
Therapeutic Prospects of Exon Skipping for Epidermolysis Bullosa


Abstract
1. Epidermolysis Bullosa
2. Exon Skipping
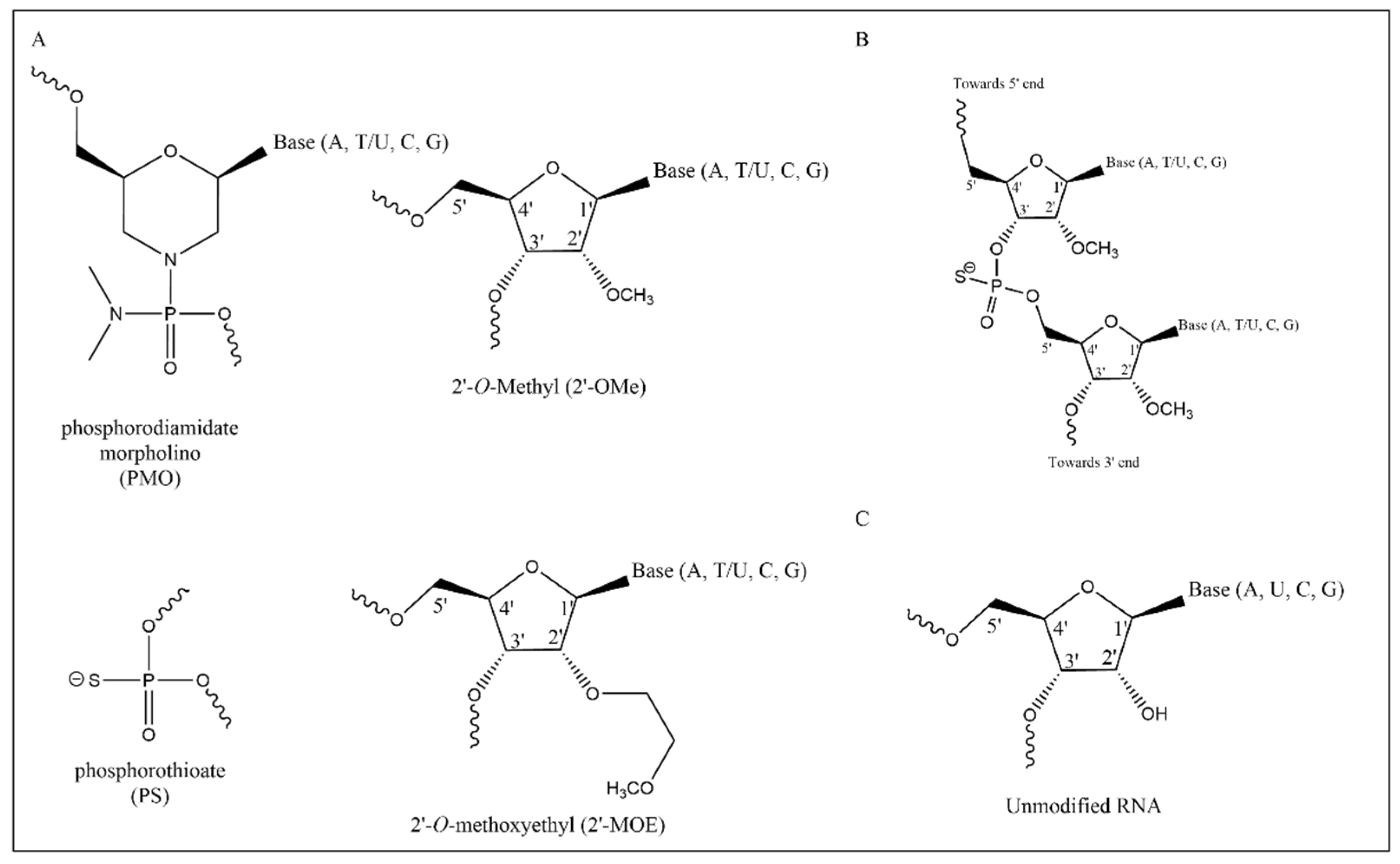
3. Antisense Oligonucleotides
4. ASOs Targeting Mutated Exons in Epidermolysis Bullosa
5. Lessons from Natural Exon Skipping in Epidermolysis Bullosa
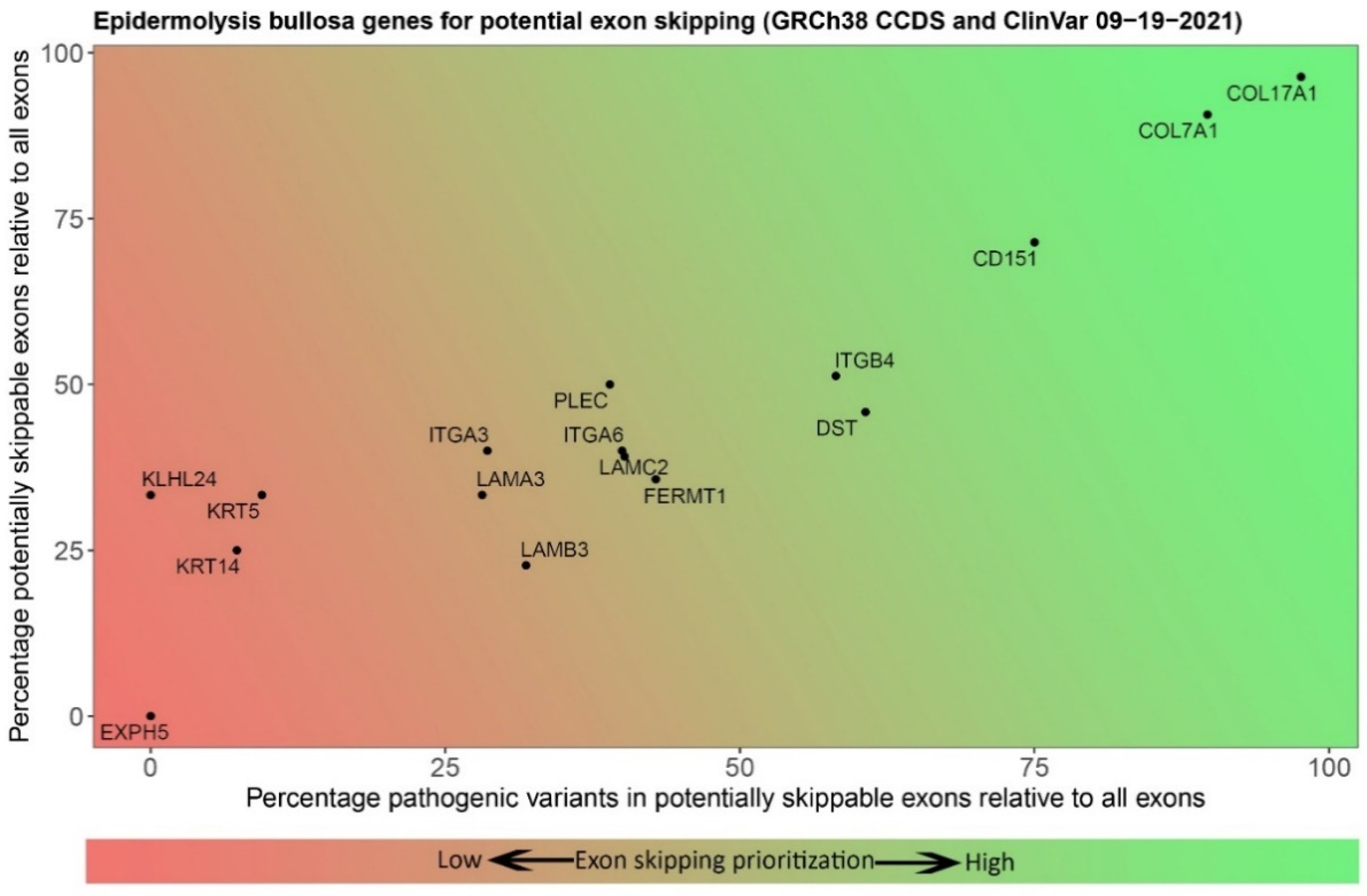
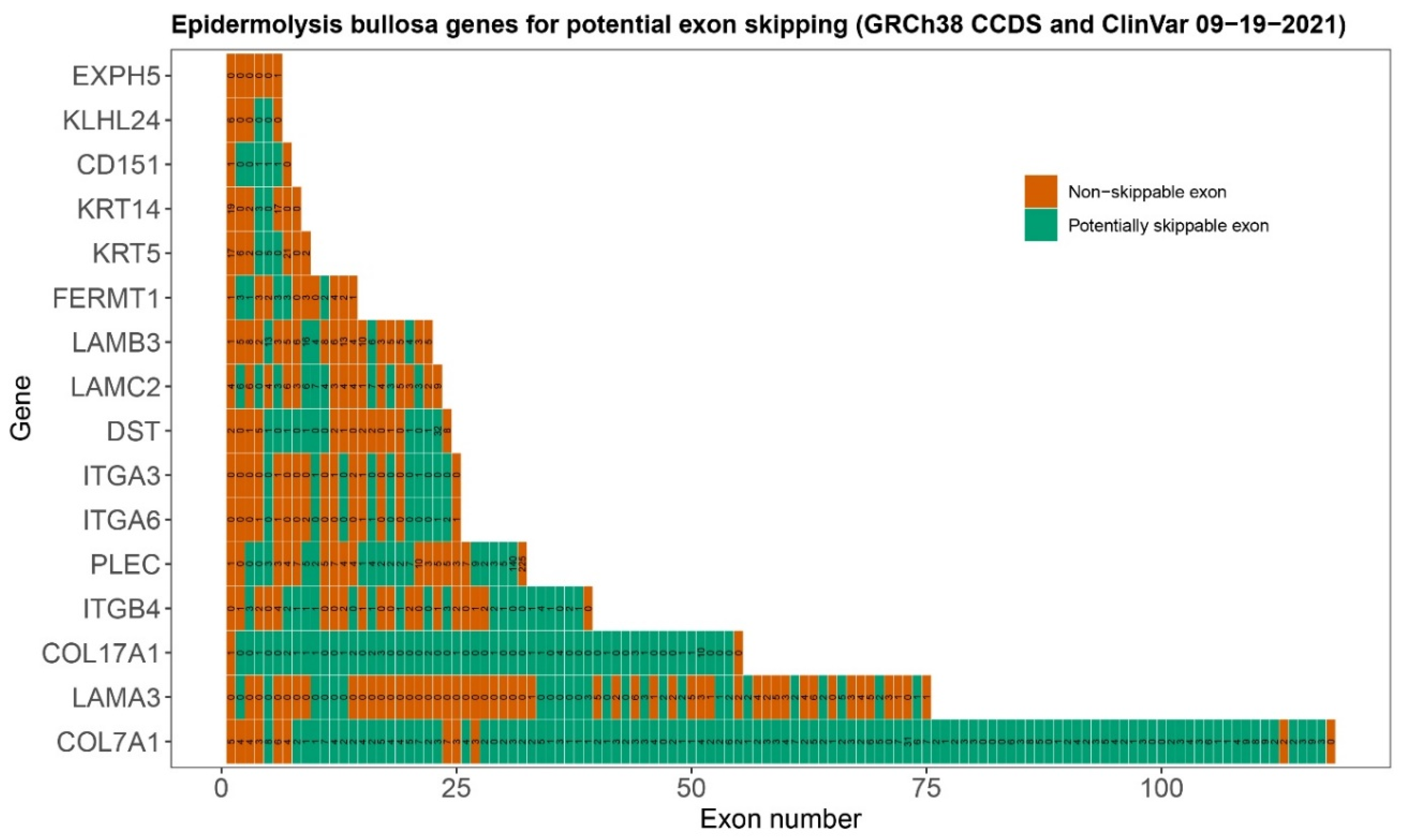
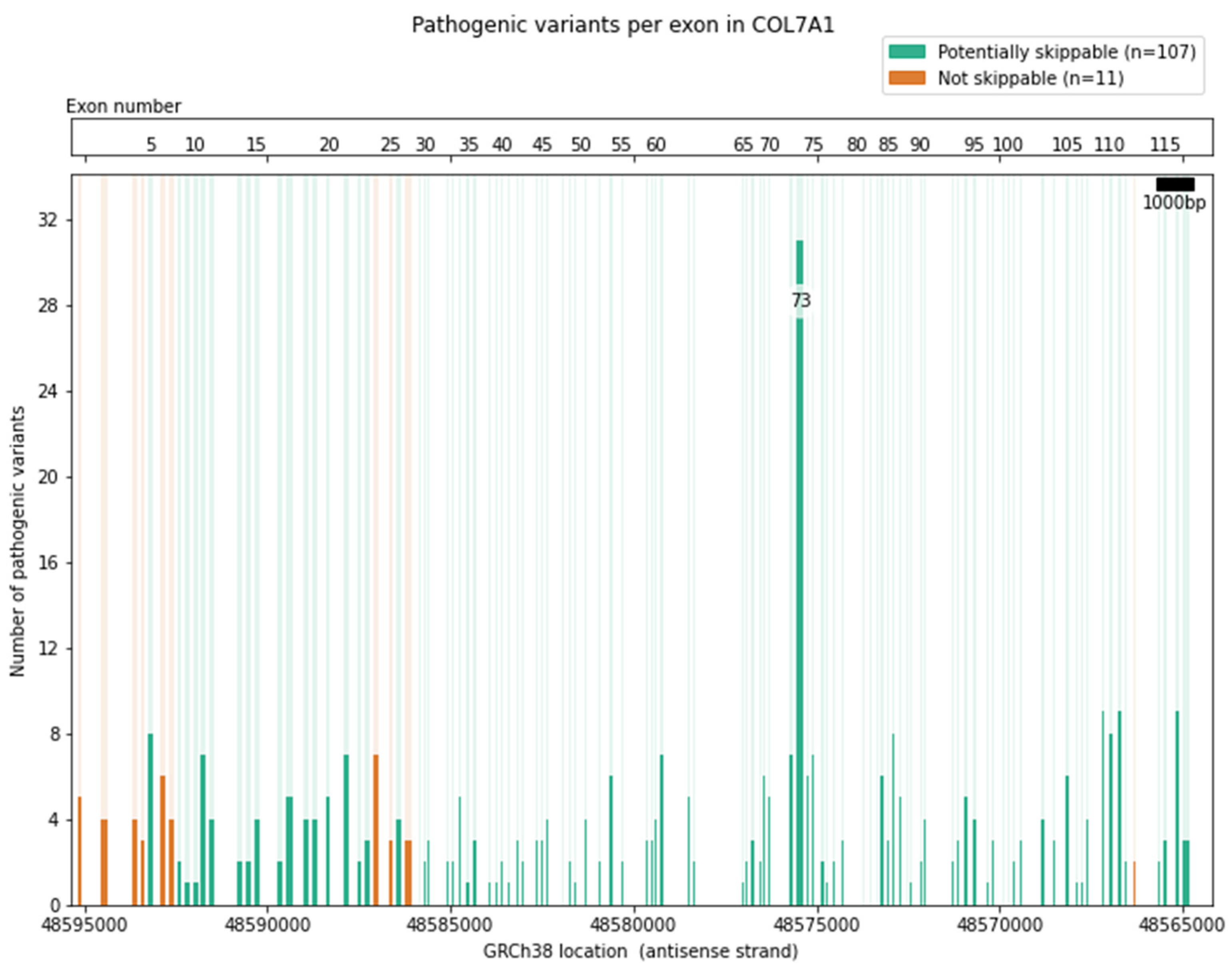
6. Selection of Epidermolysis Bullosa Genes and Exons for Targeting with Exon Skipping
7. Development of ASOs for Targeting Exon Skipping in Epidermolysis Bullosa
8. Delivery of ASO Therapies
9. Concluding Remarks and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.J.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Li, K.P.; Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef]
- Almaani, N.; Liu, L.; Dopping-Hepenstal, P.J.; Lai-Cheong, J.E.; Wong, A.; Nanda, A.; Moss, C.; Martinez, A.E.; Mellerio, J.E.; McGrath, J.A. Identical glycine substitution mutations in type VII collagen may underlie both dominant and recessive forms of dystrophic epidermolysis bullosa. Acta Derm. Venereol. 2011, 91, 262–266. [Google Scholar] [CrossRef]
- Herz, C.; Aumailley, M.; Schulte, C.; Schlötzer-Schrehardt, U.; Bruckner-Tuderman, L.; Has, C. Kindlin-1 Is a Phosphoprotein Involved in Regulation of Polarity, Proliferation, and Motility of Epidermal Keratinocytes. J. Biol. Chem. 2006, 281, 36082–36090. [Google Scholar] [CrossRef]
- Marinkovich, M.P.; Tang, J.Y. Gene Therapy for Epidermolysis Bullosa. J. Investig. Dermatol. 2019, 139, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Jacków, J.; Guo, Z.; Hansen, C.; Abaci, H.E.; Doucet, Y.S.; Shin, J.U.; Hayashi, R.; Delorenzo, D.; Kabata, Y.; Shinkuma, S.; et al. CRISPR/Cas9-based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using iPS cells. Proc. Natl. Acad. Sci. USA 2019, 116, 26846–26852. [Google Scholar] [CrossRef]
- Kocher, T.; March, O.P.; Bischof, J.; Liemberger, B.; Hainzl, S.; Klausegger, A.; Hoog, A.; Strunk, D.; Bauer, J.W.; Koller, U. Predictable CRISPR/Cas9-Mediated COL7A1 Reframing for Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2020, 140, 1985–1993.e5. [Google Scholar] [CrossRef]
- Bonafont, J.; Mencía, A.; Chacón-Solano, E.; Srifa, W.; Vaidyanathan, S.; Romano, R.; Garcia, M.; Hervás-Salcedo, R.; Ugalde, L.; Duarte, B.; et al. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol. Ther. 2021, 29, 2008–2018. [Google Scholar] [CrossRef]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef]
- Koller, U.; Bauer, J.W. Gene Replacement Therapies for Genodermatoses: A Status Quo. Front. Genet. 2021, 12, 658295. [Google Scholar] [CrossRef] [PubMed]
- Schwieger-Briel, A.; Weibel, L.; Chmel, N.; Leppert, J.; Kernland-Lang, K.; Grüninger, G.; Has, C. A COL7A1 variant leading to in-frame skipping of exon 15 attenuates disease severity in recessive dystrophic epidermolysis bullosa. Br. J. Dermatol. 2015, 173, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; Bornert, O.; Nyström, A.; Gostynski, A.; Jonkman, M.; Aartsma-Rus, A.; van den Akker, P.; Pasmooij, A. Antisense Oligonucleotide-mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef]
- Bornert, O.; Hogervorst, M.; Nauroy, P.; Bischof, J.; Swildens, J.; Athanasiou, I.; Tufa, S.; Keene, D.; Kiritsi, D.; Hainzl, S.; et al. QR-313, an Antisense Oligonucleotide, Shows Therapeutic Efficacy for Treatment of Dominant and Recessive Dystrophic Epidermolysis Bullosa: A Preclinical Study. J. Investig. Dermatol. 2021, 141, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Décha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef]
- Ablinger, M.; Lettner, T.; Friedl, N.; Potocki, H.; Palmetzhofer, T.; Koller, U.; Illmer, J.; Liemberger, B.; Hainzl, S.; Klausegger, A.; et al. Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa. Int. J. Mol. Sci. 2021, 22, 3326. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.W.M.P.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef]
- Ham, K.A.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D. Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1. Int. J. Mol. Sci. 2020, 21, 7705. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.A. A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys. Acta 1999, 1489, 69–84. [Google Scholar] [CrossRef]
- Wu, B.; Lu, P.; Benrashid, E.; Malik, S.; Ashar, J.; Doran, T.J.; Lu, Q.L. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010, 17, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Heald, A.E.; Iversen, P.L.; Saoud, J.B.; Sazani, P.; Charleston, J.S.; Axtelle, T.; Wong, M.; Smith, W.B.; Vutikullird, A.; Kaye, E. Safety and Pharmacokinetic Profiles of Phosphorodiamidate Morpholino Oligomers with Activity against Ebola Virus and Marburg Virus: Results of Two Single-Ascending-Dose Studies. Antimicrob. Agents Chemother. 2014, 58, 6639–6647. [Google Scholar] [CrossRef]
- Goto, M.; Sawamura, D.; Nishie, W.; Sakai, K.; McMillan, J.R.; Akiyama, M.; Shimizu, H. Targeted skipping of a single exon harboring a premature termination codon mutation: Implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J. Investig. Dermatol. 2006, 126, 2614–2620. [Google Scholar] [CrossRef]
- Varki, R.; Sadowski, S.; Uitto, J.; Pfendner, E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J. Med. Genet. 2006, 44, 181–192. [Google Scholar] [CrossRef]
- Bornert, O.; Kuhl, T.; Bremer, J.; van den Akker, P.C.; Pasmooij, A.M.; Nystrom, A. Analysis of the functional consequences of targeted exon deletion in COL7A1 reveals prospects for dystrophic epidermolysis bullosa therapy. Mol. Ther. 2016, 24, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Bornert, O.; Kocher, T.; Gretzmeier, C.; Liemberger, B.; Hainzl, S.; Koller, U.; Nystrom, A. Generation of rabbit polyclonal human and murine collagen VII monospecific antibodies: A useful tool for dystrophic epidermolysis bullosa therapy studies. Matrix Biol. Plus 2019, 4, 100017. [Google Scholar] [CrossRef]
- Verheul, R.C.; Van Deutekom, J.C.T.; Datson, N.A. Digital Droplet PCR for the Absolute Quantification of Exon Skipping Induced by Antisense Oligonucleotides in (Pre-)Clinical Development for Duchenne Muscular Dystrophy. PLoS ONE 2016, 11, e0162467. [Google Scholar] [CrossRef]
- Hiller, M.; Spitali, P.; Datson, N.; Aartsma-Rus, A. Exon 51 Skipping Quantification by Digital Droplet PCR in del52hDMD/mdx Mice. Methods Mol. Biol. 2018, 1828, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; van der Heijden, E.; Eichhorn, D.; Meijer, R.; Lemmink, H.; Scheffer, H.; Sinke, R.; Jonkman, M.; Pasmooij, A.; Van den Akker, P. Natural Exon Skipping Sets the Stage for Exon Skipping as Therapy for Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2019, 18, 465–475. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A.; Ashton, G.H.; Mellerio, J.E.; Salas-Alanis, J.C.; Swensson, O.; McMillan, J.R.; Eady, R.A. Moderation of phenotypic severity in dystrophic and junctional forms of epidermolysis bullosa through in-frame skipping of exons containing non-sense or frameshift mutations. J. Investig. Dermatol. 1999, 113, 314–321. [Google Scholar] [CrossRef]
- Kowalewski, C.; Bremer, J.; Gostynski, A.; Wertheim-Tysarowska, K.; Wozniak, K.; Bal, J.; Jonkman, M.; Pasmooij, A. Amelioration of junctional epidermolysis bullosa due to exon skipping. Br. J. Dermatol. 2016, 174, 1375–1379. [Google Scholar] [CrossRef]
- Sawamura, D.; Goto, M.; Sakai, K.; Nakamura, H.; McMillan, J.R.; Akiyama, M.; Shirado, O.; Oyama, N.; Satoh, M.; Kaneko, F.; et al. Possible involvement of exon 31 alternative splicing in phenotype and severity of epidermolysis bullosa caused by mutations in PLEC1. J. Investig. Dermatol. 2007, 127, 1537–1540. [Google Scholar] [CrossRef]
- Ketema, M.; Secades, P.; Kreft, M.; Nahidiazar, L.; Janssen, H.; Jalink, K.; Pereda, J.M.D.; Sonnenberg, A. The rod domain is not essential for the function of plectin in maintaining tissue integrity. Mol. Biol. Cell 2015, 26, 2402–2417. [Google Scholar] [CrossRef]
- Gostyńska, K.; Bremer, J.; van Dijk-Bos, K.K.; Sinke, R.; Pasmooij, A.M.G.; Jonkman, M.F. In-frame Exon Skipping in KRT5 due to Novel Intronic Deletion Causes Epidermolysis Bullosa Simplex, Generalized Severe. Acta Derm. Venereol. 2017, 97, 105–107. [Google Scholar] [CrossRef][Green Version]
- Ho, S. Potent antisense oligonucleotides to the human multidrug resistance-1 mRNA are rationally selected by mapping RNA-accessible sites with oligonucleotide libraries. Nucleic Acids Res. 1996, 24, 1901–1907. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; van Vliet, L.; Hirschi, M.; Janson, A.A.; Heemskerk, H.; de Winter, C.L.; de Kimpe, S.; van Deutekom, J.C.; t Hoen, P.A.; van Ommen, G.J. Guidelines for antisense oligonucleotide design and insight into splice-modulating mechanisms. Mol. Ther. 2009, 17, 548–553. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. Overview on AON design. Methods Mol. Biol. 2012, 867, 117–129. [Google Scholar] [CrossRef]
- Chiba, S.; Lim, K.R.; Sheri, N.; Anwar, S.; Erkut, E.; Shah, M.N.; Aslesh, T.; Woo, S.; Sheikh, O.; Maruyama, R.; et al. eSkip-Finder: A machine learning-based web application and database to identify the optimal sequences of antisense oligonucleotides for exon skipping. Nucleic Acids Res. 2021, 49, W193–W198. [Google Scholar] [CrossRef]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Liebow, A.; Yasuda, M.; Gan, L.; Racie, T.; Maier, M.; Kuchimanchi, S.; Foster, D.; Milstein, S.; Charisse, K.; et al. Preclinical Development of a Subcutaneous ALAS1 RNAi Therapeutic for Treatment of Hepatic Porphyrias Using Circulating RNA Quantification. Mol. Ther. Nucleic Acids 2015, 4, e263. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Van Roon-Mom, W.M.C.; Marckmann, G.; Van Duyvenvoorde, H.A.; Graessner, H.; Schüle, R.; Aartsma-Rus, A. Preparing n-of-1 Antisense Oligonucleotide Treatments for Rare Neurological Diseases in Europe: Genetic, Regulatory, and Ethical Perspectives. Nucleic Acid Ther. 2021. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level of Blistering | Gene | Protein | No. of Exons | In-Frame Exons | Percentage Skippable 1 | No. of ClinVar Variants 2 in Skippable Exons | Percentage Variants in Skippable Exons |
|---|---|---|---|---|---|---|---|
| Basal epidermal | KRT14 | Keratin 14 | 8 | 3 | 37.5 | 20 (39) | 51.3 |
| KRT5 | Keratin 5 | 9 | 4 | 44.4 | 22 (24) | 45.8 | |
| PLEC | Plectin | 39 | 17 | 43.6 | 43 (49) | 87.8 | |
| KLHL24 | Kelch-like family member 24 | 6 | 2 | 33.3 | 0 (6) | 0.0 | |
| EXPH5 | Exophilin 5 | 6 | 0 | 0.0 | 0 (1) | 0.0 | |
| CD151 | Tetraspanin-24 | 7 | 5 | 71.4 | 2 (2) | 100.0 | |
| DST | Dystonin | 24 | 11 | 45.8 | 22 (26) | 84.6 | |
| Intralamina Lucida | COL17A1 | Type XVII Collagen | 55 | 53 | 96.4 | 37 (38) | 97.4 |
| ITGB4 | Integrin β4 | 39 | 20 | 51.3 | 15 (28) | 53.6 | |
| LAMA3 | Laminin α3 chain | 75 | 25 | 33.3 | 24 (72) | 33.3 | |
| LAMB3 | Laminin β3 chain | 22 | 5 | 22.7 | 36 (108) | 33.3 | |
| LAMC2 | Laminin γ2 chain | 23 | 9 | 39.1 | 29 (73) | 39.7 | |
| ITGA6 | Integrin α6 | 25 | 10 | 40.0 | 2 (4) | 50.0 | |
| ITGA3 | Integrin α3 | 25 | 10 | 40.0 | 2 (7) | 28.6 | |
| Sublamina Densa | COL7A1 | Type VII Collagen | 118 | 107 | 90.7 | 270 (297) | 90.9 |
| Mixed levels | FERMT1 | Kindlin-1 | 14 | 9 | 64.3 | 12 (24) | 50.0 |
| Gene | Exon | ASO Sequence | GC% of ASO | ASO Length | Start Position 1 | Predicted Efficiency % 2 | ASO Chemistry 3 | ASO Name | Reference |
|---|---|---|---|---|---|---|---|---|---|
| COL7A1 | 10 | CGGGCCUCAGGCACCAAGUUC | 66 | 21 | 64 | 26.0 (2′OMe) | 2′-OMe-PS | H10A(+65+85) | [22] |
| COL7A1 | 10 | CGGGCCUCAGGCACCAAGUUC | 66 | 21 | 64 | 7.7 (PMO) | PMO | H10A(+65+85) | [22] |
| COL7A1 | 10 | CUUCCCCCGCACUGACCAGUCUC | 65 | 23 | N.A. 4 | 2′-OMe-PS | H10D(+07-16) | [22] | |
| COL7A1 | 10 | CUUCCCCCGCACUGACCAGUCUC | 65 | 23 | N.A. 4 | PMO | H10D(+07-16) | [22] | |
| COL7A1 | 70 | CGCACACUUCCAGGC | 66 | 15 | 32 | 8.3 (2′OMe) | 2′-OMe-PS | [27] | |
| COL7A1 | 73 | CGUUCUCCAGGAAAGCCGAUG | 57 | 21 | 5 | 8.1 (2′OMe) 5 | 2′-MOE-PS | QR-313 | [17] |
| COL7A1 | 73 | TCTTGCGCCCGACTTCCCGCTGGCACCTCT | 67 | 30 | 20 | 26.6 (2′OMe) | 2′OMe | [18] | |
| COL7A1 | 73 | UUCAGCCCGCGUUCUCCAGG | 65 | 20 | 15 | 11.6 (2′OMe) | 2′-OMe-PS | H73A(+16+35) | [22] |
| COL7A1 | 73 | UUCAGCCCGCGUUCUCCAGG | 65 | 20 | 15 | 22.6 (PMO) | PMO | H73A(+16+35) | [22] |
| COL7A1 | 73 | CGCCCUUCAGCCCGCGUUCU | 70 | 20 | 20 | 21.3 (2′OMe) | 2′-OMe-PS | H73A(+21+40) | [22] |
| COL7A1 | 73 | CGCCCUUCAGCCCGCGUUCU | 70 | 20 | 20 | 47.9 (PMO) | PMO | H73A(+21+40) | [22] |
| COL7A1 | 73 | CGCCCUUCAGCCCGCGUUCUCCAGG | 72 | 25 | 15 | 49.9 (2′OMe) | 2′-OMe-PS | H73A(+16+40) | [22] |
| COL7A1 | 73 | CGCCCUUCAGCCCGCGUUCUCCAGG | 72 | 25 | 15 | 60.0 (PMO) | PMO | H73A(+16+40) | [22] |
| COL7A1 | 80 | TCCCAGACGTCCCAGGTTCTCCGG | 67 | 24 | N.A. 4 | 2′OMe | [18] | ||
| COL7A1 | 105 | GAUACCAGGCACUCCAUCCU | 55 | 20 | 13 | 15.2 (2′OMe) | 2′OMe | AON1 | [16] |
| COL7A1 | 105 | CAUGAAGCCAACAUCUCCUU | 45 | 20 | 43 | 11.3 (2′OMe) | 2′OMe | AON2 | [16] |
| COL17A1 | 7 | TTTGACTCCGTCCTCTGGTT | 50 | 20 | 10 | 3.8 (2′OMe) | 2′-OMe-PS | AON1 | [19] |
| COL17A1 | 7 | TCGTGTTTGACTCCGTCCTC | 55 | 20 | 15 | 11.7 (2′OMe) | 2′-OMe-PS | AON2 | [19] |
| COL17A1 | 7 | CTCCGTCCTCTGGTTGAAGA | 55 | 20 | 5 | 42.4 (2′OMe) | 2′-OMe-PS | AON3 | [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vermeer, F.C.; Bremer, J.; Sietsma, R.J.; Sandilands, A.; Hickerson, R.P.; Bolling, M.C.; Pasmooij, A.M.G.; Lemmink, H.H.; Swertz, M.A.; Knoers, N.V.A.M.; et al. Therapeutic Prospects of Exon Skipping for Epidermolysis Bullosa. Int. J. Mol. Sci. 2021, 22, 12222. https://doi.org/10.3390/ijms222212222
Vermeer FC, Bremer J, Sietsma RJ, Sandilands A, Hickerson RP, Bolling MC, Pasmooij AMG, Lemmink HH, Swertz MA, Knoers NVAM, et al. Therapeutic Prospects of Exon Skipping for Epidermolysis Bullosa. International Journal of Molecular Sciences. 2021; 22(22):12222. https://doi.org/10.3390/ijms222212222
Chicago/Turabian StyleVermeer, Franciscus C., Jeroen Bremer, Robert J. Sietsma, Aileen Sandilands, Robyn P. Hickerson, Marieke C. Bolling, Anna M.G. Pasmooij, Henny H. Lemmink, Morris A. Swertz, Nine V.A.M. Knoers, and et al. 2021. "Therapeutic Prospects of Exon Skipping for Epidermolysis Bullosa" International Journal of Molecular Sciences 22, no. 22: 12222. https://doi.org/10.3390/ijms222212222
APA StyleVermeer, F. C., Bremer, J., Sietsma, R. J., Sandilands, A., Hickerson, R. P., Bolling, M. C., Pasmooij, A. M. G., Lemmink, H. H., Swertz, M. A., Knoers, N. V. A. M., van der Velde, K. J., & van den Akker, P. C. (2021). Therapeutic Prospects of Exon Skipping for Epidermolysis Bullosa. International Journal of Molecular Sciences, 22(22), 12222. https://doi.org/10.3390/ijms222212222