
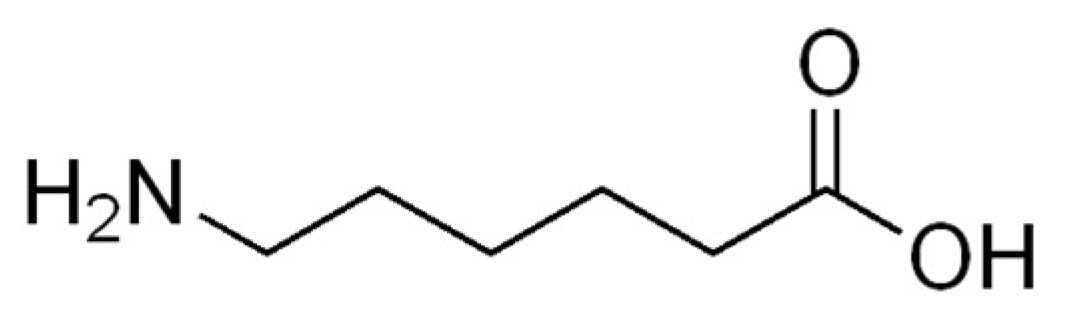
The Importance of 6-Aminohexanoic Acid as a Hydrophobic, Flexible Structural Element


Abstract
:1. Introduction
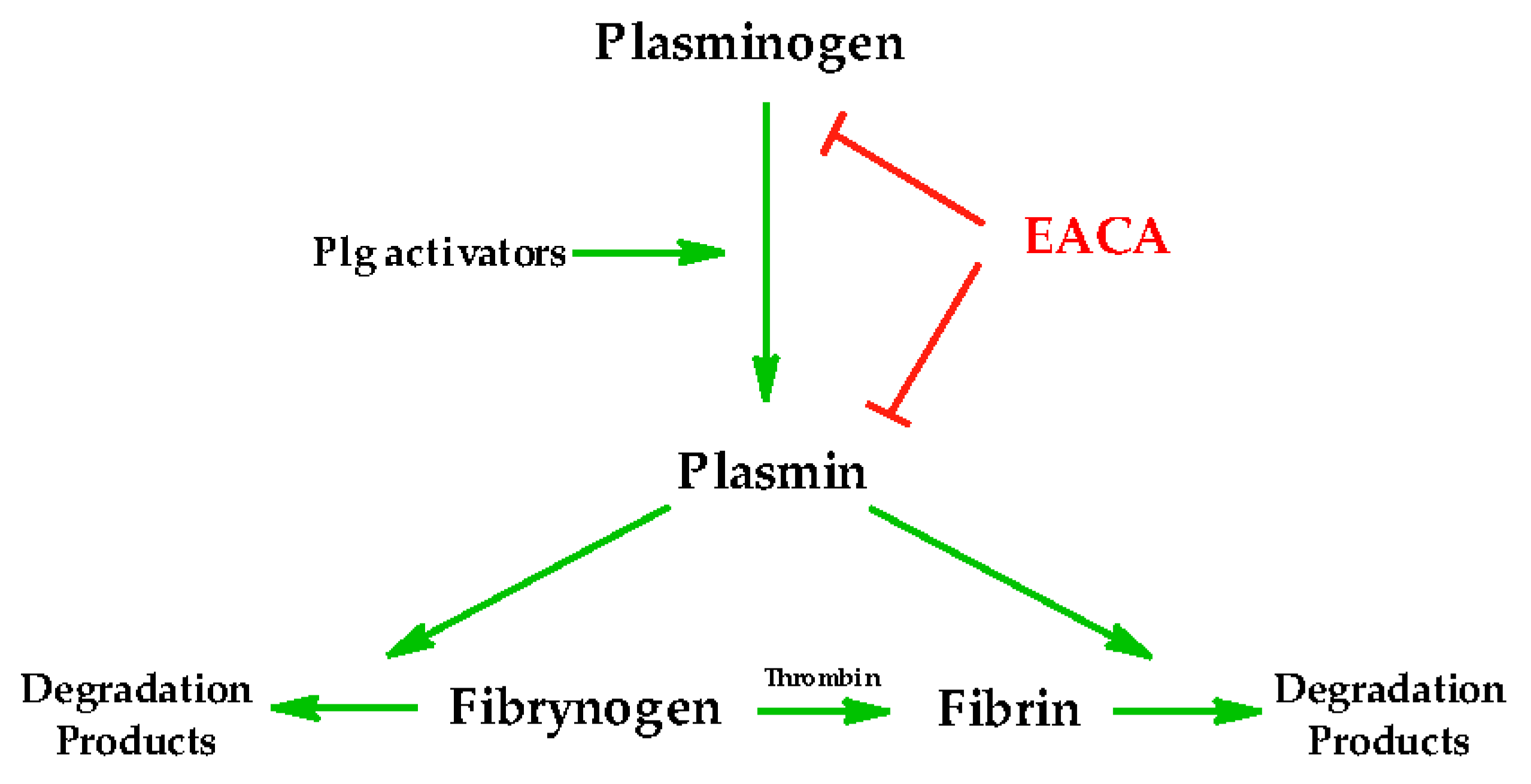
2. Ahx as an Antifibrinolytic Drug
3. Amino Acid and Short Peptide Derivatives of Ahx as Antifibrynolytics
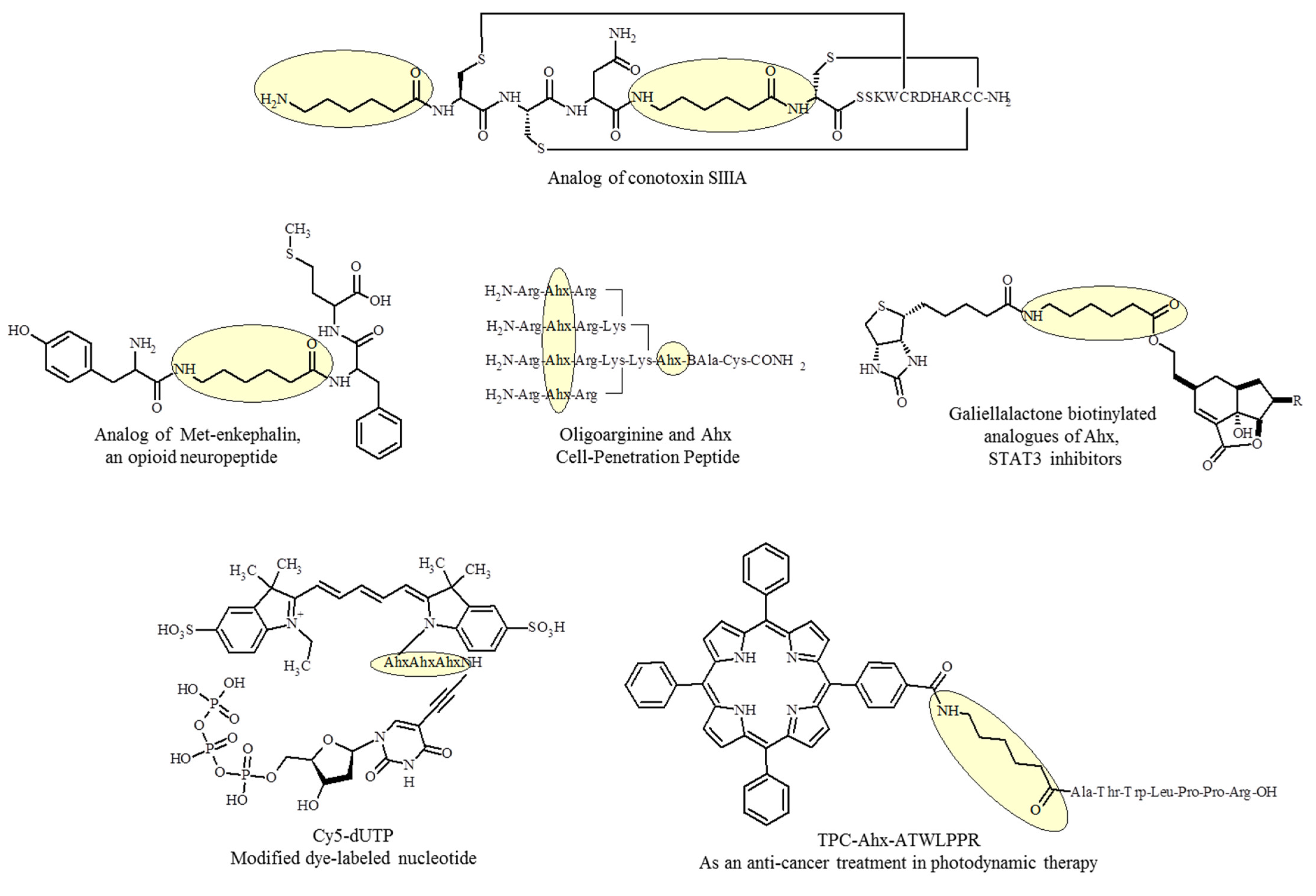
4. Introduction of Ahx into the Structure of Peptides with Biological Activity
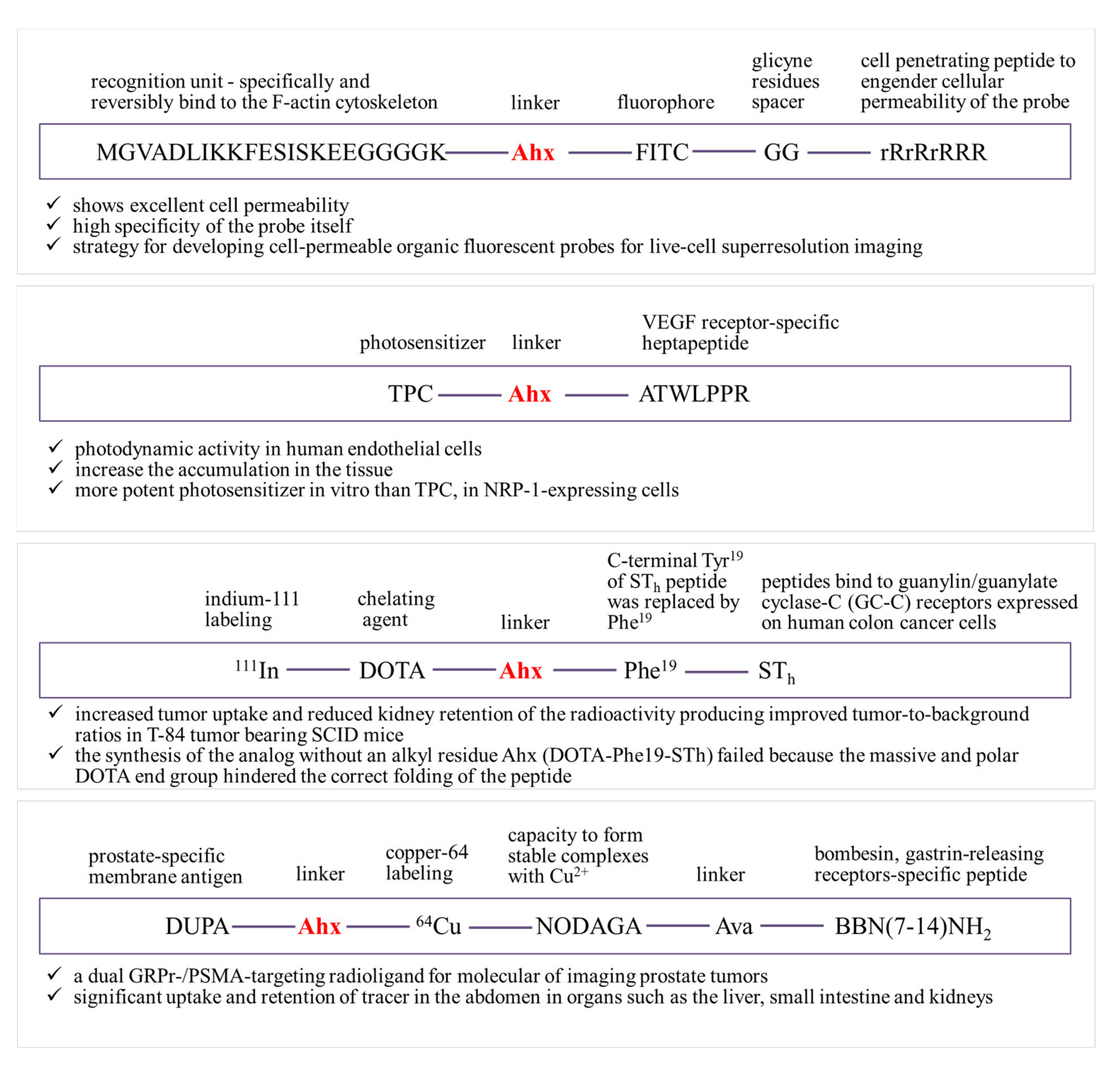
5. Ahx as a Linker
6. Ahx as an Element of Biotinylation Reagents
7. Polymers and Oligomers of Ahx
8. Other Derivatives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature and Symbolism for Amino Acids and Peptides. Corrections to Recommendations 1983. Eur. J. Biochem. 1993, 213, 2. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Desai, U.R. Recent Advances on Plasmin Inhibitors for the Treatment of Fibrinolysis-Related Disorders. Med. Res. Rev. 2014, 34, 1168–1216. [Google Scholar] [CrossRef] [PubMed]
- Bellussi, G.; Perego, C. Industrial Catalytic Aspects of the Synthesis of Monomers for Nylon Production. Cattech 2020, 4, 4–16. [Google Scholar] [CrossRef]
- Meyers, C.Y.; Miller, L.E. ε-Aminocaproic Acid. Org. Synth. 1952, 32, 13, Erratum in 1963, 4, 39. Available online: http://www.orgsyn.org/Content/pdfs/procedures/CV4P0039.pdf (accessed on 25 October 2021).
- Sattler, J.H.; Fuchs, M.; Mutti, F.G.; Grischek, B.; Engel, P.; Pfeffer, J.; Woodley, J.; Kroutil, W. Introducing an In Situ Capping Strategy in Systems Biocatalysis to Access 6-Aminohexanoic acid. Angew. Chem. Int. Ed. 2014, 53, 14153–14157. [Google Scholar] [CrossRef] [PubMed]
- Bretschneider, L.; Wegner, M.; Bühler, K.; Bühler, B.; Karande, R. One-Pot Synthesis of 6-Aminohexanoic Acid from Cyclohexane Using Mixed-Species Cultures. Microb. Biotechnol. 2021, 14, 1011–1025. [Google Scholar] [CrossRef]
- Schäfer, L.; Bühler, K.; Karande, R.; Bühler, B. Rational Engineering of a Multi-Step Biocatalytic Cascade for the Conversion of Cyclohexane to Polycaprolactone Monomers in Pseudomonas Taiwanensis. Biotechnol. J. 2020, 15, e2000091. [Google Scholar] [CrossRef]
- Ortmann, E.; Besser, M.W.; Klein, A.A. Antifibrinolytic Agents in Current Anaesthetic Practice. Br. J. Anaesth. 2013, 111, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Nakajima, T.; Okamoto, U.; Watanabe, H.; Iguchi, Y.; Igawa, T.; Chien, C.-C.; Hayashi, T. A suppressing effect of ε-amino-n-caproic acid on the bleeding of dogs, produced with the activation of plasmin in the circulatoring blood. Keio J. Med. 1959, 8, 247–266. [Google Scholar] [CrossRef] [Green Version]
- Markwardt, F. Synthetic inhibitors of fibrinolysis. In Handbook of Experimental Pharmacology: Fibrinolytics and Antifibrinolytics; Markwardt, F., Ed.; Springer: Berlin, Germany, 1978. [Google Scholar]
- Sun, Z.; Chen, Y.; Wang, P.; Zhang, J.; Gurewich, V.; Zhang, P.; Liu, J.-N. The Blockage of the High-Affinity Lysine Binding Sites of Plasminogen by EACA Significantly Inhibits Prourokinase-Induced Plasminogen Activation. Biochim. Biophys. Acta 2002, 1596, 182–192. [Google Scholar] [CrossRef]
- Winn, E.S.; Hu, S.P.; Hochschwender, S.M.; Laursen, R.A. Studies on the Lysine-Binding Sites of Human Plasminogen. The Effect of Ligand Structure on the Binding of Lysine Analogs to Plasminogen. Eur. J. Biochem. 1980, 104, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.; Jerath, A.; Wąsowicz, M. Tranexamic Acid: A Clinical Review. Anaesthesiol. Intensive Ther. 2015, 47, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Tengborn, L.; Blombäck, M.; Berntorp, E. Tranexamic Acid—An Old Drug Still Going Strong and Making a Revival. Thromb. Res. 2015, 135, 231–242. [Google Scholar] [CrossRef]
- LeCleir, L.K. Comparison of tranexamic acid and aminocaproic acid in coronary bypass surgery. Butl. J. Undergrad. Res. 2016, 2, 107–116. [Google Scholar]
- Martin, K.; Breuer, T.; Gertler, R.; Hapfelmeier, A.; Schreiber, C.; Lange, R.; Hess, J.; Wiesner, G. Tranexamic Acid versus Ɛ-Aminocaproic Acid: Efficacy and Safety in Paediatric Cardiac Surgery. Eur. J. Cardio-Thorac. Surg. 2011, 39, 892–897. [Google Scholar] [CrossRef] [Green Version]
- Riaz, O.; Aqil, A.; Asmar, S.; Vanker, R.; Hahnel, J.; Brew, C.; Grogan, R.; Radcliffe, G. Epsilon-Aminocaproic Acid versus Tranexamic Acid in Total Knee Arthroplasty: A Meta-Analysis Study. J. Orthop. Traumatol. 2019, 20, 28. [Google Scholar] [CrossRef] [Green Version]
- Broadwin, M.; Grant, P.E.; Robich, M.P.; Palmeri, M.L.; Lucas, F.L.; Rappold, J.; Kramer, R.S. Comparison of intraoperative tranexamic acid and epsilon-aminocaproic acid in cardiopulmonary bypass patients. JTCVS Open 2020, 3, 114–125. [Google Scholar] [CrossRef]
- Borst, A.J.; Bonfield, C.M.; Deenadayalan, P.S.; Le, C.H.; Xu, M.; Sobey, J.H.; Reddy, S.K. ε-Aminocaproic Acid versus Tranexamic Acid in Children Undergoing Complex Cranial Vault Reconstruction for Repair of Craniosynostosis. Pediatr. Blood Cancer 2021, 68, e29093. [Google Scholar] [CrossRef]
- Midura-Nowaczek, K.; Bruzgo, I.; Dubis, E.; Roszkowska-Jakimiec, W.; Worowski, K. Antifibrinolytic Activity of Epsilon-Aminocaproyl Derivatives of Amino Acids. Pharmazie 1996, 51, 775–777. [Google Scholar]
- Bruzgo, I.; Tomasiak, M.; Stelmach, H.; Midura-Nowaczek, K. The Effect of Some Epsilon-Aminocaproic Acid Derivatives on Platelet Responses. Acta Biochim. Pol. 2004, 51, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Bruzgo, I.; Midura-Nowaczek, K.; Bruzgo, M.; Kaczyńska, J.; Roszkowska-Jakimiec, W. Effect of Epsilon-Aminocaproylamino Acids on Fibrin Formation. Acta Pol. Pharm. 2006, 63, 149–152. [Google Scholar]
- Midura-Nowaczek, K.; Bruzgo, I.; Roszkowska-Jakimiec, W.; Markowska, A. Effects of Epsilon-Aminocaproiloaminoacids on the Amidolytic Activity of Tissue Plasminogen Activator, Urokinase and Kallikrein. Acta Pol. Pharm. 2004, 61, 75–76. [Google Scholar]
- Bruzgo, I.; Midura-Nowaczek, K.; Kaczyńska, J.; Krajewska, D. Effect of Epsilon-Aminocaproyl-S-Benzyl-L-Cysteine on the Activity of Plasminogen Activators. Acta Pol. Pharm. 2009, 66, 37–40. [Google Scholar]
- Midura-Nowaczek, K.; Kaczyńska, J.; Bruzgo, I.; Markowska, A.; Drozdowska, D. The Effect of ε-Aminocaproyl-S-Benzyl-L-Cysteine on the t-PA Activity of Human Saliva. Adv. Med. Sci. 2011, 56, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Midura-Nowaczek, K.; Bruzgo, I.; Popławski, J.; Roszkowska-Jakimiec, W.; Worowski, K. Effects of Epsilon-Aminocaproylaminoacids on Fibrinolytic and Caseinolytic Activity of Euglobulin Fraction. Acta Pol. Pharm. 1998, 55, 159–161. [Google Scholar] [PubMed]
- Midura-Nowaczek, K.; Bruzgo, I.; Popławski, J.; Roszkowska-Jakimiec, W.; Worowski, K. Synthesis and Activity of N Alpha-(Epsilon-Aminocaproyl)-Alanines and N Epsilon-(Epsilon-Aminocaproyl)-Caproic Acid. Acta Pol. Pharm. 1995, 52, 505–507. [Google Scholar]
- Purwin, M.; Bruzgo, I.; Markowska, A.; Midura-Nowaczek, K. Short Peptides Containing L-Lysine and Epsilon-Aminocaproic Acid as Potential Plasmin Inhibitors. Pharmazie 2009, 64, 765–767. [Google Scholar]
- Purwin, M.; Markowska, A.; Bruzgo, I.; Rusak, T.; Surażyński, A.; Jaworowska, U.; Midura-Nowaczek, K. Peptides with 6-Aminohexanoic Acid: Synthesis and Evaluation as Plasmin Inhibitors. Int. J. Pept. Res. Ther. 2017, 23, 235–245. [Google Scholar] [CrossRef]
- Midura-Nowaczek, K.; Markowska, A.; Krajewska, D.; Wołczyński, S. Cytotoxic Activity of Epsilon-Aminocaproylamino Acids in Breast Cancer MCF-7 and Fibroblast Cell Lines. Acta Pol. Pharm. 2010, 67, 201–204. [Google Scholar]
- Midura-Nowaczek, K.; Purwin, M.; Markowska, A.; Drozdowska, D.; Bruzgo, M. Effect of Short Peptides Containing Lysine and Epsilon-Aminocaproic Acid on Fibrinolytic Activity of Plasmin and Topoisomerase II Action on Supercoiled DNA. Acta Pol. Pharm. 2013, 70, 431–434. [Google Scholar] [PubMed]
- Mac Donald, M.; Aubé, J. Approaches to cyclic peptide beta turn mimics. Curr. Org. Chem. 2001, 5, 417–438. [Google Scholar] [CrossRef]
- Banerjee, A.; Maji, S.K.; Drew, M.G.B.; Haldar, D.; Das, A.K.; Banerjee, A. Hydrogen-bonded dimer can mediate supramolecular β-sheet formation and subsequent amyloid-like fibril formation: A model study. Tetrahedron 2004, 60, 5935–5944. [Google Scholar] [CrossRef]
- Cheng, C.T.; Lo, V.; Chen, J.; Chen, W.C.; Lin, C.Y.; Lin, H.C.; Yang, C.H.; Sheh, L. Synthesis and DNA Nicking Studies of a Novel Cyclic Peptide: Cyclo[Lys-Trp-Lys-Ahx-]. Bioorg. Med. Chem. 2001, 9, 1493–1498. [Google Scholar] [CrossRef]
- Tamura, K.; Agrios, K.A.; Vander Velde, D.; Aubé, J.; Borchardt, R.T. Effect of Stereochemistry on the Transport of Aca-Linked Beta-Turn Peptidomimetics across a Human Intestinal Cell Line. Bioorg. Med. Chem. 1997, 5, 1859–1866. [Google Scholar] [CrossRef]
- Xie, M.; Aubé, J.; Borchardt, R.T.; Morton, M.; Topp, E.M.; Vander Velde, D.; Schowen, R.L. Reactivity toward Deamidation of Asparagine Residues in Beta-Turn Structures. J. Pept. Res. 2000, 56, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R.; Catlin, P.; Zhang, M.-M.; Fiedler, B.; Bayudan, W.; Morrison, A.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M.; et al. Conotoxins Containing Nonnatural Backbone Spacers: Cladistic-Based Design, Chemical Synthesis, and Improved Analgesic Activity. Chem. Biol. 2007, 14, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-K.; Smith, M.D.; Smith, B.J.; Grussendorf, J.; Xu, L.; Gillies, R.J.; White, H.S.; Bulaj, G. Anticonvulsant Met-Enkephalin Analogues Containing Backbone Spacers Reveal Alternative Non-Opioid Signaling in the Brain. ACS Chem. Biol. 2009, 4, 659–671. [Google Scholar] [CrossRef]
- Rist, B.; Wieland, H.A.; Willim, K.D.; Beck-Sickinger, A.G. A Rational Approach for the Development of Reduced-Size Analogues of Neuropeptide Y with High Affinity to the Y1 Receptor. J. Pept. Sci. 1995, 1, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Abes, R.; Arzumanov, A.; Moulton, H.; Abes, S.; Ivanova, G.; Gait, M.J.; Iversen, P.; Lebleu, B. Arginine-Rich Cell Penetrating Peptides: Design, Structure-Activity, and Applications to Alter Pre-MRNA Splicing by Steric-Block Oligonucleotides. J. Pept. Sci. 2008, 14, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Abes, R.; Moulton, H.M.; Clair, P.; Yang, S.-T.; Abes, S.; Melikov, K.; Prevot, P.; Youngblood, D.S.; Iversen, P.L.; Chernomordik, L.V.; et al. Delivery of Steric Block Morpholino Oligomers by (R-X-R)4 Peptides: Structure-Activity Studies. Nucleic Acids Res. 2008, 36, 6343–6354. [Google Scholar] [CrossRef] [PubMed]
- Lebleu, B.; Moulton, H.M.; Abes, R.; Ivanova, G.D.; Abes, S.; Stein, D.A.; Iversen, P.L.; Arzumanov, A.A.; Gait, M.J. Cell Penetrating Peptide Conjugates of Steric Block Oligonucleotides. Adv. Drug Deliv. Rev. 2008, 60, 517–529. [Google Scholar] [CrossRef]
- Yu, G.S.; Yu, H.N.; Choe, Y.H.; Son, S.J.; Ha, T.H.; Choi, J.S. Sequential conjugation of 6-aminohexanoic acids and L-arginines to poly(amidoamine) dendrimer to modify hydrophobicity and flexibility of the polymeric gene carrier. Bull. Korean Chem. Soc. 2011, 32, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Saleh, A.F.; Arzumanov, A.; Abes, R.; Owen, D.; Lebleu, B.; Gait, M.J. Synthesis and Splice-Redirecting Activity of Branched, Arginine-Rich Peptide Dendrimer Conjugates of Peptide Nucleic Acid Oligonucleotides. Bioconjug. Chem. 2010, 21, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Pyun, J.C.; Cheong, M.Y.; Park, S.H.; Kim, H.Y.; Park, J.S. Modification of Short Peptides Using Epsilon-Aminocaproic Acid for Improved Coating Efficiency in Indirect Enzyme-Linked Immunosorbent Assays (ELISA). J. Immunol. Methods 1997, 208, 141–149. [Google Scholar] [CrossRef]
- Doyle, M.E.; Greig, N.H.; Holloway, H.W.; Betkey, J.A.; Bernier, M.; Egan, J.M. Insertion of an N-Terminal 6-Aminohexanoic Acid after the 7 Amino Acid Position of Glucagon-like Peptide-1 Produces a Long-Acting Hypoglycemic Agent. Endocrinology 2001, 142, 4462–4468. [Google Scholar] [CrossRef]
- Winiecka, I.; Jaworski, P.; Mazurek, A.P.; Marszałek, D.; Goldnik, A.; Sokulski, D. Novel Renin Inhibitors Containing Derivatives of N-Alkylleucyl-β-Hydroxy-γ-Amino. Acids. J. Pept. Sci. 2016, 22, 106–115. [Google Scholar] [CrossRef]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the Structural Biology of Protein-Protein Interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Jakab, A.; Schlosser, G.; Feijlbrief, M.; Welling-Wester, S.; Manea, M.; Vila-Perello, M.; Andreu, D.; Hudecz, F.; Mezo, G. Synthesis and Antibody Recognition of Cyclic Epitope Peptides, Together with Their Dimer and Conjugated Derivatives Based on Residues 9-22 of Herpes Simplex Virus Type 1 Glycoprotein D. Bioconjug. Chem. 2009, 20, 683–692. [Google Scholar] [CrossRef]
- Heupel, W.-M.; Efthymiadis, A.; Schlegel, N.; Müller, T.; Baumer, Y.; Baumgartner, W.; Drenckhahn, D.; Waschke, J. Endothelial Barrier Stabilization by a Cyclic Tandem Peptide Targeting VE-Cadherin Transinteraction in Vitro and in Vivo. J. Cell. Sci. 2009, 122, 1616–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaltin, P.; Borgions, F.; Rozenski, J.; Van Aerschot, A.; Herdewijn, P. New dsDNA-Binding Hybrid Molecules Combining an Unnatural Peptide and an Intercalating Moiety. Helv. Chim. Acta 2003, 86, 533–547. [Google Scholar] [CrossRef]
- Borgions, F.; Ghyssels, D.; Van Aerschot, A.; Rozenski, J.; Herdewijn, P. Synthetic dsDNA-Binding Peptides Using Natural Compounds as Model. Helv. Chim. Acta 2006, 89, 1194–1219. [Google Scholar] [CrossRef]
- Howl, J.; Langel, U.; Hawtin, S.R.; Valkna, A.; Yarwood, N.J.; Saar, K.; Wheatley, M. Chimeric Strategies for the Rational Design of Bioactive Analogs of Small Peptide Hormones. FASEB J. 1997, 11, 582–590. [Google Scholar] [CrossRef]
- Höfliger, M.M.; Castejón, G.L.; Kiess, W.; Beck Sickinger, A.G. Novel Cell Line Selectively Expressing Neuropeptide Y-Y2 Receptors. J. Recept. Signal Transduct. Res. 2003, 23, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Raghothama, S.; Balaram, P. Peptide design. Helix-helix motifs in synthetic sequences. J. Chem. Soc. Perkin Trans. 1997, 2, 2087–2094. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, L.; Finetti, F.; Diana, D.; Di Stasi, R.; Auriemma, S.; Romanelli, A.; Fattorusso, R.; Ziche, M.; Morbidelli, L.; D’Andrea, L.D. Miniaturizing VEGF: Peptides Mimicking the Discontinuous VEGF Receptor-Binding Site Modulate the Angiogenic Response. Sci. Rep. 2016, 6, 31295. [Google Scholar] [CrossRef] [Green Version]
- Torrent, M.; Pulido, D.; de la Torre, B.G.; García-Mayoral, M.F.; Nogués, M.V.; Bruix, M.; Andreu, D.; Boix, E. Refining the Eosinophil Cationic Protein Antibacterial Pharmacophore by Rational Structure Minimization. J. Med. Chem. 2011, 54, 5237–5244. [Google Scholar] [CrossRef]
- Pulido, D.; Moussaoui, M.; Andreu, D.; Nogués, M.V.; Torrent, M.; Boix, E. Antimicrobial Action and Cell Agglutination by the Eosinophil Cationic Protein Are Modulated by the Cell Wall Lipopolysaccharide Structure. Antimicrob. Agents Chemother. 2012, 56, 2378–2385. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Wei, D.; Liu, J.; Su, W. An Antisense Oligodeoxynucleotide-Doxorubicin Conjugate: Preparation and Its Reversal Multidrug Resistance of Human Carcinoma Cell Line in Vitro. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1595–1607. [Google Scholar] [CrossRef]
- Baskaran, R.; Lee, J.; Yang, S.-G. Clinical Development of Photodynamic Agents and Therapeutic Applications. Biomater. Res. 2018, 22, 25. [Google Scholar] [CrossRef]
- Tirand, L.; Frochot, C.; Vanderesse, R.; Thomas, N.; Trinquet, E.; Pinel, S.; Viriot, M.-L.; Guillemin, F.; Barberi-Heyob, M. A Peptide Competing with VEGF165 Binding on Neuropilin-1 Mediates Targeting of a Chlorin-Type Photosensitizer and Potentiates Its Photodynamic Activity in Human Endothelial Cells. J. Control. Release 2006, 111, 153–164. [Google Scholar] [CrossRef]
- Tirand, L.; Thomas, N.; Dodeller, M.; Dumas, D.; Frochot, C.; Maunit, B.; Guillemin, F.; Barberi-Heyob, M. Metabolic Profile of a Peptide-Conjugated Chlorin-Type Photosensitizer Targeting Neuropilin-1: An in Vivo and in Vitro Study. Drug Metab. Dispos. 2007, 35, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Gali, H.; Sieckman, G.L.; Hoffman, T.J.; Owen, N.K.; Mazuru, D.G.; Forte, L.R.; Volkert, W.A. Chemical Synthesis of Escherichia Coli ST(h) Analogues by Regioselective Disulfide Bond Formation: Biological Evaluation of an (111)In-DOTA-Phe(19)-ST(h) Analogue for Specific Targeting of Human Colon Cancers. Bioconjug. Chem. 2002, 13, 224–231. [Google Scholar] [CrossRef]
- Jackson, A.B.; Nanda, P.K.; Rold, T.L.; Sieckman, G.L.; Szczodroski, A.F.; Hoffman, T.J.; Chen, X.; Smith, C.J. 64Cu-NO2A-RGD-Glu-6-Ahx-BBN(7-14)NH2: A Heterodimeric Targeting Vector for Positron Emission Tomography Imaging of Prostate Cancer. Nucl. Med. Biol. 2012, 39, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Bandari, R.P.; Jiang, Z.; Reynolds, T.S.; Bernskoetter, N.E.; Szczodroski, A.F.; Bassuner, K.J.; Kirkpatrick, D.L.; Rold, T.L.; Sieckman, G.L.; Hoffman, T.J.; et al. Synthesis and Biological Evaluation of Copper-64 Radiolabeled [DUPA-6-Ahx-(NODAGA)-5-Ava-BBN(7-14)NH2], a Novel Bivalent Targeting Vector Having Affinity for Two Distinct Biomarkers (GRPr/PSMA) of Prostate Cancer. Nucl. Med. Biol. 2014, 41, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, P.K.; Wienhoff, B.E.; Rold, T.L.; Sieckman, G.L.; Szczodroski, A.F.; Hoffman, T.J.; Rogers, B.E.; Smith, C.J. Positron-Emission Tomography (PET) Imaging Agents for Diagnosis of Human Prostate Cancer: Agonist vs. Antagonist Ligands. In Vivo 2012, 26, 583–592. [Google Scholar] [PubMed]
- Pan, D.; Hu, Z.; Qiu, F.; Huang, Z.-L.; Ma, Y.; Wang, Y.; Qin, L.; Zhang, Z.; Zeng, S.; Zhang, Y.-H. A General Strategy for Developing Cell-Permeable Photo-Modulatable Organic Fluorescent Probes for Live-Cell Super-Resolution Imaging. Nat. Commun. 2014, 5, 5573. [Google Scholar] [CrossRef] [PubMed]
- Wilchek, M.; Bayer, E.A. The Avidin-Biotin Complex in Bioanalytical Applications. Anal. Biochem. 1988, 171, 1–32. [Google Scholar] [CrossRef]
- Elia, G. Biotinylation Reagents for the Study of Cell Surface Proteins. Proteomics 2008, 8, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Bar, D.Z.; Atkatsh, K.; Tavarez, U.; Erdos, M.R.; Gruenbaum, Y.; Collins, F.S. Biotinylation by Antibody Recognition-a Method for Proximity Labeling. Nat. Methods 2018, 15, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Nitecki, D.E.; Kindler, H.; Goodman, J.W. Immunogenicity of Biotinylated Hapten-Avidin Complexes. Mol. Immunol. 1984, 21, 1055–1060. [Google Scholar] [CrossRef]
- Scarborough, J.H.; Brusoski, K.; Brewer, S.; Rodich, S.; Chatley, K.S.; Nguyen, T.; Green, K.N. Development of Low Molecular Weight Ferrocene−Biotin Bioconjugates as Electrochemical Sensors. Organometallics 2015, 34, 918–925. [Google Scholar] [CrossRef]
- Gong, M.; Li, T.; Wu, L.; Zhang, Z.; Ren, L.; Duan, X.; Cao, H.; Pei, M.; Li, J.; Du, Y. Liquid-Phase and Ultrahigh-Frequency-Acoustofluidics-Based Solid-Phase Synthesis of Biotin-Tagged 6′/3′-Sialyl-N-Acetylglucosamine by Sequential One-Pot Multienzyme System. Catalysts 2020, 10, 1347. [Google Scholar] [CrossRef]
- Huttunen, S.; Toivanen, M.; Liu, C.; Tikkanen-Kaukanen, C. Novel Anti-Infective Potential of Salvianolic Acid B against Human Serious Pathogen Neisseria Meningitidis. BMC Res. Notes 2016, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, E.A.; Fishman, J.B. Labeling Antibodies with N-Hydroxysuccinimide-Long Chain (NHS-LC)-Biotin. Cold Spring Harb. Protoc. 2020, 2020, 099259. [Google Scholar] [CrossRef] [PubMed]
- Babos, F.; Szarka, E.; Nagy, G.; Majer, Z.; Sármay, G.; Magyar, A.; Hudecz, F. Role of N- or C-Terminal Biotinylation in Autoantibody Recognition of Citrullin Containing Filaggrin Epitope Peptides in Rheumatoid Arthritis. Bioconjug. Chem. 2013, 24, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Yellepeddi, V.K.; Kumar, A.; Palakurthi, S. Biotinylated Poly(Amido)Amine (PAMAM) Dendrimers as Carriers for Drug Delivery to Ovarian Cancer Cells in Vitro. Anticancer. Res. 2009, 29, 2933–2943. [Google Scholar]
- Peccerella, T.; Lukan, N.; Hofheinz, R.; Schadendorf, D.; Kostrezewa, M.; Neumaier, M.; Findeisen, P. Endoprotease Profiling with Double-Tagged Peptide Substrates: A New Diagnostic Approach in Oncology. Clin. Chem. 2010, 56, 272–280. [Google Scholar] [CrossRef]
- Matsumura, S.; Uemura, S.; Mihara, H. Construction of Biotinylated Peptide Nanotubes for Arranging Proteins. Mol. Biosyst. 2005, 1, 146–148. [Google Scholar] [CrossRef]
- Balogh, A.; Horváti, K.; Mezo, G.; Derzbach, L.; Szebeni, B.; Nagy, L.; Prechl, J.; Vásárhelyi, B.; Hudecz, F.; Bosze, S. Synthesis of Hepcidin Derivatives in Order to Develop Standards for Immune Adsorption Method. J. Pept. Sci. 2009, 15, 285–295. [Google Scholar] [CrossRef]
- Lalmanach, G.; Mayer, R.; Serveau, C.; Scharfstein, J.; Gauthier, F. Biotin-Labelled Peptidyl Diazomethane Inhibitors Derived from the Substrate-like Sequence of Cystatin: Targeting of the Active Site of Cruzipain, the Major Cysteine Proteinase of Trypanosoma Cruzi. Biochem. J. 1996, 318 Pt 2, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Finsinger, D.; Peter, K.; Guttenberg, Z.; Bärmann, M.; Kessler, H.; Escherich, A.; Moroder, L.; Böhm, J.; Baumeister, W.; et al. Intervesicle Cross-Linking with Integrin Alpha IIb Beta 3 and Cyclic-RGD-Lipopeptide. A Model of Cell-Adhesion Processes. Biochemistry 2000, 39, 12284–12294. [Google Scholar] [CrossRef] [PubMed]
- Padeste, C.; Grubelnik, A.; Tiefenauer, L. Ferrocene-Avidin Conjugates for Bioelectrochemical Applications. Biosens. Bioelectron. 2000, 15, 431–438. [Google Scholar] [CrossRef]
- Lacenere, C.; Garg, M.K.; Stoltz, B.M.; Quake, S.R. Effects of a Modified Dye-Labeled Nucleotide Spacer Arm on Incorporation by Thermophilic DNA Polymerases. Nucleosides Nucleotides Nucleic Acids 2006, 25, 9–15. [Google Scholar] [CrossRef]
- Don-Doncow, N.; Escobar, Z.; Johansson, M.; Kjellström, S.; Garcia, V.; Munoz, E.; Sterner, O.; Bjartell, A.; Hellsten, R. Galiellalactone Is a Direct Inhibitor of the Transcription Factor STAT3 in Prostate Cancer Cells. J. Biol. Chem. 2014, 289, 15969–15978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, M.; Franco, L.; Rodríguez-Galán, A.; Puiggali, J. Poly(Ester Amides)s Derived from 1,4-Butanediol, Adipic Acid and 6-Aminohexanoic Acid. Part III: Substitution of Adipic Acid Units by Terephthalic Acid Units. Polym. Degrad. Stab. 2004, 85, 595–604. [Google Scholar] [CrossRef]
- Botines, E.; Puiggali, J. Crystallization kinetics of poly(glycolic acid-alt-6-aminohexanoic acid). Eur. Polym. J. 2006, 42, 595–1608. [Google Scholar] [CrossRef]
- Kvadra, J.; Prokopova, I.; Kondelı´k, P. Reactivity of 6-Hexanelactam Cyclic Oligomers, 1 Acidolysis. Macromol. Chem. Phys. 2001, 202, 133–138. [Google Scholar] [CrossRef]
- Negoro, S.; Ohki, T.; Shibata, N.; Sasa, K.; Hayashi, H.; Nakano, H.; Yasuhira, K.; Kato, D.; Takeo, M.; Higuchi, Y. Nylon-Oligomer Degrading Enzyme/Substrate Complex: Catalytic Mechanism of 6-Aminohexanoate-Dimer Hydrolase. J. Mol. Biol. 2007, 370, 142–156. [Google Scholar] [CrossRef]
- Yasuhira, K.; Shibata, N.; Mongami, G.; Uedo, Y.; Atsumi, Y.; Kawashima, Y.; Hibino, A.; Tanaka, Y.; Lee, Y.-H.; Kato, D.; et al. X-ray Crystallographic Analysis of the 6-Aminohexanoate Cyclic Dimer Hydrolase: Catalytic Mechanism and Evolution of an Enzyme Responsible for Nylon-6 Byproduct Degradation. J. Biol. Chem. 2010, 285, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Storozhakova, N.A.; Akhmed, K.K.N.; Rakhimov, A.I. Catalytic Synthesis of Low- Molecular- Weight Oligomers of N-Adamantanoyl-ε-Aminocaproic Acid. Russ. J. Appl. Chem. 2005, 78, 1119–1121. [Google Scholar] [CrossRef]
- Rakhimov, A.I.; Storozhakova, N.A.; Akhmed, K.K.N.; Fedunov, R.G. Catalytic synthesis of N-benzyl (N-nitrobenzyl) derivatives of ε-aminocaproic acid oligomers. Russ. J. Appl. Chem. 2006, 79, 421–424. [Google Scholar] [CrossRef]
- Zhang, W. Synthesis and characterization of biodegradable copolymers based on 6-aminocaproic acid and α-L-alanine. Polym. Bull. 2008, 60, 323–330. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Y. Biodegradable Copoly(Amino Acid)s Based on 6-Aminocaproic Acid and L-Leucine. J. Polym. Environ. 2011, 19, 177–181. [Google Scholar] [CrossRef]
- Jiang, J.C.; Zhao, Z.T.; Lv, G.Y.; Yan, Y.G.; Wu, D.X. Syntheses and evaluation of copolymer 6-aminohexanoic acid and 4R-hydroxy-L-proline for bone repair. Adv. Mater. Res. 2012, 562–564, 506–511. [Google Scholar] [CrossRef]
- Brychtova, K.; Jampilek, J.; Opatrilova, R.; Raich, I.; Farsa, O.; Csollei, J. Synthesis, Physico-Chemical Properties and Penetration Activity of Alkyl-6-(2,5-Dioxopyrrolidin-1-Yl)-2-(2-Oxopyrrolidin-1-Yl)Hexanoates as Potential Transdermal Penetration Enhancers. Bioorg. Med. Chem. 2010, 18, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Farsa, O.; Doležal, P.; Hrabálek, A. Esters and amides of hexanoic acid substituted with tertiary amino group in terminal position and their activity as transdermal permeation enhancers. J. Serb. Chem. Soc. 2010, 75, 595–603. [Google Scholar] [CrossRef]
- Hrabálek, A.; Vávrová, K.; Dolezal, P.; Machácek, M. Esters of 6-Aminohexanoic Acid as Skin Permeation Enhancers: The Effect of Branching in the Alkanol Moiety. J. Pharm. Sci. 2005, 94, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Vávrová, K.; Hrabálek, A.; Dolezal, P.; Holas, T.; Klimentová, J. Biodegradable Derivatives of Tranexamic Acid as Transdermal Permeation Enhancers. J. Control. Release 2005, 104, 41–49. [Google Scholar] [CrossRef]
- Lu, J.; Chen, Z.P.; Yan, Y.P.; Knapp, S.; Schugar, H.; Chen, K.Y. Aminohexanoic Hydroxamate Is a Potent Inducer of the Differentiation of Mouse Neuroblastoma Cells. Cancer Lett. 2000, 160, 59–66. [Google Scholar] [CrossRef]
- Valverde, L.F.; Cedillo, F.D.; Lopez-Ramos, M.; Garcia-Cevera, E. Synthesis and characterization of antibacterial activity from danazol-aminocaproic derivative on both gram negative and gram positive bacteria. Int. J. PharmTech Res. 2010, 2, 334–340. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Antifibrynolytic Activity IC50 [mM] | Antiamidolytic Activity/S-2251 IC50 [mM] | References |
|---|---|---|---|
| EACA | 0.2 | - | [20] |
| H-EACA-NLeu-OH | <0.02 | 0.12 | [21,22,23,24,25] |
| HCl × H-EACA-Leu-OH | 0.08 | 13 | |
| HCl × H-EACA-Leu-OEt | - | 0.2 | |
| HCl × H-EACA-Cys(S-Bzl)-OH | 0.04 | 18 | |
| HCl × H-EACA-Cys(S-Bzl)-OEt | - | 12 | |
| H-EACA-EACA-OMe | - | 0.16 | [26,27] |
| HCl × H-EACA-Gly-OH | 0.8 | 8 | |
| HCl × H-EACA-Glu-OH | - | 0.08 | |
| Boc-EACA-Lys(Z)-EACA-NH2 | 20 | 20 | [28] |
| Boc-EACA-Lys-EACA-NH2 | 19 | 0.02 | |
| H-EACA-Lys-EACA-NH2 | 8.1 | 9 | |
| Boc-EACA-Lys(Z)-EACA-OMe | 1.2 | >20 | |
| H-EACA-Lys-EACA-OMe | 18 | 0.8 | |
| Boc-Lys(Z)-EACA-NH2 | <0.2 | 8 | |
| Boc-Lys-EACA-NH2 | <0.2 | - | |
| H-D-Ala-Phe-Lys-EACA-NH2 | - | 0.02 | [29] |
| Structure with Ahx | Biological Activity Benefits of Inserting of Ahx | References |
|---|---|---|
| Boc-Acp-Aib-Phe-OMe | Peptides form hydrogen bonded dimers that gives supramolecular β-sheets on self-assembly. | [34] |
| cyklo[Lys-Tyr-Lys-Ahx-] cyklo[Lys-Trp-Lys-Ahx-] | Higher DNA binding constant than linear analog. | [35] |
| cyclo(L-Ala-L-Ala-Aca) cyclo(L-Ala-D-Ala-Aca) | Cyclization facilitates penetration through the Caco-2 human epithelial cancer cell line monolayer. | [36] |
| cyclo-[L-Asn-Gly-Aca] cyclo-[Gly-Asn-Aca] | Cyclic peptides assume predominantly β-turn structures in solution, faster deamidation than linear analogs. | [37] |
| Ahx-SIIIA | Improve bioavailability by reducing susceptibility to proteolysis, and reducing hydrogen bond donors/acceptors. | [38] |
| Y-(6-Ahx)-Phe-Met | Reduction of affinity and agonist activity towards δ- and μ-opioid receptors, while maintaining strong antinociceptive and anticonvulsant properties. | [39] |
| (Ahx)2T127FIQFKKDLKEW137(Ahx)2 | Improvement of coating efficiency in ELISA immunoassay procedures. | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markowska, A.; Markowski, A.R.; Jarocka-Karpowicz, I. The Importance of 6-Aminohexanoic Acid as a Hydrophobic, Flexible Structural Element. Int. J. Mol. Sci. 2021, 22, 12122. https://doi.org/10.3390/ijms222212122
Markowska A, Markowski AR, Jarocka-Karpowicz I. The Importance of 6-Aminohexanoic Acid as a Hydrophobic, Flexible Structural Element. International Journal of Molecular Sciences. 2021; 22(22):12122. https://doi.org/10.3390/ijms222212122
Chicago/Turabian StyleMarkowska, Agnieszka, Adam Roman Markowski, and Iwona Jarocka-Karpowicz. 2021. "The Importance of 6-Aminohexanoic Acid as a Hydrophobic, Flexible Structural Element" International Journal of Molecular Sciences 22, no. 22: 12122. https://doi.org/10.3390/ijms222212122
APA StyleMarkowska, A., Markowski, A. R., & Jarocka-Karpowicz, I. (2021). The Importance of 6-Aminohexanoic Acid as a Hydrophobic, Flexible Structural Element. International Journal of Molecular Sciences, 22(22), 12122. https://doi.org/10.3390/ijms222212122