Deletions in Mitochondrial DNA of the Murine MPV17 Disease Model. Nucleic Acids Res. 2017, 45, 12808–12815. [Google Scholar] [CrossRef]
- Bulst, S.; Holinski-Feder, E.; Payne, B.; Abicht, A.; Krause, S.; Lochmüller, H.; Chinnery, P.F.; Walter, M.C.; Horvath, R. In Vitro Supplementation with Deoxynucleoside Monophosphates Rescues Mitochondrial DNA Depletion. Mol. Genet. Metab. 2012, 107, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Cámara, Y.; González-Vioque, E.; Scarpelli, M.; Torres-Torronteras, J.; Caballero, A.; Hirano, M.; Martí, R. Administration of Deoxyribonucleosides or Inhibition of Their Catabolism as a Pharmacological Approach for Mitochondrial DNA Depletion Syndrome. Hum. Mol. Genet. 2014, 23, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Blázquez-Bermejo, C.; Molina-Granada, D.; Vila-Julià, F.; Jiménez-Heis, D.; Zhou, X.; Torres-Torronteras, J.; Karlsson, A.; Martí, R.; Cámara, Y. Age-Related Metabolic Changes Limit Efficacy of Deoxynucleoside-Based Therapy in Thymidine Kinase 2-Deficient Mice. EBioMedicine 2019, 46, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Pursell, Z.F.; Copeland, W.C.; Longley, M.J.; Kunkel, T.A.; Mathews, C.K. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc. Natl. Acad. Sci. USA 2005, 102, 4990–4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazu, T.K.; Bricker, B.A.; Flores-Rozas, H.; Ablordeppey, S.Y. The Mechanistic Targets of Antifungal Agents: An Overview. Mini Rev. Med. Chem. 2016, 16, 555–578. [Google Scholar] [CrossRef]
- Zinser, E.; Sperka-Gottlieb, C.D.; Fasch, E.V.; Kohlwein, S.D.; Paltauf, F.; Daum, G. Phospholipid Synthesis and Lipid Composition of Subcellular Membranes in the Unicellular Eukaryote Saccharomyces cerevisiae. J. Bacteriol. 1991, 173, 2026–2034. [Google Scholar] [CrossRef] [Green Version]
- Souza, C.M.; Pichler, H. Lipid Requirements for Endocytosis in Yeast. Biochim. Biophys. Acta 2007, 1771, 442–454. [Google Scholar] [CrossRef]
- Gimpl, G.; Fahrenholz, F. Cholesterol as Stabilizer of the Oxytocin Receptor. Biochim. Biophys. Acta 2002, 1564, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Dufourc, E.J. Sterols and Membrane Dynamics. J. Chem. Biol. 2008, 1, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Dold, M.; Samara, M.T.; Li, C.; Tardy, M.; Leucht, S. Haloperidol versus First-Generation Antipsychotics for the Treatment of Schizophrenia and Other Psychotic Disorders. Cochrane Database Syst. Rev. 2015, 1, CD009831. [Google Scholar] [CrossRef]
- Moebius, F.F.; Bermoser, K.; Reiter, R.J.; Hanner, M.; Glossmann, H. Yeast Sterol C8-C7 Isomerase: Identification and Characterization of a High-Affinity Binding Site for Enzyme Inhibitors. Biochemistry 1996, 35, 16871–16878. [Google Scholar] [CrossRef]
- Lum, P.Y.; Armour, C.D.; Stepaniants, S.B.; Cavet, G.; Wolf, M.K.; Butler, J.S.; Hinshaw, J.C.; Garnier, P.; Prestwich, G.D.; Leonardson, A.; et al. Discovering Modes of Action for Therapeutic Compounds Using a Genome-Wide Screen of Yeast Heterozygotes. Cell 2004, 116, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.J.; Low, C.; Bottema, C.D.; Parks, L.W. Multiple Functions for Sterols in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1985, 837, 336–343. [Google Scholar] [CrossRef]
- Parks, L.W.; Casey, W.M. Physiological Implications of Sterol Biosynthesis in Yeast. Annu. Rev. Microbiol. 1995, 49, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Weete, J.D.; Abril, M.; Blackwell, M. Phylogenetic Distribution of Fungal Sterols. PLoS ONE 2010, 5, e10899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haslam, J.M.; Astin, A.M.; Nichols, W.W. The Effects of Altered Sterol Composition on the Mitochondrial Adenine Nucleotide Transporter of Saccharomyces cerevisiae. Biochem. J. 1977, 166, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, O.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Sensitivity of the 2-Oxoglutarate Carrier to Alcohol Intake Contributes to Mitochondrial Glutathione Depletion. Hepatol. Baltim. Md. 2003, 38, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Parlo, R.A.; Coleman, P.S. Enhanced Rate of Citrate Export from Cholesterol-Rich Hepatoma Mitochondria. The Truncated Krebs Cycle and Other Metabolic Ramifications of Mitochondrial Membrane Cholesterol. J. Biol. Chem. 1984, 259, 9997–10003. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Dinoi, P. Decreased Activity of the Phosphate Carrier and Modification of Lipids in Cardiac Mitochondria from Senescent Rats. Int. J. Biochem. 1992, 24, 783–787. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Gadaleta, M.N.; Ruggiero, F.M. The Effect of Aging and Acetyl-L-Carnitine on the Pyruvate Transport and Oxidation in Rat Heart Mitochondria. FEBS Lett. 1999, 454, 207–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Sivakumaran, S.; Yazlovitskaya, E.M.; Hiebert, S.W.; Guengerich, F.P.; Waterman, M.R.; Lepesheva, G.I. Human Sterol 14α-Demethylase as a Target for Anticancer Chemotherapy: Towards Structure-Aided Drug Design. J. Lipid Res. 2016, 57, 1552–1563. [Google Scholar] [CrossRef] [Green Version]
- Friggeri, L.; Hargrove, T.Y.; Wawrzak, Z.; Guengerich, F.P.; Lepesheva, G.I. Validation of Human Sterol 14α-Demethylase (CYP51) Druggability: Structure-Guided Design, Synthesis, and Evaluation of Stoichiometric, Functionally Irreversible Inhibitors. J. Med. Chem. 2019, 62, 10391–10401. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-Y.; Prudom, C.; Raines, S.M.; Charkhzarrin, S.; Melman, S.D.; De Haro, L.P.; Allen, C.; Lee, S.A.; Sklar, L.A.; Parra, K.J. Inhibitors of V-ATPase Proton Transport Reveal Uncoupling Functions of Tether Linking Cytosolic and Membrane Domains of V0 Subunit a (Vph1p). J. Biol. Chem. 2012, 287, 10236–10250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siles, S.A.; Srinivasan, A.; Pierce, C.G.; Lopez-Ribot, J.L.; Ramasubramanian, A.K. High-Throughput Screening of a Collection of Known Pharmacologically Active Small Compounds for Identification of Candida Albicans Biofilm Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 3681–3687. [Google Scholar] [CrossRef] [Green Version]
- Mamouei, Z.; Alqarihi, A.; Singh, S.; Xu, S.; Mansour, M.K.; Ibrahim, A.S.; Uppuluri, P. Alexidine Dihydrochloride Has Broad-Spectrum Activities against Diverse Fungal Pathogens. mSphere 2018, 3, e00539-18. [Google Scholar] [CrossRef] [Green Version]
- Serrano, R.; Bernal, D.; Simón, E.; Ariño, J. Copper and Iron Are the Limiting Factors for Growth of the Yeast Saccharomyces cerevisiae in an Alkaline Environment. J. Biol. Chem. 2004, 279, 19698–19704. [Google Scholar] [CrossRef] [Green Version]
- Schreier, S.; Malheiros, S.V.; de Paula, E. Surface Active Drugs: Self-Association and Interaction with Membranes and Surfactants. Physicochemical and Biological Aspects. Biochim. Biophys. Acta 2000, 1508, 210–234. [Google Scholar] [CrossRef] [Green Version]
- Uesono, Y.; Araki, T.; Toh-E, A. Local Anesthetics, Antipsychotic Phenothiazines, and Cationic Surfactants Shut down Intracellular Reactions through Membrane Perturbation in Yeast. Biosci. Biotechnol. Biochem. 2008, 72, 2884–2894. [Google Scholar] [CrossRef]
- Halliwell, W.H. Cationic Amphiphilic Drug-Induced Phospholipidosis. Toxicol. Pathol. 1997, 25, 53–60. [Google Scholar] [CrossRef]
- Thomas, B.J.; Rothstein, R. Elevated Recombination Rates in Transcriptionally Active DNA. Cell 1989, 56, 619–630. [Google Scholar] [CrossRef]
- Bonneaud, N.; Ozier-Kalogeropoulos, O.; Li, G.Y.; Labouesse, M.; Minvielle-Sebastia, L.; Lacroute, F. A Family of Low and High Copy Replicative, Integrative and Single-Stranded, S. cerevisiae/E. coli Shuttle Vectors. Yeast 1991, 7, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. High-Efficiency Yeast Transformation Using the LiAc/SS Carrier DNA/PEG Method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, R.J. One-Step Gene Disruption in Yeast. Methods Enzymol. 1983, 101, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Wach, A.; Brachat, A.; Pöhlmann, R.; Philippsen, P. New Heterologous Modules for Classical or PCR-Based Gene Disruptions in Saccharomyces cerevisiae. Yeast 1994, 10, 1793–1808. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-Directed Mutagenesis by Overlap Extension Using the Polymerase Chain Reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Baruffini, E.; Ferrero, I.; Foury, F. In Vivo Analysis of MtDNA Replication Defects in Yeast. Methods 2010, 51, 426–436. [Google Scholar] [CrossRef]
- Baruffini, E.; Ruotolo, R.; Bisceglie, F.; Montalbano, S.; Ottonello, S.; Pelosi, G.; Buschini, A.; Lodi, T. Mechanistic Insights on the Mode of Action of an Antiproliferative Thiosemicarbazone-Nickel Complex Revealed by an Integrated Chemogenomic Profiling Study. Sci. Rep. 2020, 10, 10524. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.K.; Wheeler, L.J. Measuring DNA Precursor Pools in Mitochondria. Methods Mol. Biol. 2009, 554, 371–381. [Google Scholar] [CrossRef]
- Pontarin, G.; Gallinaro, L.; Ferraro, P.; Reichard, P.; Bianchi, V. Origins of Mitochondrial Thymidine Triphosphate: Dynamic Relations to Cytosolic Pools. Proc. Natl. Acad. Sci. USA 2003, 100, 12159–12164. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Roy, B.; Beuneu, C.; Roux, P.; Buc, H.; Lemaire, G.; Lepoivre, M. Simultaneous Determination of Pyrimidine or Purine Deoxyribonucleoside Triphosphates Using a Polymerase Assay. Anal. Biochem. 1999, 269, 403–409. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target in Yeast | Target in Mammals |
|---|---|---|
| POS | Lanosterol 14-α-demethylase (ergosterol pathway) | Lanosterol 14-α-demethylase (cholesterol pathway) |
| FEN | Lanosterol 14-α-demethylase (ergosterol pathway) | Lanosterol 14-α-demethylase (cholesterol pathway) |
| IMA | Lanosterol 14-α-demethylase (ergosterol pathway) | Lanosterol 14-α-demethylase (cholesterol pathway) |
| ITR | Lanosterol 14-α-demethylase (ergosterol pathway) | Lanosterol 14-α-demethylase (cholesterol pathway) |
| SEC | Lanosterol 14-α-demethylase (ergosterol pathway) | Lanosterol 14-α-demethylase (cholesterol pathway) |
| HAL | Sterol C8-C7 isomerase (ergosterol pathway) | Dopamine receptors; sigma-1 receptor; 3β-hydroxysterol-∆8, ∆7 isomerase (cholesterol pathway) |
| THO | Vacuolar ATPase proton transporter | Vacuolar ATPase proton transporter |
| ALE | Vacuolar ATPase proton transporter | Vacuolar ATPase proton transporter |
| BEN | unknown | hERG K-channel |
| DOM | unknown | hERG K-channel |
| OTI | unknown | Muscarinic receptor, VD Ca-channel, NK receptor |
| SER | Phospholipid membranes | Serotonin 5-HT transporter |
| Rescue on mtDNA Instability | |||
|---|---|---|---|
| Drug | sym1R51W | rnr2L362V | mip1G651S |
| POS | +++ | ++ | + |
| FEN | ++ | + | ++ |
| IMA | +++ | ++ | +++ |
| HAL | ++ | +++ | +++ |
| THO | + | ++ | n.e. |
| ALE | + | + | n.e |
| BEN | +++ | +++ | ++ |
| DOM | ++ | + | n.e. |
| OTI | ++ | ++ | n.e |
| SER | ++ | n.e | ++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
di Punzio, G.; Gilberti, M.; Baruffini, E.; Lodi, T.; Donnini, C.; Dallabona, C. A Yeast-Based Repurposing Approach for the Treatment of Mitochondrial DNA Depletion Syndromes Led to the Identification of Molecules Able to Modulate the dNTP Pool. Int. J. Mol. Sci. 2021, 22, 12223. https://doi.org/10.3390/ijms222212223
di Punzio G, Gilberti M, Baruffini E, Lodi T, Donnini C, Dallabona C. A Yeast-Based Repurposing Approach for the Treatment of Mitochondrial DNA Depletion Syndromes Led to the Identification of Molecules Able to Modulate the dNTP Pool. International Journal of Molecular Sciences. 2021; 22(22):12223. https://doi.org/10.3390/ijms222212223
Chicago/Turabian Styledi Punzio, Giulia, Micol Gilberti, Enrico Baruffini, Tiziana Lodi, Claudia Donnini, and Cristina Dallabona. 2021. "A Yeast-Based Repurposing Approach for the Treatment of Mitochondrial DNA Depletion Syndromes Led to the Identification of Molecules Able to Modulate the dNTP Pool" International Journal of Molecular Sciences 22, no. 22: 12223. https://doi.org/10.3390/ijms222212223
APA Styledi Punzio, G., Gilberti, M., Baruffini, E., Lodi, T., Donnini, C., & Dallabona, C. (2021). A Yeast-Based Repurposing Approach for the Treatment of Mitochondrial DNA Depletion Syndromes Led to the Identification of Molecules Able to Modulate the dNTP Pool. International Journal of Molecular Sciences, 22(22), 12223. https://doi.org/10.3390/ijms222212223