
Intramolecular Charge Transfer of 1-Aminoanthraquinone and Ultrafast Solvation Dynamics of Dimethylsulfoxide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
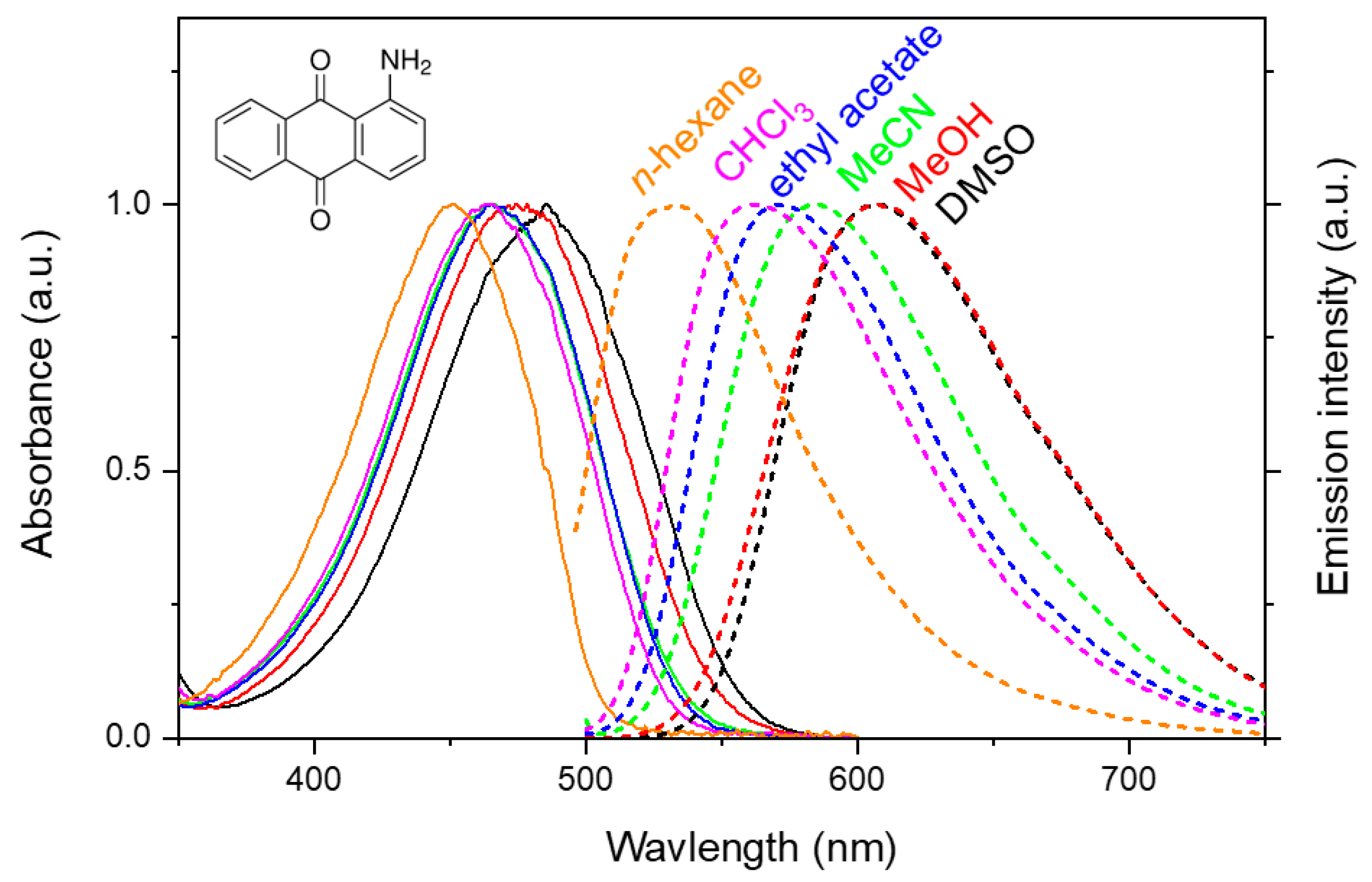
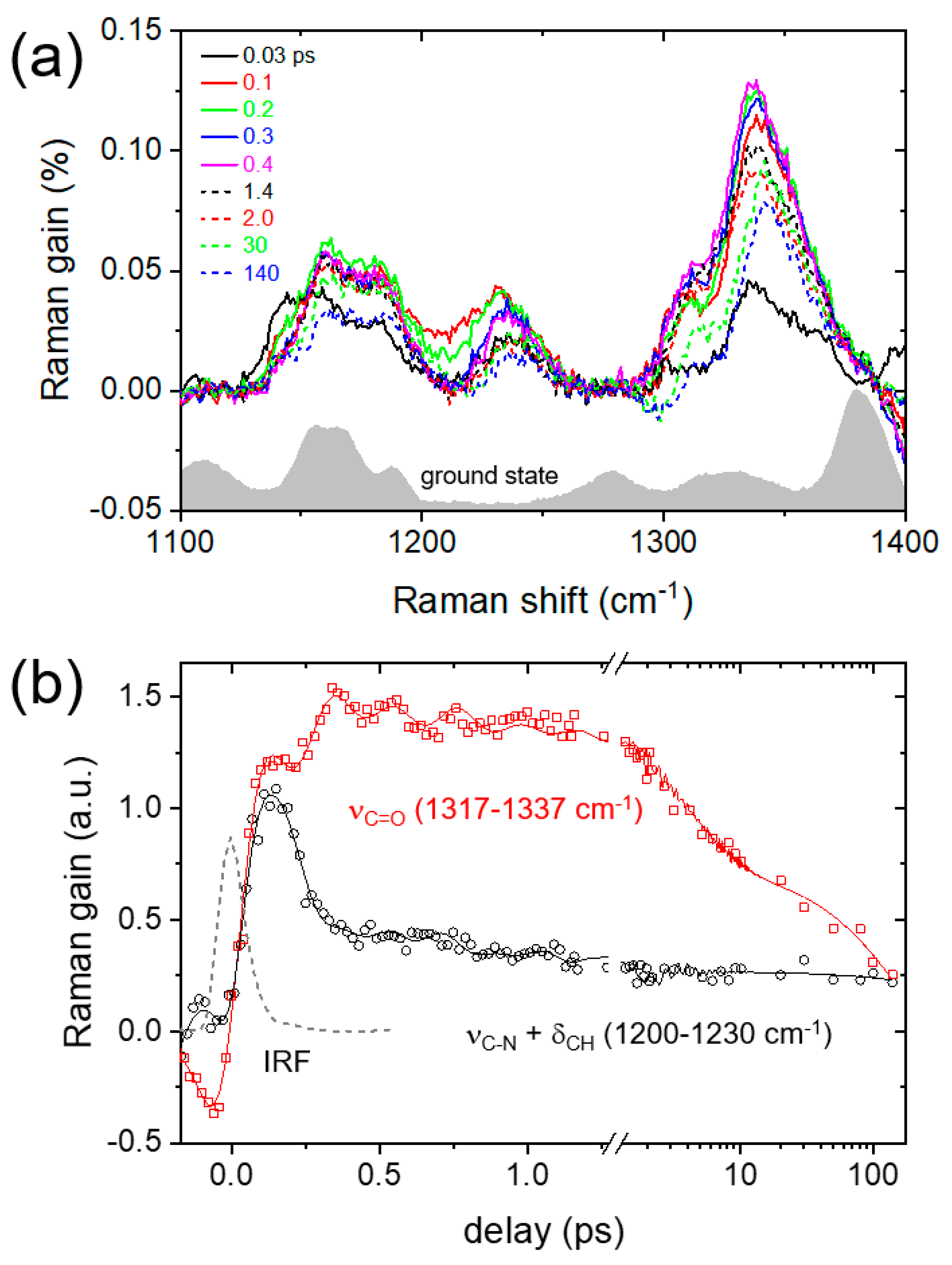
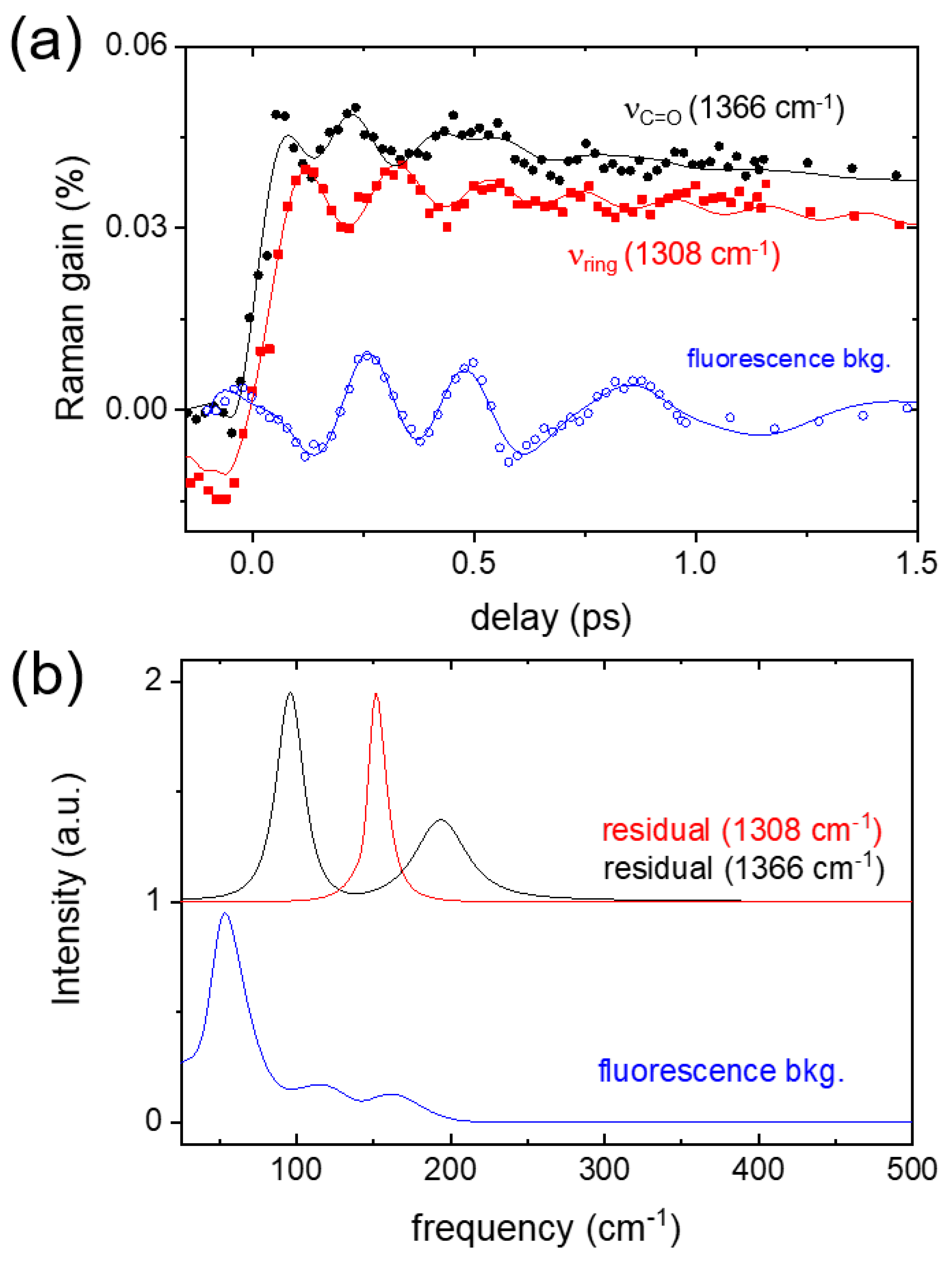
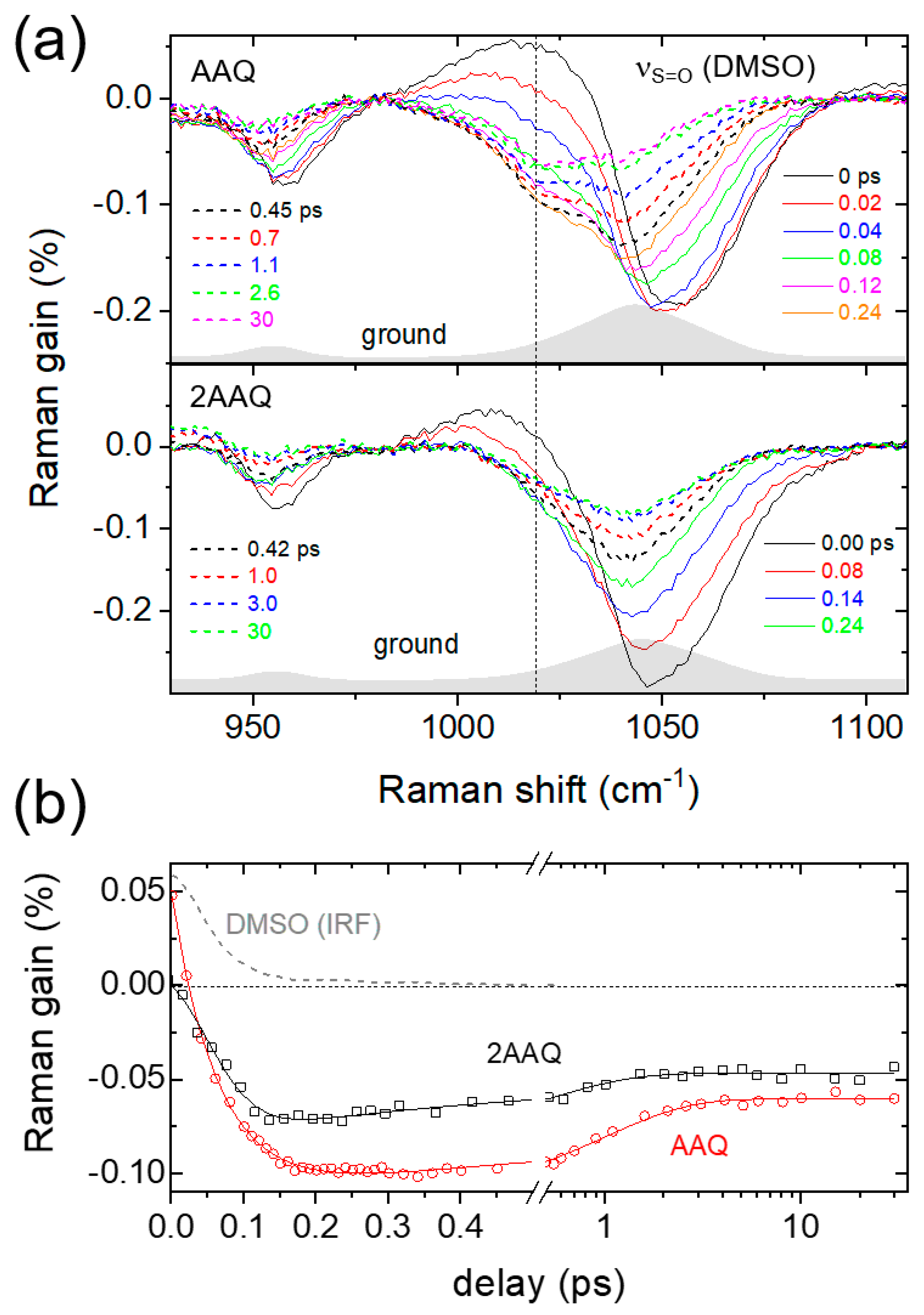
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Femtosecond Stimulated Raman Setup
3.3. Femtosecond Transient Absorption Setup
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Coropceanu, V.; Chen, X.-K.; Wang, T.; Zheng, Z.; Brédas, J.-L. Charge-transfer electronic states in organic solar cells. Nat. Rev. Mater. 2019, 4, 689–707. [Google Scholar] [CrossRef]
- Goetz, K.P.; Vermeulen, D.; Payne, M.E.; Kloc, C.; McNeil, L.E.; Jurchescu, O.D. Charge-transfer complexes: New perspectives on an old class of compounds. J. Mater. Chem. C 2014, 2, 3065–3076. [Google Scholar] [CrossRef]
- Vandewal, K. Interfacial Charge Transfer States in Condensed Phase Systems. Annu. Rev. Phys. Chem. 2016, 67, 113–133. [Google Scholar] [CrossRef]
- Wahadoszamen, M.; Margalit, I.; Ara, A.M.; van Grondelle, R.; Noy, D. The role of charge-transfer states in energy transfer and dissipation within natural and artificial bacteriochlorophyll proteins. Nat. Commun. 2014, 5, 5287. [Google Scholar] [CrossRef] [PubMed]
- Koslowski, T.; Burggraf, F.; Krapf, S.; Steinbrecher, T.; Wittekindt, C. Recent progress in biological charge transfer: Theory and simulation. Biochim. Biophys. Acta 2012, 1817, 1955–1957. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beratan, D.N.; Liu, C.; Migliore, A.; Polizzi, N.F.; Skourtis, S.S.; Zhang, P.; Zhang, Y. Charge Transfer in Dynamical Biosystems, or The Treachery of (Static) Images. Acc. Chem. Res. 2015, 48, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Chen, K.-S.; Yip, H.-L.; Hau, S.K.; Acton, O.; Zhang, Y.; Luo, J.; Jen, A.K.Y. Development of New Conjugated Polymers with Donor−π-Bridge−Acceptor Side Chains for High Performance Solar Cells. J. Am. Chem. Soc. 2009, 131, 13886–13887. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, T.; Liu, H.; Tian, M.-Z.; Li, Y. Self-Assembly of Intramolecular Charge-Transfer Compounds into Functional Molecular Systems. Acc. Chem. Res. 2014, 47, 1186–1198. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.P.; Kong, X.X.; Jenekhe, S.A. High-performance organic light-emitting diodes based on intramolecular charge-transfer emission from donor-acceptor molecules: Significance of electron-donor strength and molecular geometry. Adv. Funct. Mater. 2006, 16, 1057–1066. [Google Scholar] [CrossRef]
- Moon, C.-K.; Suzuki, K.; Shizu, K.; Adachi, C.; Kaji, H.; Kim, J.-J. Combined Inter- and Intramolecular Charge-Transfer Processes for Highly Efficient Fluorescent Organic Light-Emitting Diodes with Reduced Triplet Exciton Quenching. Adv. Mater. 2017, 29, 1606448. [Google Scholar] [CrossRef]
- Chen, X.-K.; Coropceanu, V.; Brédas, J.-L. Assessing the nature of the charge-transfer electronic states in organic solar cells. Nat. Commun. 2018, 9, 5295. [Google Scholar] [CrossRef] [PubMed]
- Bakulin, A.A.; Dimitrov, S.D.; Rao, A.; Chow, P.C.Y.; Nielsen, C.B.; Schroeder, B.C.; McCulloch, I.; Bakker, H.J.; Durrant, J.R.; Friend, R.H. Charge-Transfer State Dynamics Following Hole and Electron Transfer in Organic Photovoltaic Devices. J. Phys. Chem. Lett. 2013, 4, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Drummen, G.P.C.; Konishi, G.-i. Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry. J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar] [CrossRef]
- Guido, C.A.; Mennucci, B.; Jacquemin, D.; Adamo, C. Planar vs. twisted intramolecular charge transfer mechanism in Nile Red: New hints from theory. Phys. Chem. Chem. Phys. 2010, 12, 8016–8023. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Druzhinin, S.I.; Zachariasse, K.A. Fast Intramolecular Charge Transfer with a Planar Rigidized Electron Donor/Acceptor Molecule. J. Am. Chem. Soc. 2004, 126, 8535–8539. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Kwok, W.M.; Matousek, P.; Parker, A.W.; Phillips, D.; Toner, W.T.; Towrie, M. Excited States of 4-Aminobenzonitrile (ABN) and 4-Dimethylaminobenzonitrile (DMABN): Time-resolved Resonance Raman, Transient Absorption, Fluorescence, and ab Initio Calculations. J. Phys. Chem. A 2002, 106, 3294–3305. [Google Scholar] [CrossRef]
- Inoue, H.; Hida, M.; Nakashima, N.; Yoshihara, K. Picosecond fluorescence lifetimes of anthraquinone derivatives. Radiationless deactivation via intra- and intermolecular hydrogen bonds. J. Phys. Chem. 1982, 86, 3184–3188. [Google Scholar] [CrossRef]
- Srivatsavoy, V.J.P.; Venkataraman, B.; Periasamy, N. The non-radiative processes from the S1 state of aminoanthraquinones: A steady state and time-resolved study. J. Photochem. Photobiol. A 1992, 68, 169–184. [Google Scholar] [CrossRef]
- Dahiya, P.; Kumbhakar, M.; Maity, D.K.; Mukherjee, T.; Tripathi, A.B.R.; Chattopadhyay, N.; Pal, H. Solvent polarity and intramolecular hydrogen bonding effects on the photophysical properties of 1-amino-9,10-anthraquinone dye. J. Photochem. Photobiol. A 2006, 181, 338–347. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, M.; Zhou, P.; Yang, S.; Liu, Y.; Yang, C.; Yang, Y. Mechanism of Fluorescence Quenching by Acylamino Twist in the Excited State for 1-(Acylamino)anthraquinones. J. Phys. Chem. A 2018, 122, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Schroeder, J.; Troe, J. Intramolecular Hydrogen Bonding in 1,8-Dihydroxyanthraquinone, 1-Aminoanthraquinone, and 9-Hydroxyphenalenone Studied by Picosecond Time-Resolved Fluorescence Spectroscopy in a Supersonic Jet. J. Phys. Chem. B 2006, 110, 19820–19832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sun, S.; Zhou, M.; Wang, L.; Zhang, B. Ultrafast investigation of photoinduced charge transfer in aminoanthraquinone pharmaceutical product. Sci. Rep. 2017, 7, 43419. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Qin, C.; Liu, H.; Jiang, C. Excitation wavelength dependent ICT character and ISC efficiency in a photocleavage agent of 1-aminoanthraquinone. Spectrochim. Acta A 2020, 234, 118200. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Tahara, T. Coherent Nuclear Wavepacket Motions in Ultrafast Excited-State Intramolecular Proton Transfer: Sub-30-fs Resolved Pump−Probe Absorption Spectroscopy of 10-Hydroxybenzo[h]quinoline in Solution. J. Phys. Chem. A 2005, 109, 10199–10207. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Frontiera, R.R.; Tran, R.; Mathies, R.A. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200. [Google Scholar] [CrossRef]
- Wang, Q.; Schoenlein, R.W.; Peteanu, L.A.; Mathies, R.A.; Shank, C.V. Vibrationally coherent photochemistry in the femtosecond primary event of vision. Science 1994, 266, 422. [Google Scholar] [CrossRef]
- Mitsunaga, M.; Tang, C.L. Theory of quantum beats in optical transmission-correlation and pump-probe measurements. Phys. Rev. A 1987, 35, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Tahara, T. Femtosecond absorption study of photodissociation of diphenylcyclopropenone in solution: Reaction dynamics and coherent nuclear motion. J. Chem. Phys. 2004, 120, 4768–4776. [Google Scholar] [CrossRef] [PubMed]
- Schriever, C.; Barbatti, M.; Stock, K.; Aquino, A.J.A.; Tunega, D.; Lochbrunner, S.; Riedle, E.; de Vivie-Riedle, R.; Lischka, H. The interplay of skeletal deformations and ultrafast excited-state intramolecular proton transfer: Experimental and theoretical investigation of 10-hydroxybenzo[h]quinoline]. Chem. Phys. 2008, 347, 446–461. [Google Scholar] [CrossRef]
- Schriever, C.; Lochbrunner, S.; Ofial, A.R.; Riedle, E. The origin of ultrafast proton transfer: Multidimensional wave packet motion vs. tunneling. Chem. Phys. Lett. 2011, 503, 61–65. [Google Scholar] [CrossRef]
- Kim, C.H.; Joo, T. Coherent excited state intramolecular proton transfer probed by time-resolved fluorescence. Phys. Chem. Chem. Phys. 2009, 11, 10266–10269. [Google Scholar] [CrossRef]
- Kim, J.; Kim, D.E.; Joo, T. Excited-State Dynamics of Thioflavin T: Planar Stable Intermediate Revealed by Nuclear Wave Packet Spectroscopies. J. Phys. Chem. A 2018, 122, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, C.H.; Joo, T. Active Role of Proton in Excited State Intramolecular Proton Transfer Reaction. J. Phys. Chem. A 2013, 117, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Kim, J.; Kim, S.Y.; Kim, D.E.; Joo, T. Vibrational Spectrum of an Excited State and Huang–Rhys Factors by Coherent Wave Packets in Time-Resolved Fluorescence Spectroscopy. ChemPhysChem 2017, 18, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Heo, W.; Uddin, N.; Park, J.W.; Rhee, Y.M.; Choi, C.H.; Joo, T. Coherent intermolecular proton transfer in the acid–base reaction of excited state pyranine. Phys. Chem. Chem. Phys. 2017, 19, 18243–18251. [Google Scholar] [CrossRef]
- Hoffman, D.P.; Mathies, R.A. Photoexcited structural dynamics of an azobenzene analog 4-nitro-4′-dimethylamino-azobenzene from femtosecond stimulated Raman. Phys. Chem. Chem. Phys. 2012, 14, 6298–6306. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.P.; Lee, O.P.; Millstone, J.E.; Chen, M.S.; Su, T.A.; Creelman, M.; Fréchet, J.M.J.; Mathies, R.A. Electron Transfer Dynamics of Triphenylamine Dyes Bound to TiO 2 Nanoparticles from Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. C 2013, 117, 6990–6997. [Google Scholar] [CrossRef]
- Hoffman, D.P.; Ellis, S.R.; Mathies, R.A. Characterization of a Conical Intersection in a Charge-Transfer Dimer with Two-Dimensional Time-Resolved Stimulated Raman Spectroscopy. J. Phys. Chem. A 2014, 118, 4955–4965. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.P.; Mathies, R.A. Femtosecond Stimulated Raman Exposes the Role of Vibrational Coherence in Condensed-Phase Photoreactivity. Acc. Chem. Res. 2016, 49, 616–625. [Google Scholar] [CrossRef]
- Frontiera, R.R.; Fang, C.; Dasgupta, J.; Mathies, R.A. Probing structural evolution along multidimensional reaction coordinates with femtosecond stimulated Raman spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhu, L.; Taylor, A.M.; Wang, Y.; Remington, J.S.; Fang, C. Excited State Structural Evolution of a GFP Single-Site Mutant Tracked by Tunable Femtosecond-Stimulated Raman Spectroscopy. Molecules 2018, 23, 2226. [Google Scholar] [CrossRef]
- Bagchi, B.; Jana, B. Solvation dynamics in dipolar liquids. Chem. Soc. Rev. 2010, 39, 1936–1954. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, I. Chemical Reactions and Solvation at Liquid Interfaces: A Microscopic Perspective. Chem. Rev. 1996, 96, 1449–1476. [Google Scholar] [CrossRef] [PubMed]
- Oscar, B.G.; Liu, W.; Rozanov, N.D.; Fang, C. Ultrafast intermolecular proton transfer to a proton scavenger in an organic solvent. Phys. Chem. Chem. Phys. 2016, 18, 26151–26160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.; Tang, L.; Oscar, B.; Han, F.; Fang, C. Early Time Excited-State Structural Evolution of Pyranine in Methanol Revealed by Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. A 2013, 117, 6024–6042. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Han, F.; Smith, C.; Fang, C. Ultrafast Conformational Dynamics of Pyranine during Excited State Proton Transfer in Aqueous Solution Revealed by Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. B 2012, 116, 10535–10550. [Google Scholar] [CrossRef]
- Jen, M.; Jeon, K.; Lee, S.; Hwang, S.; Chung, W.J.; Pang, Y. Ultrafast intramolecular proton transfer reactions and solvation dynamics of DMSO. Struct. Dyn. 2019, 6, 064901. [Google Scholar] [CrossRef] [PubMed]
- Siddlingeshwar, B.; Hanagodimath, S.M. Estimation of first excited singlet-state dipole moments of aminoanthraquinones by solvatochromic method. Spectrochim. Acta A 2009, 72, 490–495. [Google Scholar] [CrossRef]
- Easter, D.C.; Baronavski, A.P. Ultrafast relaxation in the fluorescent state of the laser dye DCM. Chem. Phys. Lett. 1993, 201, 153–158. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.; Seger, R.; Mullen, K.M.; van Stokkum, I.H.M. Glotaran: A Java-based graphical user interface for the R package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef]
- Van Stokkum, I.H.M.; Larsen, D.S.; Van Grondelle, R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta 2004, 1657, 82–104. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.A.A.; Lewis, N.H.C.; Fleming, G.R. Correlating the motion of electrons and nuclei with two-dimensional electronic–vibrational spectroscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 10061. [Google Scholar] [CrossRef] [PubMed]
- Rhinehart, J.M.; Challa, J.R.; McCamant, D.W. Multimode charge-transfer dynamics of 4-(dimethylamino)benzonitrile probed with ultraviolet femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 10522–10534. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jen, M.; Pang, Y. Twisted intramolecular charge transfer state of a “push-pull” emitter. Int. J. Mol. Sci. 2020, 21, 7999. [Google Scholar] [CrossRef]
- Jen, M.; Lee, S.; Jeon, K.; Hussain, S.; Pang, Y. Ultrafast Intramolecular Proton Transfer of Alizarin Investigated by Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. B 2017, 121, 4129–4136. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Jen, M.; Pang, Y. Metal-enhanced fluorescence: Wavelength-dependent ultrafast energy transfer. J. Phys. Chem. C 2015, 119, 23285–23291. [Google Scholar] [CrossRef]
- Lee, G.; Jang, T.; Lee, S.; Oh, H.; Lee, H.; Pang, Y. Excited-state dynamics of 4-dimethylamino-4′-nitrobiphenyl confined in AOT reverse micelles. J. Mol. Liq. 2020, 305, 112873. [Google Scholar] [CrossRef]
- Wise, F.; Rosker, M.; Millhauser, G.; Tang, C. Application of linear prediction least-squares fitting to time-resolved optical spectroscopy. IEEE J. Quantum Electron. 1987, 23, 1116–1121. [Google Scholar] [CrossRef]
- Barkhuijsen, H.; de Beer, R.; Bovée, W.M.M.J.; van Ormondt, D. Retrieval of frequencies, amplitudes, damping factors, and phases from time-domain signals using a linear least-squares procedure. J. Magn. Reson. 1985, 61, 465–481. [Google Scholar] [CrossRef]
- Madsen, D.; Stenger, J.; Dreyer, J.; Nibbering, E.T.J.; Hamm, P.; Elsaesser, T. Coherent vibrational ground-state dynamics of an intramolecular hydrogen bond. Chem. Phys. Lett. 2001, 341, 56–62. [Google Scholar] [CrossRef]
- Stenger, J.; Madsen, D.; Dreyer, J.; Nibbering, E.T.J.; Hamm, P.; Elsaesser, T. Coherent Response of Hydrogen Bonds in Liquids Probed by Ultrafast Vibrational Spectroscopy. J. Phys. Chem. A 2001, 105, 2929–2932. [Google Scholar] [CrossRef]
- Marco, L.D.; Thämer, M.; Reppert, M.; Tokmakoff, A. Direct observation of intermolecular interactions mediated by hydrogen bonding. J. Chem. Phys. 2014, 141, 034502. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.B.; Roberts, S.T.; Ramasesha, K.; Nocera, D.G.; Tokmakoff, A. Ultrafast N−H Vibrational Dynamics of Cyclic Doubly Hydrogen-Bonded Homo- and Heterodimers. J. Phys. Chem. B 2008, 112, 13167–13171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kukura, P.; McCamant, D.W.; Yoon, S.; Wandschneider, D.B.; Mathies, R.A. Chemistry: Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science 2005, 310, 1006–1009. [Google Scholar] [CrossRef]
- Batignani, G.; Fumero, G.; Pontecorvo, E.; Ferrante, C.; Mukamel, S.; Scopigno, T. Genuine Dynamics vs Cross Phase Modulation Artifacts in Femtosecond Stimulated Raman Spectroscopy. ACS Photonics 2019, 6, 492–500. [Google Scholar] [CrossRef]
- Oh, K.-I.; Rajesh, K.; Stanton, J.F.; Baiz, C.R. Quantifying Hydrogen-Bond Populations in Dimethyl Sulfoxide/Water Mixtures. Angew. Chem. Int. Ed. 2017, 56, 11375–11379. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Liu, W.; Zhu, L.; Wang, Y.; Fang, C. Initial hydrogen-bonding dynamics of photoexcited coumarin in solution with femtosecond stimulated Raman spectroscopy. J. Mater. Chem. C 2016, 4, 2954–2963. [Google Scholar] [CrossRef]
- Chudoba, C.; Nibbering, E.; Elsaesser, T. Site-specific excited-state solute-solvent interactions probed by femtosecond vibrational spectroscopy. Phys. Rev. Lett. 1998, 81, 3010. [Google Scholar] [CrossRef]
- Chudoba, C.; Nibbering, E.T.J.; Elsaesser, T. Ultrafast Structural Response of Hydrogen Bonded Complexes to Electronic Excitation in the Liquid Phase. J. Phys. Chem. A 1999, 103, 5625–5628. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Pang, Y. Excited state intramolecular proton transfer of 1,2-dihydroxyanthraquinone by femtosecond transient absorption spectroscopy. Curr. Appl. Phys. 2015, 15, 1492–1499. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, K.; Jen, M.; Lee, S.; Jang, T.; Pang, Y. Intramolecular Charge Transfer of 1-Aminoanthraquinone and Ultrafast Solvation Dynamics of Dimethylsulfoxide. Int. J. Mol. Sci. 2021, 22, 11926. https://doi.org/10.3390/ijms222111926
Jeon K, Jen M, Lee S, Jang T, Pang Y. Intramolecular Charge Transfer of 1-Aminoanthraquinone and Ultrafast Solvation Dynamics of Dimethylsulfoxide. International Journal of Molecular Sciences. 2021; 22(21):11926. https://doi.org/10.3390/ijms222111926
Chicago/Turabian StyleJeon, Kooknam, Myungsam Jen, Sebok Lee, Taehyung Jang, and Yoonsoo Pang. 2021. "Intramolecular Charge Transfer of 1-Aminoanthraquinone and Ultrafast Solvation Dynamics of Dimethylsulfoxide" International Journal of Molecular Sciences 22, no. 21: 11926. https://doi.org/10.3390/ijms222111926
APA StyleJeon, K., Jen, M., Lee, S., Jang, T., & Pang, Y. (2021). Intramolecular Charge Transfer of 1-Aminoanthraquinone and Ultrafast Solvation Dynamics of Dimethylsulfoxide. International Journal of Molecular Sciences, 22(21), 11926. https://doi.org/10.3390/ijms222111926