
Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway


Abstract
:1. Introduction
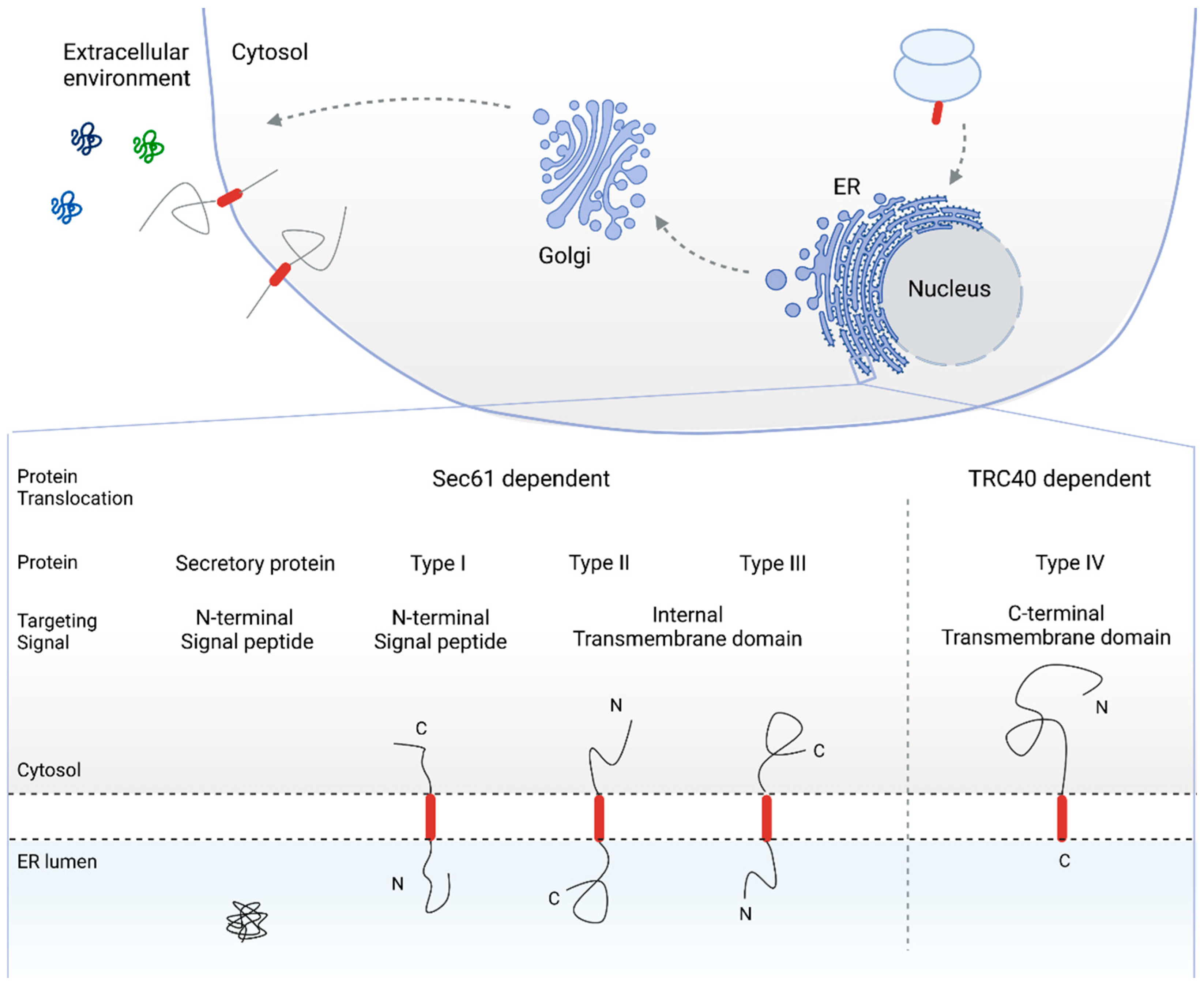
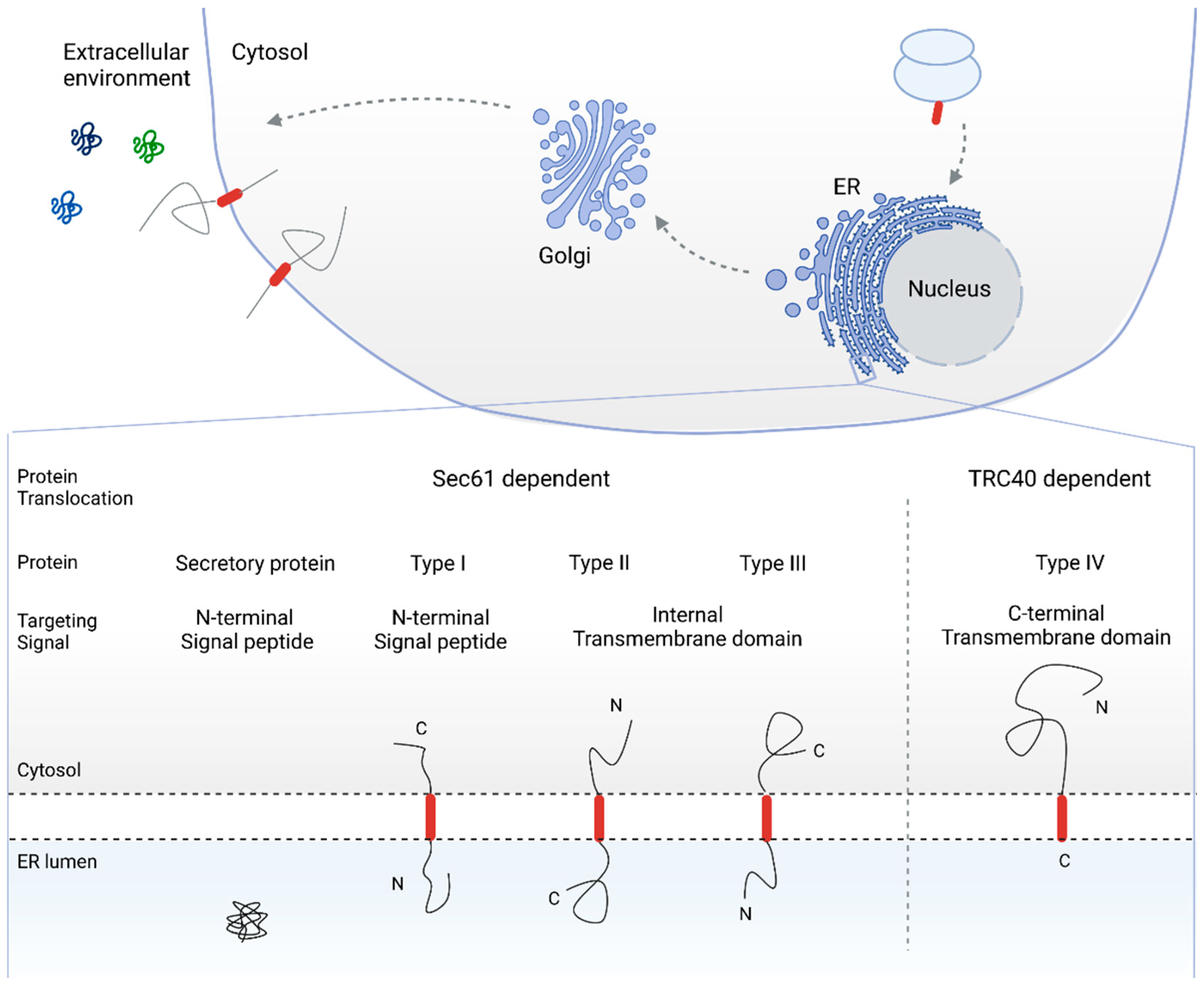
2. The Sec61 Dependent Pathway for Co- and Post-Translational Protein Translocation
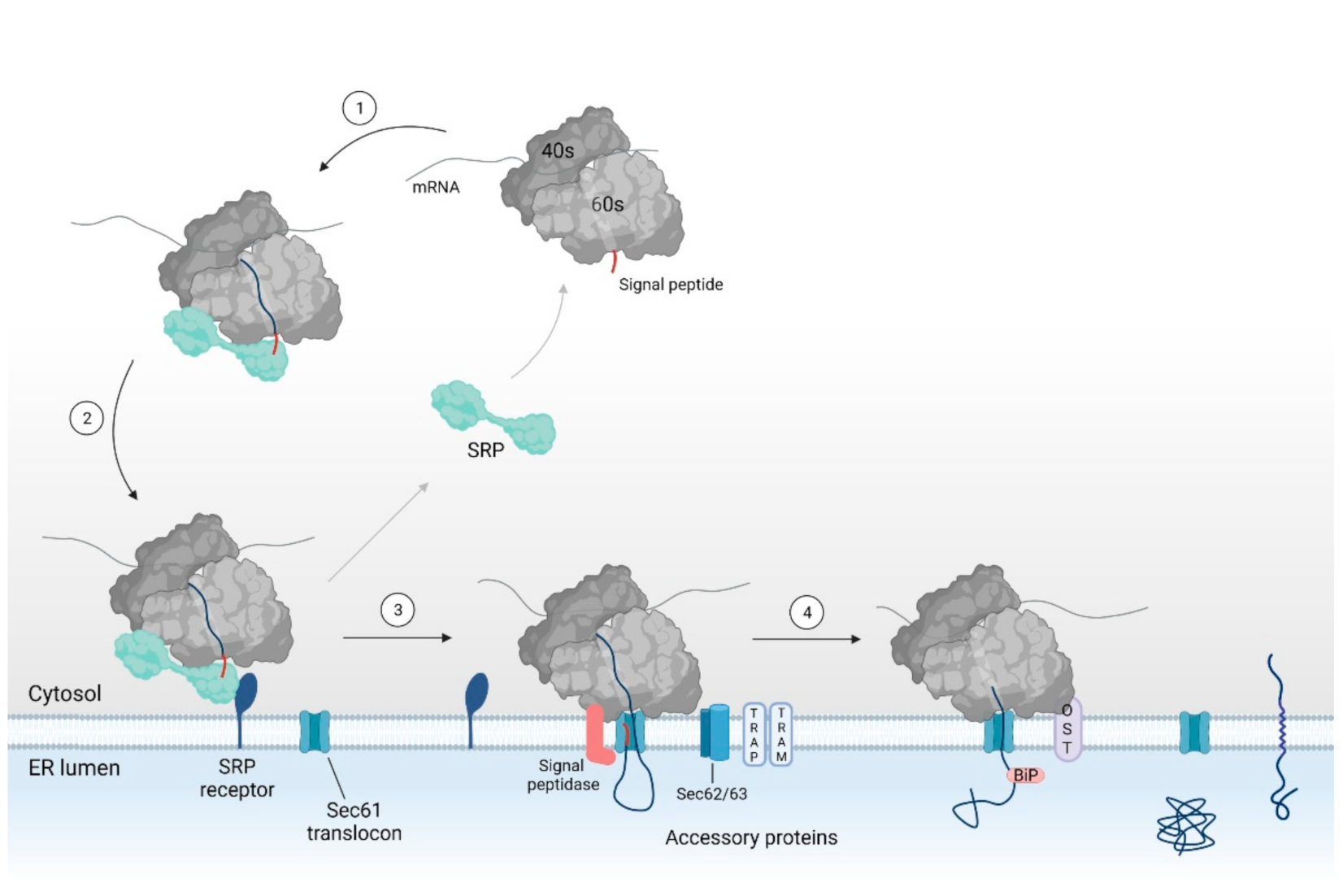
2.1. SRP Dependent Protein Targeting to the ER Membrane Keeps the Protein in a Translocation Competent State
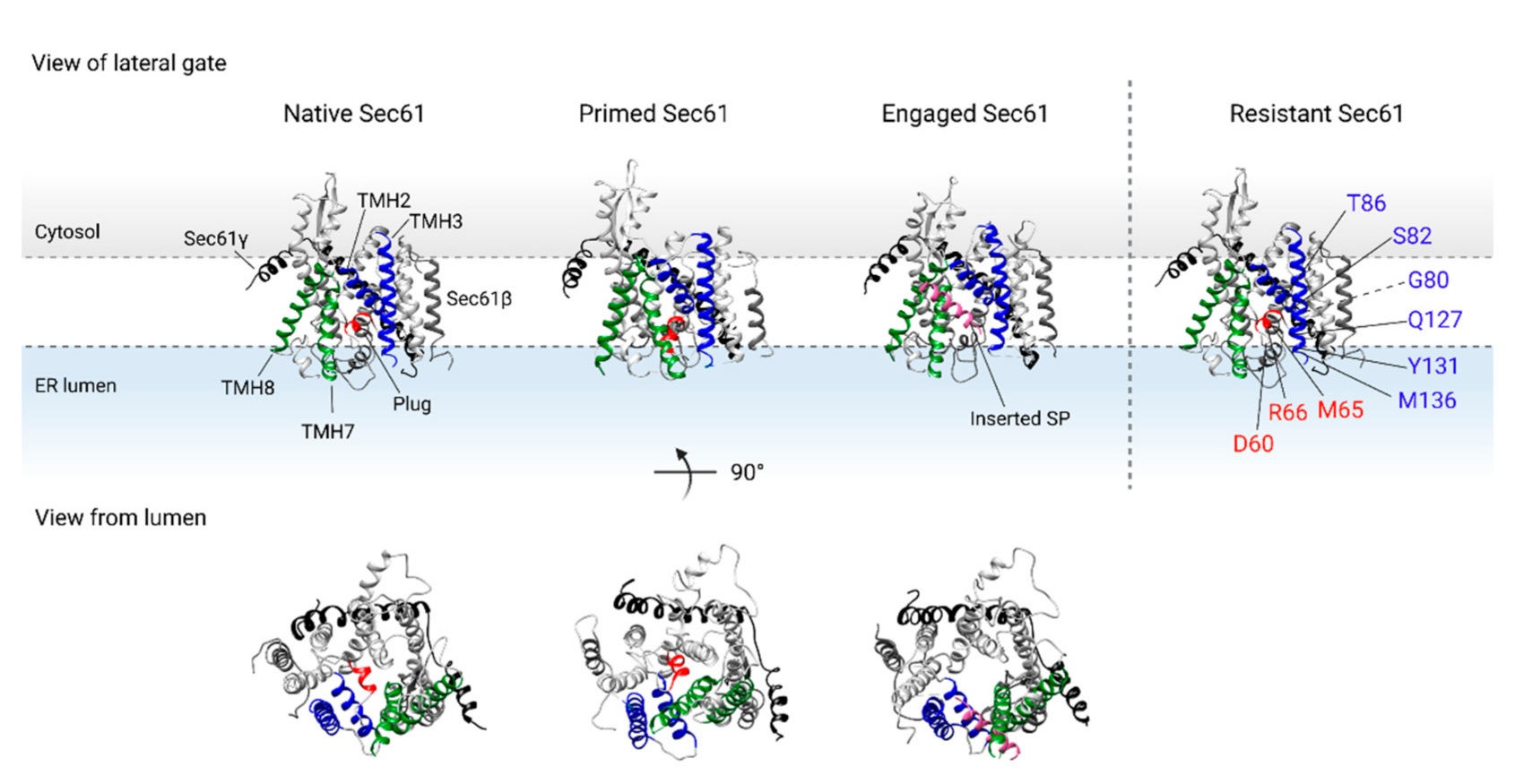
2.2. Binding of the RNC Complex Induces Dynamic Conformational Changes in the Translocon
2.3. Assisted Opening of the Sec61 Translocon
2.4. Chaperone Mediated Completion of Protein Translocation and Post-Translational Modifications in the ER Lumen
3. Translocation Inhibitors of the Sec61 Dependent Protein Translocation Pathway
3.1. Sec61 Inhibitors of Natural Origin
3.1.1. HUN7293, CAM741, and Cotransin
3.1.2. Decatransin
3.1.3. Apratoxin A and Coibamide A
3.1.4. Mycolactone
3.1.5. Ipomoeassin F
3.2. Synthetic Sec61 Inhibitors
3.2.1. Cyclotriazadisulfonamide
3.2.2. Eeyarestatin
3.2.3. KZR-261 and KZR-834
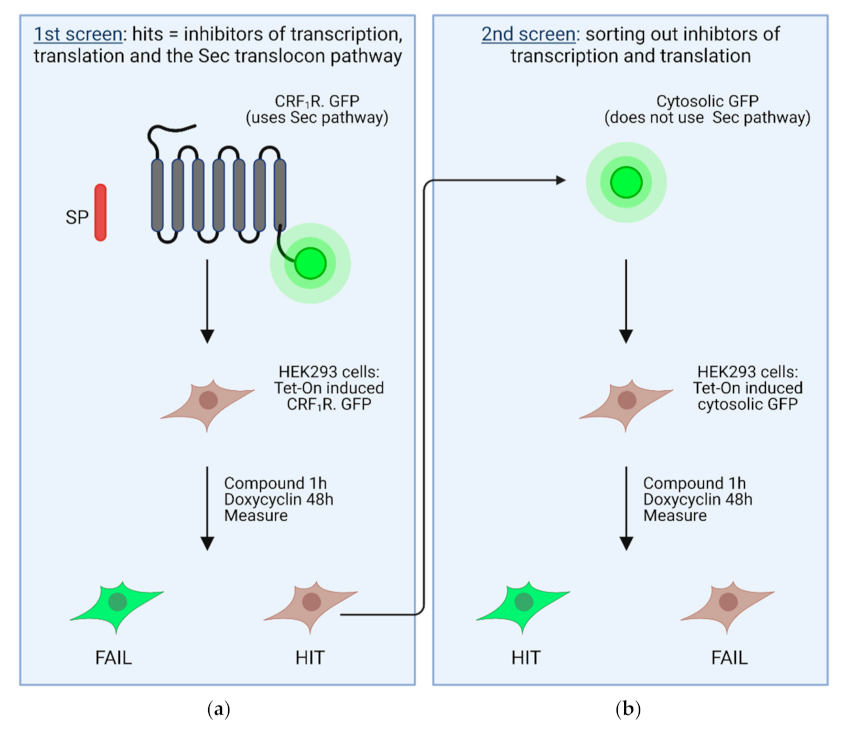
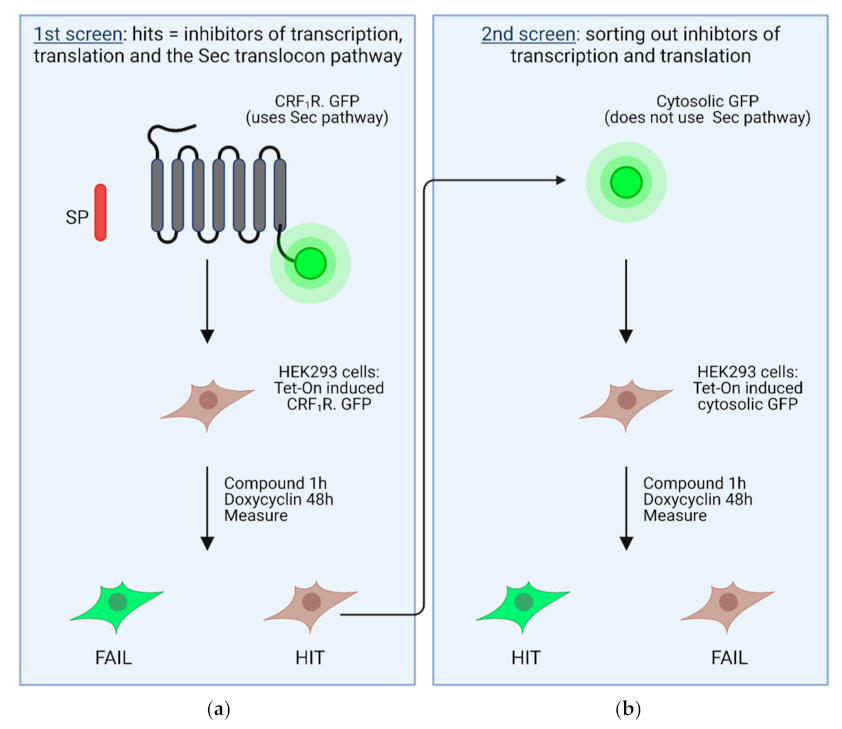
4. High Throughput Screening Assays to Define Novel Inhibitors of the Sec61 Complex
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Siekevitz, P.; Palade, G.E. A cytochemical study on the pancreas of the guinea pig. 5. In vivo incorporation of leucine-1-C14 into the chymotrypsinogen of various cell fractions. J. Cell Biol. 1958, 7, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, L.G.; Palade, G.E. Protein synthesis, storage, and discharge in the pancreatic exocrine cell. An autoradiographic study. J. Cell Biol. 1964, 20, 473–495. [Google Scholar] [PubMed]
- Jamieson, J.D.; Palade, G.E. Intracellular transport of secretory proteins in the pancreatic endocrine cell: II. Transport to Condensing Vacuoles and Zymogen Granules. J. Cell Biol. 1967, 34, 597–615. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, J.D.; Palade, G.E. Intracellular transport of secretory proteins in the pancreatic endocrine cell: I. Role of the Peripheral Elements of the Golgi Complex. J. Cell Biol. 1967, 34, 577–596. [Google Scholar] [CrossRef] [Green Version]
- Matlin, K.S.; Caplan, M.J. The secretory pathway at 50: A golden anniversary for some momentous grains of silver. Mol. Biol. Cell 2017, 28, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Gilmore, R.; Müller, M.; Blobel, G. The protein translocation machinery of the endoplasmic reticulum. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1982, 300, 225–228. [Google Scholar] [CrossRef]
- Walter, P.; Gilmore, R.; Blobel, G. Protein translocation across the endoplasmic reticulum. Cell 1984, 38, 5–8. [Google Scholar] [CrossRef]
- Von Heijne, G. Signal sequences. The limits of variation. J. Mol. Biol. 1985, 184, 99–105. [Google Scholar] [CrossRef]
- Cross, B.C.; Sinning, I.; Luirink, J.; High, S. Delivering proteins for export from the cytosol. Nat. Rev. Mol. Cell Biol. 2009, 10, 255–264. [Google Scholar] [CrossRef]
- Mateja, A.; Keenan, R.J. A structural perspective on tail-anchored protein biogenesis by the GET pathway. Curr. Opin. Struct. Biol. 2018, 51, 195–202. [Google Scholar] [CrossRef]
- Ast, T.; Cohen, G.; Schuldiner, M. A network of cytosolic factors targets SRP-independent proteins to the endoplasmic reticulum. Cell 2013, 152, 1134–1145. [Google Scholar] [CrossRef] [Green Version]
- Aviram, N.; Ast, T.; Costa, E.A.; Arakel, E.C.; Chuartzman, S.G.; Jan, C.H.; Hassdenteufel, S.; Dudek, J.; Jung, M.; Schorr, S.; et al. The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 2016, 540, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, J.J.; High, S. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 1999, 112 Pt 10, 1477–1486. [Google Scholar] [PubMed]
- Kim, S.J.; Mitra, D.; Salerno, J.R.; Hegde, R.S. Signal sequences control gating of the protein translocation channel in a substrate-specific manner. Dev. Cell 2002, 2, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Kraut-Cohen, J.; Afanasieva, E.; Haim-Vilmovsky, L.; Slobodin, B.; Yosef, I.; Bibi, E.; Gerst, J.E. Translation- and SRP-independent mRNA targeting to the endoplasmic reticulum in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2013, 24, 3069–3084. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Powis, K.; High, S. Post-translational translocation into the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2403–2409. [Google Scholar] [CrossRef]
- Mandon, E.C.; Trueman, S.F.; Gilmore, R. Translocation of proteins through the Sec61 and SecYEG channels. Curr. Opin. Cell Biol. 2009, 21, 501–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgese, N.; Coy-Vergara, J.; Colombo, S.F.; Schwappach, B. The Ways of Tails: The GET Pathway and more. Protein J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Denic, V. A portrait of the GET pathway as a surprisingly complicated young man. Trends Biochem. Sci. 2012, 37, 411–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartron, J.W.; Clemons, W.M., Jr.; Suloway, C.J. The complex process of GETting tail-anchored membrane proteins to the ER. Curr. Opin. Struct. Biol. 2012, 22, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Hassdenteufel, S.; Nguyen, D.; Helms, V.; Lang, S.; Zimmermann, R. ER import of small human presecretory proteins: Components and mechanisms. FEBS Lett. 2019, 593, 2506–2524. [Google Scholar] [CrossRef]
- Jadhav, B.; McKenna, M.; Johnson, N.; High, S.; Sinning, I.; Pool, M.R. Mammalian SRP receptor switches the Sec61 translocase from Sec62 to SRP-dependent translocation. Nat. Commun. 2015, 6, 10133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef]
- Aviram, N.; Schuldiner, M. Embracing the void—How much do we really know about targeting and translocation to the endoplasmic reticulum? Curr. Opin. Cell Biol. 2014, 29, 8–17. [Google Scholar] [CrossRef]
- Johnson, N.; Hassdenteufel, S.; Theis, M.; Paton, A.W.; Paton, J.C.; Zimmermann, R.; High, S. The signal sequence influences post-translational ER translocation at distinct stages. PLoS ONE 2013, 8, e75394. [Google Scholar] [CrossRef] [Green Version]
- Kriegler, T.; Magoulopoulou, A.; Amate Marchal, R.; Hessa, T. Measuring Endoplasmic Reticulum Signal Sequences Translocation Efficiency Using the Xbp1 Arrest Peptide. Cell Chem. Biol. 2018, 25, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, R.M.; Hegde, R.S. Structure of the Sec61 channel opened by a signal sequence. Science 2016, 351, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Hassdenteufel, S.; Schauble, N.; Cassella, P.; Leznicki, P.; Muller, A.; High, S.; Jung, M.; Zimmermann, R. Ca2+-calmodulin inhibits tail-anchored protein insertion into the mammalian endoplasmic reticulum membrane. FEBS Lett. 2011, 585, 3485–3490. [Google Scholar] [CrossRef] [Green Version]
- Casson, J.; McKenna, M.; Hassdenteufel, S.; Aviram, N.; Zimmerman, R.; High, S. Multiple pathways facilitate the biogenesis of mammalian tail-anchored proteins. J. Cell Sci. 2017, 130, 3851–3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, S.F.; Cardani, S.; Maroli, A.; Vitiello, A.; Soffientini, P.; Crespi, A.; Bram, R.F.; Benfante, R.; Borgese, N. Tail-anchored Protein Insertion in Mammals: Function and reciprocal interactions of the two subunits of the TRC40 receptor. J. Biol. Chem. 2016, 291, 15292–15306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coy-Vergara, J.; Rivera-Monroy, J.; Urlaub, H.; Lenz, C.; Schwappach, B. A trap mutant reveals the physiological client spectrum of TRC40. J. Cell Sci. 2019, 132, jcs230094. [Google Scholar] [CrossRef] [Green Version]
- Borgese, N.; Fasana, E. Targeting pathways of C-tail-anchored proteins. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Sakisaka, T. Molecular Machinery for Insertion of Tail-Anchored Membrane Proteins into the Endoplasmic Reticulum Membrane in Mammalian Cells. Mol. Cell 2012, 48, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.; Marques, D.; Funk, C.; Bailer, S.M. Asna1/TRC40 that mediates membrane insertion of tail-anchored proteins is required for efficient release of Herpes simplex virus 1 virions. Virol. J. 2016, 13, 175. [Google Scholar] [CrossRef]
- Lang, S.; Benedix, J.; Fedeles, S.V.; Schorr, S.; Schirra, C.; Schauble, N.; Jalal, C.; Greiner, M.; Hassdenteufel, S.; Tatzelt, J.; et al. Different effects of Sec61alpha, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 2012, 125, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, S.; Pfeffer, S.; Lee, P.H.; Cavalie, A.; Helms, V.; Forster, F.; Zimmermann, R. An Update on Sec61 Channel Functions, Mechanisms, and Related Diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2017, 2, 17002. [Google Scholar] [CrossRef] [PubMed]
- Kalies, K.U.; Romisch, K. Inhibitors of Protein Translocation across the ER Membrane. Traffic 2015, 16, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Van Puyenbroeck, V.; Vermeire, K. Inhibitors of protein translocation across membranes of the secretory pathway: Novel antimicrobial and anticancer agents. Cell. Mol. Life Sci. CMLS 2018, 75, 1541–1558. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, R.; Eyrisch, S.; Ahmad, M.; Helms, V. Protein translocation across the ER membrane. Biochim. Biophys. Acta 2011, 1808, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Matlack, K.E.S.; Mothes, W.; Rapoport, T.A. Protein Translocation: Tunnel Vision. Cell 1998, 92, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Luesch, H.; Paavilainen, V.O. Natural products as modulators of eukaryotic protein secretion. Nat. Prod. Rep. 2020, 37, 717–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakkaraju, A.K.; Thankappan, R.; Mary, C.; Garrison, J.L.; Taunton, J.; Strub, K. Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Mol. Biol. Cell 2012, 23, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Denks, K.; Vogt, A.; Sachelaru, I.; Petriman, N.A.; Kudva, R.; Koch, H.G. The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol. Membr. Biol. 2014, 31, 58–84. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Zimmermann, R.; Forster, F. Organization of the native ribosome-translocon complex at the mammalian endoplasmic reticulum membrane. Biochim. Biophys. Acta 2016, 1860, 2122–2129. [Google Scholar] [CrossRef]
- Luirink, J.; Sinning, I. SRP-mediated protein targeting: Structure and function revisited. Biochim. Biophys. Acta 2004, 1694, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, A.K.; Mary, C.; Scherrer, A.; Johnson, A.E.; Strub, K. SRP keeps polypeptides translocation-competent by slowing translation to match limiting ER-targeting sites. Cell 2008, 133, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Wild, K.; Juaire, K.D.; Soni, K.; Shanmuganathan, V.; Hendricks, A.; Segnitz, B.; Beckmann, R.; Sinning, I. Reconstitution of the human SRP system and quantitative and systematic analysis of its ribosome interactions. Nucleic Acids Res. 2019, 47, 3184–3196. [Google Scholar] [CrossRef]
- Dudek, J.; Pfeffer, S.; Lee, P.H.; Jung, M.; Cavalie, A.; Helms, V.; Forster, F.; Zimmermann, R. Protein transport into the human endoplasmic reticulum. J. Mol. Biol. 2015, 427, 1159–1175. [Google Scholar] [CrossRef]
- Nyathi, Y.; Wilkinson, B.M.; Pool, M.R. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2392–2402. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Hegde, R.S. Structures of the scanning and engaged states of the mammalian SRP-ribosome complex. eLife 2015, 4, e07975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorhees, R.M.; Fernandez, I.S.; Scheres, S.H.; Hegde, R.S. Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 2014, 157, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, R.M.; Hegde, R.S. Toward a structural understanding of co-translational protein translocation. Curr. Opin. Cell Biol. 2016, 41, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Schaffer, M.; Ng, B.G.; Albert, S.; Plitzko, J.M.; Baumeister, W.; Zimmermann, R.; Freeze, H.H.; Engel, B.D.; et al. Dissecting the molecular organization of the translocon-associated protein complex. Nat. Commun. 2017, 8, 14516. [Google Scholar] [CrossRef]
- Braunger, K.; Pfeffer, S.; Shrimal, S.; Gilmore, R.; Berninghausen, O.; Mandon, E.C.; Becker, T.; Forster, F.; Beckmann, R. Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 2018, 360, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Wang, T.; Zhao, G.; Kovach, A.; Li, H. The atomic structure of a eukaryotic oligosaccharyltransferase complex. Nature 2018, 555, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Forster, F. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaci, A.M.; Steigenberger, B.; Telles de Souza, P.C.; Tamara, S.; Grollers-Mulderij, M.; Ogrissek, P.; Marrink, S.J.; Scheltema, R.A.; Forster, F. Structure of the human signal peptidase complex reveals the determinants for signal peptide cleavage. Mol. Cell 2021, 81, 3934–3948. [Google Scholar] [CrossRef] [PubMed]
- Auclair, S.M.; Bhanu, M.K.; Kendall, D.A. Signal peptidase I: Cleaving the way to mature proteins. Protein Sci. 2012, 21, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Jomaa, A.; Jaskolowski, M.; Yang, C.I.; Ban, N.; Shan, S.O. The molecular mechanism of cotranslational membrane protein recognition and targeting by SecA. Nat. Struct. Mol. Biol. 2019, 26, 919–929. [Google Scholar] [CrossRef]
- Hassdenteufel, S.; Sicking, M.; Schorr, S.; Aviram, N.; Fecher-Trost, C.; Schuldiner, M.; Jung, M.; Zimmermann, R.; Lang, S. hSnd2 protein represents an alternative targeting factor to the endoplasmic reticulum in human cells. FEBS Lett. 2017, 591, 3211–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strzyz, P. The third route to the ER. Nat. Rev. Mol. Cell Biol. 2017, 18, 3. [Google Scholar] [CrossRef]
- Ast, T.; Schuldiner, M. All roads lead to Rome (but some may be harder to travel): SRP-independent translocation into the endoplasmic reticulum. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 273–288. [Google Scholar] [CrossRef]
- Egea, P.F.; Stroud, R.M.; Walter, P. Targeting proteins to membranes: Structure of the signal recognition particle. Curr. Opin. Struct. Biol. 2005, 15, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Halic, M.; Beckmann, R. The signal recognition particle and its interactions during protein targeting. Curr. Opin. Struct. Biol. 2005, 15, 116–125. [Google Scholar] [CrossRef]
- Hwang Fu, Y.H.; Chandrasekar, S.; Lee, J.H.; Shan, S.O. A molecular recognition feature mediates ribosome-induced SRP-receptor assembly during protein targeting. J. Cell Biol. 2019, 218, 3307–3319. [Google Scholar] [CrossRef] [Green Version]
- Kellogg, M.K.; Miller, S.C.; Tikhonova, E.B.; Karamyshev, A.L. SRPassing Co-translational Targeting: The Role of the Signal Recognition Particle in Protein Targeting and mRNA Protection. Int. J. Mol. Sci. 2021, 22, 6284. [Google Scholar] [CrossRef]
- Saraogi, I.; Shan, S.O. Molecular mechanism of co-translational protein targeting by the signal recognition particle. Traffic 2011, 12, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Burbaum, L.; Unverdorben, P.; Pech, M.; Chen, Y.; Zimmermann, R.; Beckmann, R.; Forster, F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 8403. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Brandt, F.; Hrabe, T.; Lang, S.; Eibauer, M.; Zimmermann, R.; Forster, F. Structure and 3D arrangement of endoplasmic reticulum membrane-associated ribosomes. Structure 2012, 20, 1508–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, B.J.; Devaraneni, P.K.; Yang, Z.; David, L.L.; Skach, W.R. Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 2015, 58, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.; Stutz, R.; Schorr, S.; Lang, S.; Pfeffer, S.; Freeze, H.H.; Forster, F.; Helms, V.; Dudek, J.; Zimmermann, R. Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat. Commun. 2018, 9, 3765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fons, R.D.; Bogert, B.A.; Hegde, R.S. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 2003, 160, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.C.; Lerner, M.; Nguyen, D.; Pfeffer, S.; Dudek, J.; Forster, F.; Helms, V.; Lang, S.; Zimmermann, R. TRAM1 protein may support ER protein import by modulating the phospholipid bilayer near the lateral gate of the Sec61-channel. Channels 2020, 14, 28–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, S.; Vilar, M.; Martinez-Gil, L.; Johnson, A.E.; Mingarro, I. Membrane insertion and topology of the translocating chain-associating membrane protein (TRAM). J. Mol. Biol. 2011, 406, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Russo, A. Understanding the mammalian TRAP complex function(s). Open Biol. 2020, 10, 190244. [Google Scholar] [CrossRef]
- Klein, W.; Rutz, C.; Eckhard, J.; Provinciael, B.; Specker, E.; Neuenschwander, M.; Kleinau, G.; Scheerer, P.; von Kries, J.P.; Nazare, M.; et al. Use of a sequential high throughput screening assay to identify novel inhibitors of the eukaryotic SRP-Sec61 targeting/translocation pathway. PLoS ONE 2018, 13, e0208641. [Google Scholar] [CrossRef]
- Hegde, R.S.; Voigt, S.; Rapoport, T.A.; Lingappa, V.R. TRAM Regulates the Exposure of Nascent Secretory Proteins to the Cytosol during Translocation into the Endoplasmic Reticulum. Cell 1998, 92, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Voigt, S.; Jungnickel, B.; Hartmann, E.; Rapoport, T.A. Signal sequence-dependent function of the TRAM protein during early phases of protein transport across the endoplasmic reticulum membrane. J. Cell Biol. 1996, 134, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Alder, N.N.; Shen, Y.; Brodsky, J.L.; Hendershot, L.M.; Johnson, A.E. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 389–399. [Google Scholar] [CrossRef]
- Alfaro-Valdés, H.; Burgos-Bravo, F.; Casanova-Morales, N.; Quiroga-Roger, D.; Wilson, C. Mechanical Properties of Chaperone BiP, the Master Regulator of the Endoplasmic Reticulum; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Law, D.T.; Williams, D.B. Binding protein BiP is required for translocation of secretory proteins into the endoplasmic reticulum in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1991, 88, 1565–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matlack, K.E.; Misselwitz, B.; Plath, K.; Rapoport, T.A. BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell 1999, 97, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [Green Version]
- Comyn, S.A.; Chan, G.T.; Mayor, T. False start: Cotranslational protein ubiquitination and cytosolic protein quality control. J. Proteom. 2014, 100, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Vazquez-Laslop, N.; Mankin, A.S. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell 2012, 151, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verchot, J. The ER quality control and ER associated degradation machineries are vital for viral pathogenesis. Front. Plant Sci. 2014, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Sicking, M.; Lang, S.; Bochen, F.; Roos, A.; Drenth, J.P.H.; Zakaria, M.; Zimmermann, R.; Linxweiler, M. Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex. Cells 2021, 10, 1036. [Google Scholar] [CrossRef]
- Lu, Z.; Zhou, L.; Killela, P.; Rasheed, A.B.; Di, C.; Poe, W.E.; McLendon, R.E.; Bigner, D.D.; Nicchitta, C.; Yan, H. Glioblastoma proto-oncogene SEC61gamma is required for tumor cell survival and response to endoplasmic reticulum stress. Cancer Res. 2009, 69, 9105–9111. [Google Scholar] [CrossRef] [Green Version]
- Jung, V.; Kindich, R.; Kamradt, J.; Jung, M.; Müller, M.; Schulz, W.A.; Engers, R.; Unteregger, G.; Stöckle, M.; Zimmermann, R.; et al. Genomic and expression analysis of the 3q25-q26 amplification unit reveals TLOC1/SEC62 as a probable target gene in prostate cancer. Mol. Cancer Res. 2006, 4, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Greiner, M.; Kreutzer, B.; Jung, V.; Grobholz, R.; Hasenfus, A.; Stöhr, R.F.; Tornillo, L.; Dudek, J.; Stöckle, M.; Unteregger, G.; et al. Silencing of the SEC62 gene inhibits migratory and invasive potential of various tumor cells. Int. J. Cancer 2011, 128, 2284–2295. [Google Scholar] [CrossRef]
- Linxweiler, M.; Linxweiler, J.; Barth, M.; Benedix, J.; Jung, V.; Kim, Y.J.; Bohle, R.M.; Zimmermann, R.; Greiner, M. Sec62 bridges the gap from 3q amplification to molecular cell biology in non-small cell lung cancer. Am. J. Pathol. 2012, 180, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.S.L.; Pfohler, C.; Wahl, M.; Bochen, F.; Korner, S.; Kuhn, J.P.; Bozzato, A.; Schick, B.; Linxweiler, M. Expression of SEC62 Oncogene in Benign, Malignant and Borderline Melanocytic Tumors-Unmasking the Wolf in Sheep’s Clothing? Cancers 2021, 13, 1645. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Jiang, X.; Wang, J.; Sang, Z.; Guo, L.; Yin, G.; Wang, Y. SEC61G is upregulated and required for tumor progression in human kidney cancer. Mol. Med. Rep. 2021, 23, 427. [Google Scholar] [CrossRef] [PubMed]
- Witham, C.M.; Paxman, A.L.; Baklous, L.; Steuart, R.F.L.; Schulz, B.L.; Mousley, C.J. Cancer associated mutations in Sec61γ alter the permeability of the ER translocase. PLoS Genet. 2021, 17, e1009780. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S.; Bagchi, P.; Cunningham, C.N.; Tsai, B. Opportunistic intruders: How viruses orchestrate ER functions to infect cells. Nat. Rev. Microbiol. 2016, 14, 407–420. [Google Scholar] [CrossRef]
- Vermeire, K.; Schols, D. Specific CD4 down-modulating compounds with potent anti-HIV activity. J. Leukoc. Biol. 2003, 74, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Moshkina, N.; Fenouil, R.; Gardner, T.J.; Aguirre, S.; Shah, P.S.; Zhao, N.; Manganaro, L.; Hultquist, J.F.; Noel, J.; et al. Targeting Viral Proteostasis Limits Influenza Virus, HIV, and Dengue Virus Infection. Immunity 2016, 44, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Gillece, P.; Pilon, M.; Römisch, K. The protein translocation channel mediates glycopeptide export across the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2000, 97, 4609–4614. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, K.; Bell, T.W.; Choi, H.-J.; Jin, Q.; Samala, M.F.; Sodoma, A.; De Clercq, E.; Schols, D. The Anti-HIV Potency of Cyclotriazadisulfonamide Analogs Is Directly Correlated with Their Ability to Down-Modulate the CD4 Receptor. Mol. Pharmacol. 2003, 63, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeire, K.; Zhang, Y.; Princen, K.; Hatse, S.; Samala, M.F.; Dey, K.; Choi, H.J.; Ahn, Y.; Sodoma, A.; Snoeck, R.; et al. CADA inhibits human immunodeficiency virus and human herpesvirus 7 replication by down-modulation of the cellular CD4 receptor. Virology 2002, 302, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Foster, C.A.; Dreyfuss, M.; Mandak, B.; Meingassner, J.G.; Naegeli, H.U.; Nussbaumer, A.; Oberer, L.; Scheel, G.; Swoboda, E.-M. Pharmacological Modulation of Endothelial Cell-associated Adhesion Molecule Expression: Implications for Future Treatment of Dermatological Diseases. J. Dermatol. 1994, 21, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Hommel, U.; Weber, H.P.; Oberer, L.; Naegeli, H.U.; Oberhauser, B.; Foster, C.A. The 3D-structure of a natural inhibitor of cell adhesion molecule expression. FEBS Lett. 1996, 379, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Boger, D.L.; Chen, Y.; Foster, C.A. Synthesis and evaluation of aza HUN-7293. Bioorg. Med. Chem. Lett. 2000, 10, 1741–1744. [Google Scholar] [CrossRef]
- Chen, Y.; Bilban, M.; Foster, C.A.; Boger, D.L. Solution-phase parallel synthesis of a pharmacophore library of HUN-7293 analogues: A general chemical mutagenesis approach to defining structure-function properties of naturally occurring cyclic (depsi)peptides. J. Am. Chem. Soc. 2002, 124, 5431–5440. [Google Scholar] [CrossRef]
- Harant, H.; Lettner, N.; Hofer, L.; Oberhauser, B.; de Vries, J.E.; Lindley, I.J. The translocation inhibitor CAM741 interferes with vascular cell adhesion molecule 1 signal peptide insertion at the translocon. J. Biol. Chem. 2006, 281, 30492–30502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, J.L.; Kunkel, E.J.; Hegde, R.S.; Taunton, J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 2005, 436, 285–289. [Google Scholar] [CrossRef]
- Klein, W.; Westendorf, C.; Schmidt, A.; Conill-Cortes, M.; Rutz, C.; Blohs, M.; Beyermann, M.; Protze, J.; Krause, G.; Krause, E.; et al. Defining a conformational consensus motif in cotransin-sensitive signal sequences: A proteomic and site-directed mutagenesis study. PLoS ONE 2015, 10, e0120886. [Google Scholar] [CrossRef]
- Schreiner, E.P.; Kern, M.; Steck, A.; Foster, C.A. Synthesis of ether analogues derived from HUN-7293 and evaluation as inhibitors of VCAM-1 expression. Bioorg. Med. Chem. Lett. 2004, 14, 5003–5006. [Google Scholar] [CrossRef]
- Besemer, J.; Harant, H.; Wang, S.; Oberhauser, B.; Marquardt, K.; Foster, C.A.; Schreiner, E.P.; de Vries, J.E.; Dascher-Nadel, C.; Lindley, I.J. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature 2005, 436, 290–293. [Google Scholar] [CrossRef]
- Harant, H.; Wolff, B.; Schreiner, E.P.; Oberhauser, B.; Hofer, L.; Lettner, N.; Maier, S.; de Vries, J.E.; Lindley, I.J. Inhibition of vascular endothelial growth factor cotranslational translocation by the cyclopeptolide CAM741. Mol. Pharm. 2007, 71, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, C.; Schmidt, A.; Coin, I.; Furkert, J.; Ridelis, I.; Zampatis, D.; Rutz, C.; Wiesner, B.; Rosenthal, W.; Beyermann, M.; et al. Inhibition of biosynthesis of human endothelin B receptor by the cyclodepsipeptide cotransin. J. Biol. Chem. 2011, 286, 35588–35600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maifeld, S.V.; MacKinnon, A.L.; Garrison, J.L.; Sharma, A.; Kunkel, E.J.; Hegde, R.S.; Taunton, J. Secretory protein profiling reveals TNF-alpha inactivation by selective and promiscuous Sec61 modulators. Chem. Biol. 2011, 18, 1082–1088. [Google Scholar] [CrossRef] [Green Version]
- MacKinnon, A.L.; Paavilainen, V.O.; Sharma, A.; Hegde, R.S.; Taunton, J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. eLife 2014, 3, e01483. [Google Scholar] [CrossRef] [Green Version]
- MacKinnon, A.L.; Garrison, J.L.; Hegde, R.S.; Taunton, J. Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J. Am. Chem. Soc. 2007, 129, 14560–14561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-alpha therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Saenz, A.; Sandhu, M.; Carrasco, Y.; Maglathlin, R.L.; Taunton, J.; Moasser, M.M. Targeting HER3 by interfering with its Sec61-mediated cotranslational insertion into the endoplasmic reticulum. Oncogene 2015, 34, 5288–5294. [Google Scholar] [CrossRef] [Green Version]
- Junne, T.; Wong, J.; Studer, C.; Aust, T.; Bauer, B.W.; Beibel, M.; Bhullar, B.; Bruccoleri, R.; Eichenberger, J.; Estoppey, D.; et al. Decatransin, a new natural product inhibiting protein translocation at the Sec61/SecYEG translocon. J. Cell Sci. 2015, 128, 1217–1229. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.C.; Chen, Z.; Jiang, Y.; Akare, S.; Kolber-Simonds, D.; Condon, K.; Agoulnik, S.; Tendyke, K.; Shen, Y.; Wu, K.M.; et al. Apratoxin A Shows Novel Pancreas-Targeting Activity through the Binding of Sec 61. Mol. Cancer Ther. 2016, 15, 1208–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paatero, A.O.; Kellosalo, J.; Dunyak, B.M.; Almaliti, J.; Gestwicki, J.E.; Gerwick, W.H.; Taunton, J.; Paavilainen, V.O. Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem. Biol. 2016, 23, 561–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hau, A.M.; Greenwood, J.A.; Lohr, C.V.; Serrill, J.D.; Proteau, P.J.; Ganley, I.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A induces mTOR-independent autophagy and cell death in human glioblastoma cells. PLoS ONE 2013, 8, e65250. [Google Scholar] [CrossRef] [Green Version]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Izpisúa Belmonte, J.C.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef]
- Liu, Y.; Law, B.K.; Luesch, H. Apratoxin a reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol. Pharm. 2009, 76, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Serrill, J.D.; Humphreys, I.R.; Tan, M.; McPhail, K.L.; Ganley, I.G.; Ishmael, J.E. ATG5 Promotes Death Signaling in Response to the Cyclic Depsipeptides Coibamide A and Apratoxin A. Mar. Drugs 2018, 16, 77. [Google Scholar] [CrossRef] [Green Version]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barria, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef] [Green Version]
- Serrill, J.D.; Wan, X.; Hau, A.M.; Jang, H.S.; Coleman, D.J.; Indra, A.K.; Alani, A.W.; McPhail, K.L.; Ishmael, J.E. Coibamide A, a natural lariat depsipeptide, inhibits VEGFA/VEGFR2 expression and suppresses tumor growth in glioblastoma xenografts. Investig. New Drugs 2016, 34, 24–40. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Tan, L.T.; Sitachitta, N. Nitrogen-containing metabolites from marine cyanobacteria. Alkaloids. Chem. Biol. 2001, 57, 75–184. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, D.; Roy, A.; Kar, P.; Mutanda, T.; Anandraj, A. Cyanobacterial metabolites as promising drug leads against the M(pro) and PL(pro) of SARS-CoV-2: An in silico analysis. J. Biomol. Struct. Dyn. 2021, 39, 6218–6230. [Google Scholar] [CrossRef] [PubMed]
- Tranter, D.; Paatero, A.O.; Kawaguchi, S.; Kazemi, S.; Serrill, J.D.; Kellosalo, J.; Vogel, W.K.; Richter, U.; Mattos, D.R.; Wan, X.; et al. Coibamide A Targets Sec61 to Prevent Biogenesis of Secretory and Membrane Proteins. ACS Chem. Biol. 2020, 15, 2125–2136. [Google Scholar] [CrossRef]
- Shi, W.; Lu, D.; Wu, C.; Li, M.; Ding, Z.; Li, Y.; Chen, B.; Lin, X.; Su, W.; Shao, X.; et al. Coibamide A kills cancer cells through inhibiting autophagy. Biochem. Biophys. Res. Commun. 2021, 547, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.; Simmonds, R. Pleiotropic molecular effects of the Mycobacterium ulcerans virulence factor mycolactone underlying the cell death and immunosuppression seen in Buruli ulcer. Biochem. Soc. Trans. 2014, 42, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, J.D.; Paatero, A.O.; Wei, J.; Yewdell, J.W.; Guenin-Mace, L.; Van Haver, D.; Impens, F.; Pietrosemoli, N.; Paavilainen, V.O.; Demangel, C. Proteomics Reveals Scope of Mycolactone-mediated Sec61 Blockade and Distinctive Stress Signature. Mol. Cell. Proteom. MCP 2018, 17, 1750–1765. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.; Simmonds, R.E.; High, S. Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J. Cell Sci. 2016, 129, 1404–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demangel, C.; High, S. Sec61 blockade by mycolactone: A central mechanism in Buruli ulcer disease. Biol. Cell 2018, 110, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Mve-Obiang, A.; Lee, R.E.; Umstot, E.S.; Trott, K.A.; Grammer, T.C.; Parker, J.M.; Ranger, B.S.; Grainger, R.; Mahrous, E.A.; Small, P.L.C. A newly discovered mycobacterial pathogen isolated from laboratory colonies of Xenopus species with lethal infections produces a novel form of mycolactone, the Mycobacterium ulcerans macrolide toxin. Infect. Immun. 2005, 73, 3307–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenin-Macé, L.; Ruf, M.-T.; Pluschke, G.; Demangel, C. Mycolactone: More than Just a Cytotoxin. In Buruli Ulcer: Mycobacterium Ulcerans Disease; Pluschke, G., Röltgen, K., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 117–134. [Google Scholar] [CrossRef] [Green Version]
- George, K.M.; Chatterjee, D.; Gunawardana, G.; Welty, D.; Hayman, J.; Lee, R.; Small, P.L. Mycolactone: A polyketide toxin from Mycobacterium ulcerans required for virulence. Science 1999, 283, 854–857. [Google Scholar] [CrossRef] [Green Version]
- Guenin-Macé, L.; Baron, L.; Chany, A.C.; Tresse, C.; Saint-Auret, S.; Jönsson, F.; Le Chevalier, F.; Bruhns, P.; Bismuth, G.; Hidalgo-Lucas, S.; et al. Shaping mycolactone for therapeutic use against inflammatory disorders. Sci. Transl. Med. 2015, 7, 289ra285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, L.; Paatero, A.O.; Morel, J.D.; Impens, F.; Guenin-Mace, L.; Saint-Auret, S.; Blanchard, N.; Dillmann, R.; Niang, F.; Pellegrini, S.; et al. Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J. Exp. Med. 2016, 213, 2885–2896. [Google Scholar] [CrossRef] [PubMed]
- Ogbechi, J.; Hall, B.S.; Sbarrato, T.; Taunton, J.; Willis, A.E.; Wek, R.C.; Simmonds, R.E. Inhibition of Sec61-dependent translocation by mycolactone uncouples the integrated stress response from ER stress, driving cytotoxicity via translational activation of ATF4. Cell Death Dis. 2018, 9, 397. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.; Simmonds, R.E.; High, S. Mycolactone reveals the substrate-driven complexity of Sec61-dependent transmembrane protein biogenesis. J. Cell Sci. 2017, 130, 1307–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerard, S.F.; Hall, B.S.; Zaki, A.M.; Corfield, K.A.; Mayerhofer, P.U.; Costa, C.; Whelligan, D.K.; Biggin, P.C.; Simmonds, R.E.; Higgins, M.K. Structure of the Inhibited State of the Sec Translocon. Mol. Cell 2020, 79, 406–415.e7. [Google Scholar] [CrossRef] [PubMed]
- Zong, G.; Barber, E.; Aljewari, H.; Zhou, J.; Hu, Z.; Du, Y.; Shi, W.Q. Total Synthesis and Biological Evaluation of Ipomoeassin F and Its Unnatural 11R-Epimer. J. Org. Chem. 2015, 80, 9279–9291. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Norris, A.; Wisse, J.H.; Miller, J.S.; Evans, R.; Kingston, D.G. Ipomoeassin F, a new cytotoxic macrocyclic glycoresin from the leaves of Ipomoea squamosa from the Suriname rainforest. Nat. Prod. Res. 2007, 21, 872–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, G.; Hu, Z.; O’Keefe, S.; Tranter, D.; Iannotti, M.J.; Baron, L.; Hall, B.; Corfield, K.; Paatero, A.O.; Henderson, M.J.; et al. Ipomoeassin F Binds Sec61alpha to Inhibit Protein Translocation. J. Am. Chem. Soc. 2019, 141, 8450–8461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keefe, S.; Roboti, P.; Duah, K.B.; Zong, G.; Schneider, H.; Shi, W.Q.; High, S. Ipomoeassin-F inhibits the in vitro biogenesis of the SARS-CoV-2 spike protein and its host cell membrane receptor. J. Cell Sci. 2021, 134, jcs257758. [Google Scholar] [CrossRef]
- Roboti, P.; O’Keefe, S.; Duah, K.B.; Shi, W.Q.; High, S. Ipomoeassin-F disrupts multiple aspects of secretory protein biogenesis. Sci. Rep. 2021, 11, 11562. [Google Scholar] [CrossRef]
- Zong, G.; Hu, Z.; Duah, K.B.; Andrews, L.E.; Zhou, J.; O’Keefe, S.; Whisenhunt, L.; Shim, J.S.; Du, Y.; High, S.; et al. Ring Expansion Leads to a More Potent Analogue of Ipomoeassin F. J. Org. Chem. 2020, 85, 16226–16235. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, S.; Kawaguchi, S.; Badr, C.E.; Mattos, D.R.; Ruiz-Saenz, A.; Serrill, J.D.; Moasser, M.M.; Dolan, B.P.; Paavilainen, V.O.; Oishi, S.; et al. Targeting of HER/ErbB family proteins using broad spectrum Sec61 inhibitors coibamide A and apratoxin A. Biochem. Pharm. 2021, 183, 114317. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Pauwels, E.; Rutz, C.; Provinciael, B.; Stroobants, J.; Schols, D.; Hartmann, E.; Krause, E.; Stephanowitz, H.; Schulein, R.; Vermeire, K. A Proteomic Study on the Membrane Protein Fraction of T Cells Confirms High Substrate Selectivity for the ER Translocation Inhibitor Cyclotriazadisulfonamide. Mol. Cell. Proteom. MCP 2021, 20, 100144. [Google Scholar] [CrossRef]
- Claeys, E.; Pauwels, E.; Humblet-Baron, S.; Provinciael, B.; Schols, D.; Waer, M.; Sprangers, B.; Vermeire, K. Small Molecule Cyclotriazadisulfonamide Abrogates the Upregulation of the Human Receptors CD4 and 4-1BB and Suppresses In Vitro Activation and Proliferation of T Lymphocytes. Front. Immunol. 2021, 12, 1340. [Google Scholar] [CrossRef] [PubMed]
- Van Puyenbroeck, V.; Claeys, E.; Schols, D.; Bell, T.W.; Vermeire, K. A Proteomic Survey Indicates Sortilin as a Secondary Substrate of the ER Translocation Inhibitor Cyclotriazadisulfonamide (CADA). Mol. Cell. Proteom. MCP 2017, 16, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Cross, B.C.; McKibbin, C.; Callan, A.C.; Roboti, P.; Piacenti, M.; Rabu, C.; Wilson, C.M.; Whitehead, R.; Flitsch, S.L.; Pool, M.R.; et al. Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J. Cell Sci. 2009, 122, 4393–4400. [Google Scholar] [CrossRef] [Green Version]
- Lowe, E.; Fan, R.; Jiang, J.; Johnson, H.; Kirk, C.; McMinn, D.; Millare, B.; Muchamuel, T.; Qian, Y.; Tuch, B.; et al. Preclinical evaluation of KZR-261, a novel small molecule inhibitor of Sec61. J. Clin. Oncol. 2020, 38, 3582. [Google Scholar] [CrossRef]
- Vermeire, K.; Princen, K.; Hatse, S.; De Clercq, E.; Dey, K.; Bell, T.W.; Schols, D. CADA, a novel CD4-targeted HIV inhibitor, is synergistic with various anti-HIV drugs in vitro. AIDS 2004, 18, 2115–2125. [Google Scholar] [CrossRef]
- Berger, K.; Pauwels, E.; Parkinson, G.; Landberg, G.; Le, T.; Demillo, V.G.; Lumangtad, L.A.; Jones, D.E.; Islam, M.A.; Olsen, R.; et al. Reduction of Progranulin-Induced Breast Cancer Stem Cell Propagation by Sortilin-Targeting Cyclotriazadisulfonamide (CADA) Compounds. J. Med. Chem. 2021, 64, 12865–12876. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.W.; Anugu, S.; Bailey, P.; Catalano, V.J.; Dey, K.; Drew, M.G.; Duffy, N.H.; Jin, Q.; Samala, M.F.; Sodoma, A.; et al. Synthesis and structure-activity relationship studies of CD4 down-modulating cyclotriazadisulfonamide (CADA) analogues. J. Med. Chem. 2006, 49, 1291–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, R.; Van Puyenbroeck, V.; Pflug, N.C.; Sama, A.; Ali, R.; Schols, D.; Vermeire, K.; Bell, T.W. Tuning Side Arm Electronics in Unsymmetrical Cyclotriazadisulfonamide (CADA) Endoplasmic Reticulum (ER) Translocation Inhibitors to Improve their Human Cluster of Differentiation 4 (CD4) Receptor Down-Modulating Potencies. J. Med. Chem. 2016, 59, 2633–2647. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, K.; Lisco, A.; Grivel, J.C.; Scarbrough, E.; Dey, K.; Duffy, N.; Margolis, L.; Bell, T.W.; Schols, D. Design and cellular kinetics of dansyl-labeled CADA derivatives with anti-HIV and CD4 receptor down-modulating activity. Biochem. Pharm. 2007, 74, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Lumangtad, L.A.; Claeys, E.; Hamal, S.; Intasiri, A.; Basrai, C.; Yen-Pon, E.; Beenfeldt, D.; Vermeire, K.; Bell, T.W. Syntheses and anti-HIV and human cluster of differentiation 4 (CD4) down-modulating potencies of pyridine-fused cyclotriazadisulfonamide (CADA) compounds. Bioorg. Med. Chem. 2020, 28, 115816. [Google Scholar] [CrossRef] [PubMed]
- Demillo, V.G.; Goulinet-Mateo, F.; Kim, J.; Schols, D.; Vermeire, K.; Bell, T.W. Unsymmetrical cyclotriazadisulfonamide (CADA) compounds as human CD4 receptor down-modulating agents. J. Med. Chem. 2011, 54, 5712–5721. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.W.; Demillo, V.G.; Schols, D.; Vermeire, K. Improving potencies and properties of CD4 down-modulating CADA analogs. Expert Opin. Drug Discov. 2012, 7, 39–48. [Google Scholar] [CrossRef]
- Vermeire, K.; Bell, T.W.; Van Puyenbroeck, V.; Giraut, A.; Noppen, S.; Liekens, S.; Schols, D.; Hartmann, E.; Kalies, K.U.; Marsh, M. Signal peptide-binding drug as a selective inhibitor of co-translational protein translocation. PLoS Biol. 2014, 12, e1002011. [Google Scholar] [CrossRef] [Green Version]
- Van Puyenbroeck, V.; Pauwels, E.; Provinciael, B.; Bell, T.W.; Schols, D.; Kalies, K.U.; Hartmann, E.; Vermeire, K. Preprotein signature for full susceptibility to the co-translational translocation inhibitor cyclotriazadisulfonamide. Traffic 2020, 21, 250–264. [Google Scholar] [CrossRef]
- Fiebiger, E.; Hirsch, C.; Vyas, J.M.; Gordon, E.; Ploegh, H.L.; Tortorella, D. Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol. Biol. Cell 2004, 15, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, L.; Ye, Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 2008, 283, 7445–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.M. Could a Common Mechanism of Protein Degradation Impairment Underlie Many Neurodegenerative Diseases? J. Exp. Neurosci. 2018, 12, 1179069518794675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shinkre, B.A.; Lee, J.G.; Weniger, M.A.; Liu, Y.; Chen, W.; Wiestner, A.; Trenkle, W.C.; Ye, Y. The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group. PLoS ONE 2010, 5, e15479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, S.; Erdmann, F.; Jung, M.; Wagner, R.; Cavalie, A.; Zimmermann, R. Sec61 complexes form ubiquitous ER Ca2+ leak channels. Channels 2011, 5, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamayun, I.; O’Keefe, S.; Pick, T.; Klein, M.C.; Nguyen, D.; McKibbin, C.; Piacenti, M.; Williams, H.M.; Flitsch, S.L.; Whitehead, R.C.; et al. Eeyarestatin Compounds Selectively Enhance Sec61-Mediated Ca(2+) Leakage from the Endoplasmic Reticulum. Cell Chem. Biol. 2019, 26, 571–583.e6. [Google Scholar] [CrossRef] [Green Version]
- Ding, R.; Zhang, T.; Xie, J.; Williams, J.; Ye, Y.; Chen, L. Eeyarestatin I derivatives with improved aqueous solubility. Bioorg. Med. Chem. Lett. 2016, 26, 5177–5181. [Google Scholar] [CrossRef]
- Du, R.; Sullivan, D.K.; Azizian, N.G.; Liu, Y.; Li, Y. Inhibition of ERAD synergizes with FTS to eradicate pancreatic cancer cells. BMC Cancer 2021, 21, 237. [Google Scholar] [CrossRef] [PubMed]
- Aletrari, M.O.; McKibbin, C.; Williams, H.; Pawar, V.; Pietroni, P.; Lord, J.M.; Flitsch, S.L.; Whitehead, R.; Swanton, E.; High, S.; et al. Eeyarestatin 1 interferes with both retrograde and anterograde intracellular trafficking pathways. PLoS ONE 2011, 6, e22713. [Google Scholar] [CrossRef] [Green Version]
- McKibbin, C.; Mares, A.; Piacenti, M.; Williams, H.; Roboti, P.; Puumalainen, M.; Callan, A.C.; Lesiak-Mieczkowska, K.; Linder, S.; Harant, H.; et al. Inhibition of protein translocation at the endoplasmic reticulum promotes activation of the unfolded protein response. Biochem. J. 2012, 442, 639–648. [Google Scholar] [CrossRef]
- Auner, H.W.; Moody, A.M.; Ward, T.H.; Kraus, M.; Milan, E.; May, P.; Chaidos, A.; Driessen, C.; Cenci, S.; Dazzi, F.; et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS ONE 2013, 8, e74415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenhuis, M.; Koningstein, G.M.; Oswald, J.; Pick, T.; O’Keefe, S.; Koch, H.G.; Cavalie, A.; Whitehead, R.C.; Swanton, E.; High, S.; et al. Eeyarestatin 24 impairs SecYEG-dependent protein trafficking and inhibits growth of clinically relevant pathogens. Mol. Microbiol. 2021, 115, 28–40. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Substrate Selectivity 1 | Active Concentration 2 | Sec61α Resistance Conferring Mutations | Reference |
|---|---|---|---|---|
| HUN-7293 | VCAM-1, ICAM-1, E-selectin | IC50 1–24 nM | Undefined | [104,107] |
| CAM741 | VCAM-1, VEGF | IC50 5–200 nM | Undefined | [105,108,111,113,118] |
| Cotransin | VCAM-1, P-selectin, Angiotensinogen, β-lactamase, CRF1, ETBR, AQP2, HER-3, TNF-α | IC50 0.5–5 µM | R66, G80, S82, M136 | [105,109,110,114,115,116,119] |
| Decatransin | Broad-spectrum | CC50 30–40 nM | I41, D60, M65, R66, S71, G80, S82, M136 | [120] |
| Apratoxin A | Broad-spectrum | CC50 13 nM | T86, Y131 | [122,155] |
| Coibamide A | Broad-spectrum | CC50 10–100 nM | S71 | [135,155,156] |
| Mycolactone | Broad-spectrum | IC50 3–12 nM | R66, S71, G80, S82, T86, Q127, M136 | [137,138,139,140,141,142,145] |
| Ipomoeassin F | Broad-spectrum | IC50 50–120 µM 3 | R66, S82 | [151,152,153] |
| CADA | huCD4, SORT, CD137, DNAJC3, PTK7, ERLEC1 | IC50 0.2–2 µM | Undefined | [103,157,158,159] |
| Eeyarestatin | Broad-spectrum | IC50 70–200 µM 3 | Undefined | [160] |
| KZR-261/834 | Broad-spectrum | IC50 nanomolar range | Undefined | [161] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauwels, E.; Schülein, R.; Vermeire, K. Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway. Int. J. Mol. Sci. 2021, 22, 12007. https://doi.org/10.3390/ijms222112007
Pauwels E, Schülein R, Vermeire K. Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway. International Journal of Molecular Sciences. 2021; 22(21):12007. https://doi.org/10.3390/ijms222112007
Chicago/Turabian StylePauwels, Eva, Ralf Schülein, and Kurt Vermeire. 2021. "Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway" International Journal of Molecular Sciences 22, no. 21: 12007. https://doi.org/10.3390/ijms222112007
APA StylePauwels, E., Schülein, R., & Vermeire, K. (2021). Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway. International Journal of Molecular Sciences, 22(21), 12007. https://doi.org/10.3390/ijms222112007