
The Role of Endothelium in COVID-19


 ,
,  ,
,  ,
,  , , and
, , and 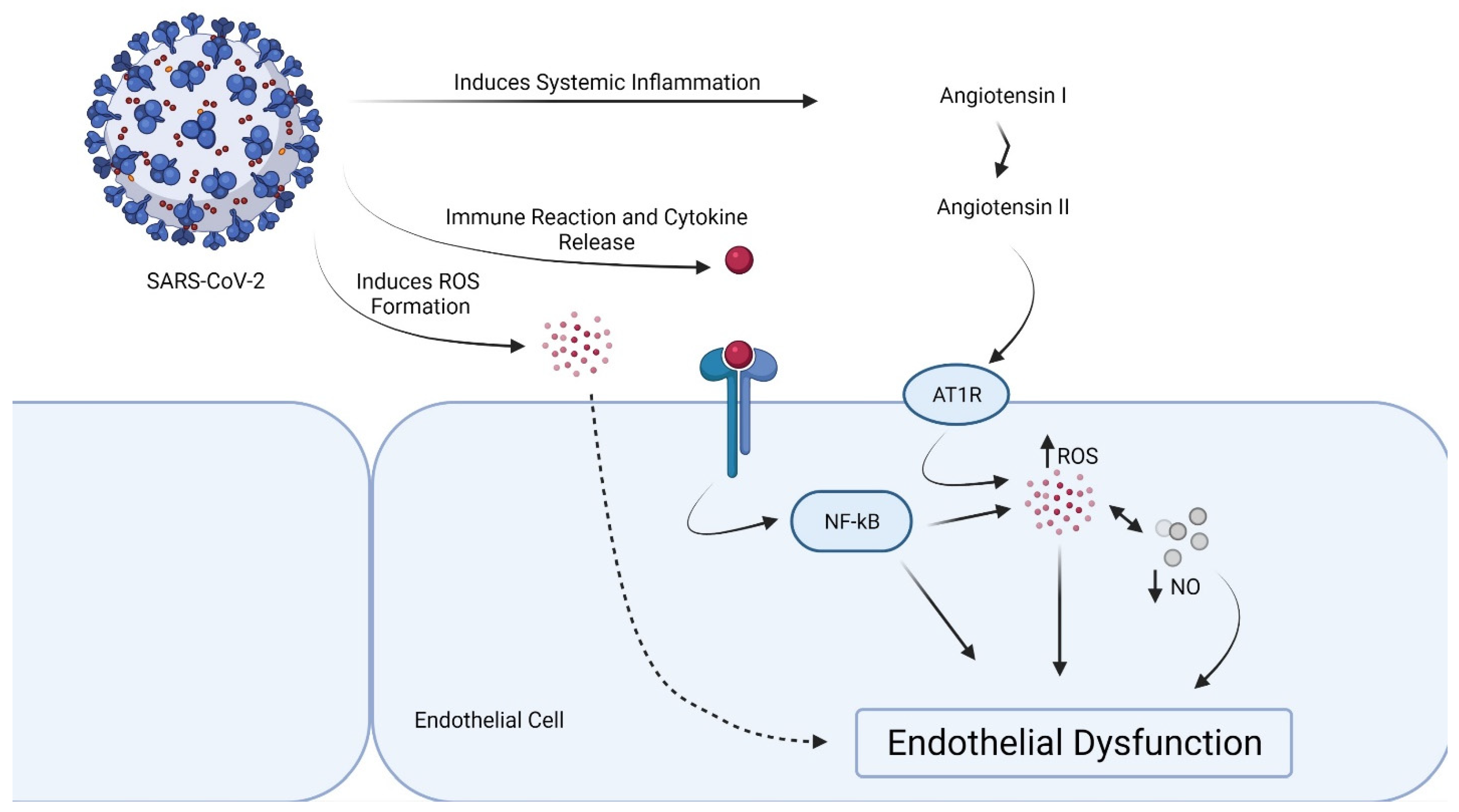{kind=link}
{kind=link}
Abstract
:1. Introduction
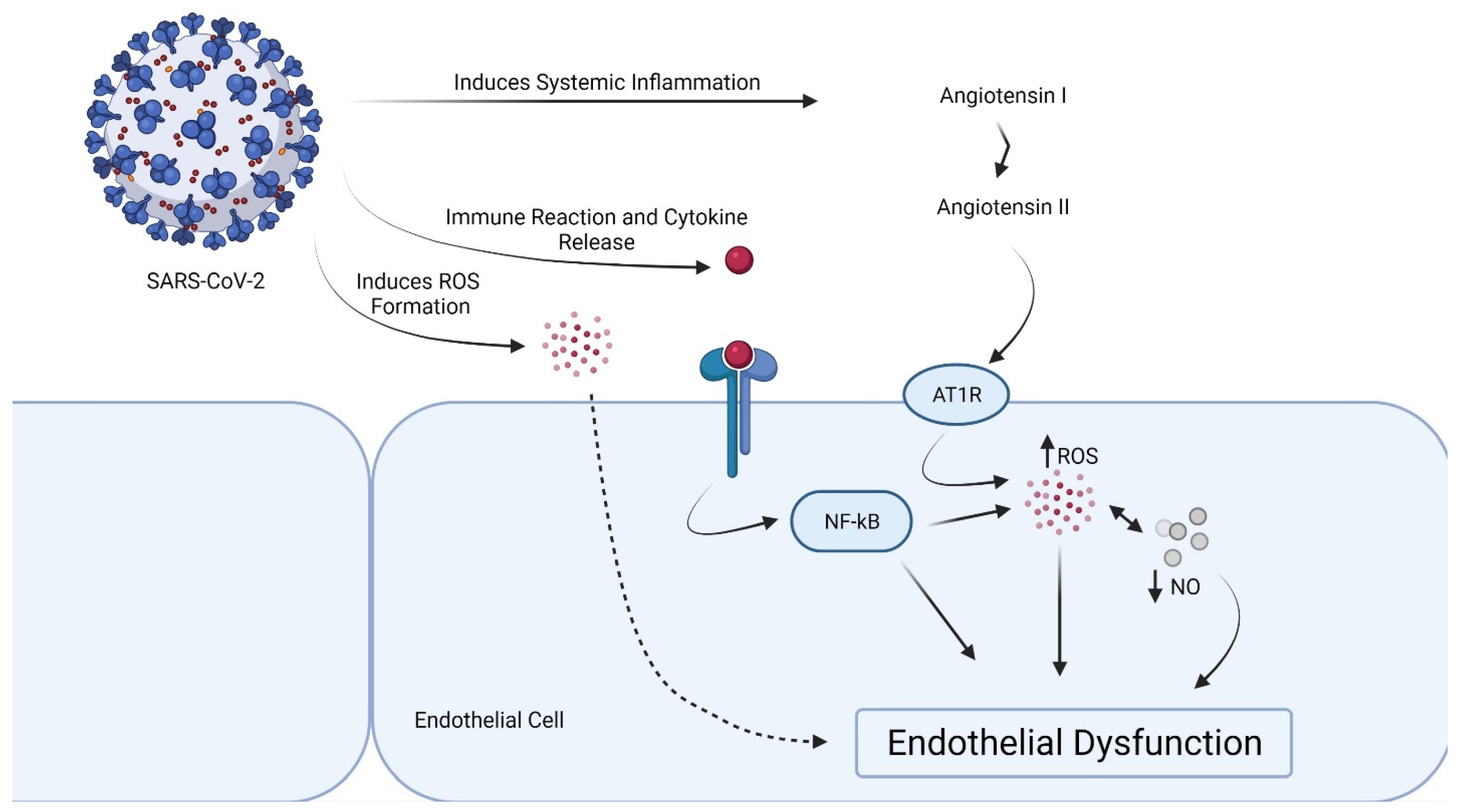
2. Effects of Cytokine Storm in the Endothelial Cells
3. Proof of Endothelial Dysfunction in COVID-19
4. Local Dysregulation of the RAS and Endothelial Dysfunction
5. Heart Failure and COVID-19
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gavriilaki, E.; Anyfanti, P.; Gavriilaki, M.; Lazaridis, A.; Douma, S.; Gkaliagkousi, E. Endothelial Dysfunction in COVID-19: Lessons Learned from Coronaviruses. Curr. Hypertens. Rep. 2020, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The vascular biology of atherosclerosis. In Braunwald’s Heart Disease, 11th ed.; Zipes, D.P., Libby, P., Bonow, R.O., Mann, D.L., Tomaselli, G.F., Eds.; Elsevier: Philadelphia, PA, USA, 2018; pp. 859–875. [Google Scholar]
- Steinberg, B.; Goldenberg, N.; Lee, W. Do viral infections mimic bacterial sepsis? The role of microvascular permeability: A review of mechanisms and methods. Antivir. Res. 2012, 93, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Goeijenbier, M.; van Wissen, M.; van de Weg, C.; Jong, E.; Gerdes, V.; Meijers, J.; Brandjes, D.; van Gorp, E. Review: Viral infections and mechanisms of thrombosis and bleeding. J. Med. Virol. 2012, 84, 1680–1696. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular Aging and Heart Failure: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 74, 804–813. [Google Scholar] [CrossRef] [PubMed]
- García-Lucio, J.; Peinado, V.I.; De Jover, L.; Del Pozo, R.; Blanco, I.; Bonjoch, C.; Coll-Bonfill, N.; Paul, T.; Tura-Ceide, O.; Barberà, J.A. Imbalance between endothelial damage and repair capacity in chronic obstructive pulmonary disease. PLoS ONE 2018, 13, e0195724. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- He, J.; Wu, B.; Chen, Y.; Tang, J.; Liu, Q.; Zhou, S.; Chen, C.; Qin, Q.; Huang, K.; Lv, J.; et al. Characteristic Electrocardiographic Manifestations in Patients with COVID-19. Can. J. Cardiol. 2020, 36, 966.e1–966.e4. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Escobar, S.; Tibiriçá, E. Systemic endothelial dysfunction: A common pathway for COVID-19, cardiovascular and metabolic diseases. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1401–1402. [Google Scholar] [CrossRef]
- Del Turco, S.; Vianello, A.; Ragusa, R.; Caselli, C.; Basta, G. COVID-19 and cardiovascular consequences: Is the endothelial dysfunction the hardest challenge? Thromb. Res. 2020, 196, 143–151. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Corrado, E.; Rizzo, M.; Coppola, G.; Muratori, I.; Carella, M.; Novo, S. Endothelial dysfunction and carotid lesions are strong predictors of clinical events in patients with early stages of atherosclerosis: A 24-month follow-up study. Coron. Artery Dis. 2008, 19, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Nikolic, D.; Patti, A.M.; Mannina, C.; Montalto, G.; McAdams, B.S.; Rizvi, A.A.; Cosentino, F. GLP-1 receptor agonists and reduction of cardiometabolic risk: Potential underlying mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 2814–2821. [Google Scholar] [CrossRef]
- Abate, N.; Sallam, H.S.; Rizzo, M.; Nikolic, D.; Obradovic, M.; Bjelogrlic, P.; Isenovic, E. Resistin: An Inflammatory Cytokine. Role in Cardiovascular Diseases, Diabetes and the Metabolic Syndrome. Curr. Pharm. Des. 2014, 20, 4961–4969. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Pernice, V.; Frasheri, A.; Berneis, K. Atherogenic lipoprotein phenotype and LDL size and subclasses in patients with peripheral arterial disease. Atherosclerosis 2008, 197, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Spinas, G.A.; Cesur, M.; Ozbalkan, Z.; Rini, G.B.; Berneis, K. Atherogenic lipoprotein phenotype and LDL size and subclasses in drug-naïve patients with early rheumatoid arthritis. Atherosclerosis 2009, 207, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Goedecke, J.H.; Utzschneider, K.; Faulenbach, M.V.; Rizzo, M.; Berneis, K.; Spinas, G.A.; Dave, J.; Levitt, N.S.; Lambert, E.V.; Olsson, T.; et al. Ethnic differences in serum lipoproteins and their determinants in South African women. Metabolism 2010, 59, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Bayram, F.; Kocer, D.; Gundogan, K.; Kaya, A.; Demir, O.; Coskun, R.; Sabuncu, T.; Karaman, A.; Cesur, M.; Rizzo, M.; et al. Prevalence of dyslipidemia and associated risk factors in Turkish adults. J. Clin. Lipidol. 2014, 8, 206–216. [Google Scholar] [CrossRef]
- Rizzo, M.; Berneis, K. Who needs to care about small, dense low-density lipoproteins? Int. J. Clin. Pract. 2007, 61, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Berneis, K.; Rizzo, M.; Stettler, C.; Chappuis, B.; Braun, M.; Diem, P.; Christ, E.R. Comparative effects of rosiglitazone and pioglitazone on fasting and postprandial low-density lipoprotein size and subclasses in patients with Type 2 diabetes. Expert Opin. Pharmacother. 2008, 9, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Rizvi, A.A.; Patti, A.M.; Nikolic, D.; Giglio, R.V.; Castellino, G.; Volti, G.L.; Caprio, M.; Montalto, G.; Provenzano, V.; et al. Liraglutide improves metabolic parameters and carotid intima-media thickness in diabetic patients with the metabolic syndrome: An 18-month prospective study. Cardiovasc. Diabetol. 2016, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, D.; Giglio, R.V.; Rizvi, A.A.; Patti, A.M.; Montalto, G.; Maranta, F.; Cianflone, D.; Stoian, A.P.; Rizzo, M. Liraglutide Reduces Carotid Intima-Media Thickness by Reducing Small Dense Low-Density Lipoproteins in a Real-World Setting of Patients with Type 2 Diabetes: A Novel Anti-Atherogenic Effect. Diabetes Ther. 2021, 12, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.P.; Barylski, M.; Nikolic, D.; Rizzo, M.; Montalto, G.; Banach, M. Should low high-density lipoprotein cholesterol (HDL-C) be treated? Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Barylski, M.; Toth, P.P.; Nikolic, D.; Banach, M.; Rizzo, M.; Montalto, G. Emerging therapies for raising high-density lipoprotein cholesterol (HDL-C) and augmenting HDL particle functionality. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 453–461. [Google Scholar] [CrossRef]
- Heitsch, H.; Brovkovych, S.; Malinski, T.; Wiemer, G. Angiotensin-(1-7)–Stimulated Nitric Oxide and Superoxide Release from Endothelial Cells. Hypertension 2001, 37, 72–76. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Dejana, E. VE-Cadherin and Endothelial Adherens Junctions: Active Guardians of Vascular Integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Chao, S.; Schwartz, S.M.; Reidy, M.A. Aortic endothelial cell death and replication in normal and lipopolysaccharide-treated rats. Am. J. Pathol. 1985, 121, 123–127. [Google Scholar]
- Kavurma, M.M.; Tan, N.Y.; Bennett, M.R. Death Receptors and Their Ligands in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2008, 28, 1694–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Huertas, A.; Guignabert, C.; Barberà, J.A.; Bärtsch, P.; Bhattacharya, J.; Bhattacharya, S.; Bonsignore, M.R.; Dewachter, L.; Dinh-Xuan, A.T.; Dorfmüller, P.; et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur. Respir. J. 2018, 51, 1700745. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pöhlmann, S.; Soilleux, E.J. Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef] [PubMed]
- Muus, C.; Luecken, M.D.; Eraslan, G.; Waghray, A.; Heimberg, G.; Sikkema, L. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Ferreira, A.J.; Verano-Braga, T.; Bader, M. Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: New players of the renin–angiotensin system. J. Endocrinol. 2012, 216, R1–R17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Tacey, A.; Apostolopoulos, V.; Levinger, I.; Rimarova, K.; Egom, E.E.; Rodrigo, L.; Kruzliak, P.; et al. The potential actions of angiotensin-converting enzyme II (ACE2) activator diminazene aceturate (DIZE) in various diseases. Clin. Exp. Pharmacol. Physiol. 2020, 47, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, R.A.; Sorg, B.S.; Wankhede, M.; DeDeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raizada, M.K.; et al. ACE2 Activation Promotes Antithrombotic Activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ottaiano, T.F.; Andrade, S.S.; de Oliveira, C.; Silva, M.C.; Buri, M.V.; Juliano, M.A.; Girão, M.J.; Sampaio, M.U.; Schmaier, A.H.; Wlodawer, A.; et al. Plasma kallikrein enhances platelet aggregation response by subthreshold doses of ADP. Biochimie 2017, 135, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biancatelli, R.M.L.C.; Solopov, P.A.; Sharlow, E.R.; Lazo, J.S.; Marik, P.E.; Catravas, J.D. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Κ18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L477–L484. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-J.; Dong, X.; Cao, Y.-Y.; Yuan, Y.-D.; Yang, Y.-B.; Yan, Y.-Q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Paul, M.; Mehr, A.P.; Kreutz, R. Physiology of Local Renin-Angiotensin Systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.P. The Pulmonary Renin-Angiotensin System. Curr. Pharm. Des. 2003, 9, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.; Penninger, J.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates a new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Amraei, R.; Rahimi, N. COVID19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Goulter, A.B.; Goddard, M.J.; Allen, J.C.; Clark, K.L. ACE2 gene expression is up-regulated in the human failing heart. BMC Med. 2004, 2, 19. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of Angiotensin-Converting Enzyme 2 After Myocardial Infarction by Blockade of Angiotensin II Receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Esler, M.; Esler, D. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic? J. Hypertens. 2020, 38, 781–782. [Google Scholar] [CrossRef]
- British and Irish Hypertension Society. BIHS on ACEi/ARB and COVID19. Available online: https://bihsoc.org/wp-content/uploads/2020/03/BIHS-Statement-on-ACEARB-and-Covid19-16-March-2020.pdf (accessed on 27 October 2021).
- American College of Cardiology. HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19. Available online: https://www.acc.org/latest-in-cardiology/articles/2020/03/17/08/59/hfsa-acc-aha-statement-addresses-concerns-re-using-raas-antagonists-in-covid-19 (accessed on 27 October 2021).
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Tan, Z.C.; Fu, L.H.; Wang, D.D.; Hong, K. Cardiac manifestations of patients with COVID-19 pneumonia and related treatment recommendations. Zhonghua Xin Xue Guan Bing Za Zhi 2020, 48, E005. [Google Scholar] [PubMed]
- Zhu, Z.W.; Tang, J.J.; Chai, X.P.; Fang, Z.F.; Liu, Q.M.; Hu, X.Q.; Xu, D.Y.; Tang, L.; Tai, S.; Wu, Y.Z.; et al. Comparison of heart failure and 2019 novel coronavirus pneumonia in chest CT features and clinical characteristics. Zhonghua Xin Xue Guan Bing Za Zhi 2020, 48, E007. [Google Scholar]
- Goldhaber, S.Z. Venous thromboembolism in heart failure patients: Pathophysiology, predictability, prevention. J. Am. Coll. Cardiol. 2020, 75, 159–162. [Google Scholar] [CrossRef]
- Fanola, C.L.; Norby, F.; Shah, A.M.; Chang, P.P.; Lutsey, P.L.; Rosamond, W.D.; Cushman, M.; Folsom, A.R. Incident Heart Failure and Long-Term Risk for Venous Thromboembolism. J. Am. Coll. Cardiol. 2020, 75, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Speed, V.; Roberts, L.N.; Patel, J.P.; Arya, R. Venous thromboembolism and women’s health. Br. J. Haematol. 2018, 183, 346–363. [Google Scholar] [CrossRef] [Green Version]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Canepa, M.; Franssen, F.M.; Olschewski, H.; Lainscak, M.; Böhm, M.; Tavazzi, L.; Rosenkranz, S. Diagnostic and Therapeutic Gaps in Patients with Heart Failure and Chronic Obstructive Pulmonary Disease. JACC Heart Fail. 2019, 7, 823–833. [Google Scholar] [CrossRef]
- Vachiéry, J.-L.; Adir, Y.; Barberà, J.A. Pulmonary hypertension due to left heart diseases. J. Am. Coll. Cardiol. 2013, 62, D100–D108. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.; Evans, T.W. Pathophysiology and pharmacological treatment of pulmonary hypertension in acute respiratory distress syndrome. Eur. Respir. J. 2003, 21, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Moss, N.; Rakita, V.; Lala, A.; Parikh, A.; Roldan, J.; Mitter, S.S.; Anyanwu, A.; Campoli, M.; Burkhoff, D.; Mancini, D.M. Hemodynamic Response to Exercise in Patients Supported by Continuous Flow Left Ventricular Assist Devices. JACC Heart Fail. 2020, 8, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.C.; Agostoni, P.; Mancini, D.M. Prognostic Significance and Measurement of Exercise-Derived Hemodynamic Variables in Patients with Heart Failure. J. Card. Fail. 2007, 13, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L. Inflammation in heart failure. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Radley, G.; Pieper, I.L.; Ali, S.; Bhatti, F.; Thornton, C.A. The Inflammatory Response to Ventricular Assist Devices. Front. Immunol. 2018, 9, 2651. [Google Scholar] [CrossRef] [PubMed]
- Grosman-Rimon, L.; McDonald, M.A.; Jacobs, I.; Tumiati, L.C.; Bar-Ziv, S.P.; Shogilev, D.J.; Mociornita, A.G.; Ghashghai, A.; Chruscinski, A.; Cherney, D.Z.I.; et al. Markers of Inflammation in Recipients of Continuous-Flow Left Ventricular Assist Devices. Asaio J. 2014, 60, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, N.E.; Mather, H.; McKendrick, K.; Gelfman, L.P.; Hutchinson, M.D.; Lampert, R.; Lipman, H.I.; Matlock, D.D.; Strand, J.J.; Swetz, K.M.; et al. Improving Communication in Heart Failure Patient Care. J. Am. Coll. Cardiol. 2019, 74, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Cambula, G.; Bacile, I.; Rizzo, M.; Galia, M.; Mangiapane, P.; Picone, P.; Giacomazza, D.; Scalisi, L. Long-Term Brain Disorders in Post Covid-19 Neurological Syndrome (PCNS) Patient. Brain Sci. 2021, 11, 454. [Google Scholar] [CrossRef]
- Nuzzo, D.; Vasto, S.; Scalisi, L.; Cottone, S.; Cambula, G.; Rizzo, M.; Giacomazza, D.; Picone, P. Post-Acute COVID-19 Neurological Syndrome: A New Medical Challenge. J. Clin. Med. 2021, 10, 1947. [Google Scholar] [CrossRef]
- Ceriello, A.; Stoian, A.P.; Rizzo, M. COVID-19 and diabetes management: What should be considered? Diabetes Res. Clin. Pract. 2020, 163, 108151. [Google Scholar] [CrossRef]
- Stoian, A.P.; Banerjee, Y.; Rizvi, A.A.; Rizzo, M. Diabetes and the COVID-19 Pandemic: How Insights from Recent Experience Might Guide Future Management. Metab. Syndr. Relat. Disord. 2020, 18, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.; Baum, S.; Gluckman, T.J.; Gulati, M.; Martin, S.S.; Michos, E.D.; Navar, A.M.; Taub, P.R.; Toth, P.P.; Virani, S.S.; et al. Continuity of care and outpatient management for patients with and at high risk for cardiovascular disease during the COVID-19 pandemic: A scientific statement from the American Society for Preventive Cardiology. Am. J. Prev. Cardiol. 2020, 1, 100009. [Google Scholar] [CrossRef] [PubMed]
- Stoian, A.P.; Pricop-Jeckstadt, M.; Pana, A.; Ileanu, B.V.; Schitea, R.; Geanta, M.; Catrinoiu, D.; Suceveanu, A.I.; Serafinceanu, C.; Pituru, S.; et al. Death by SARS-CoV 2: A Romanian COVID-19 multi-centre comorbidity study. Sci. Rep. 2020, 10, 21613. [Google Scholar] [CrossRef]
- Dascalu, A.M.; Tudosie, M.; Smarandache, G.; Serban, D. Impact of the COVID-19 pandemic upon the ophthalmological clinical practice. Rom. J. Leg. Med. 2020, 28, 96–100. [Google Scholar] [CrossRef]
- Ceriello, A.; Standl, E.; Catrinoiu, D.; Itzhak, B.; Lalic, N.M.; Rahelic, D.; Schnell, O.; Škrha, J.; Valensi, P. “Diabetes and Cardiovascular Disease (D&CVD)” Study Group of the European Association for the Study of Diabetes (EASD). Issues for the management of people with diabetes and COVID-19 in ICU. Cardiovasc. Diabetol. 2020, 19, 114. [Google Scholar] [PubMed]
- Nikolic, D.; Banach, M.; Nikfar, S.; Salari, P.; Mikhailidis, D.P.; Toth, P.P.; Abdollahi, M.; Ray, K.K.; Pencina, M.J.; Malyszko, J.; et al. A meta-analysis of the role of statins on renal outcomes in patients with chronic kidney disease. Is the duration of therapy important? Int. J. Cardiol. 2013, 168, 5437–5447. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ionescu, M.; Stoian, A.P.; Rizzo, M.; Serban, D.; Nuzzo, D.; Mazilu, L.; Suceveanu, A.I.; Dascalu, A.M.; Parepa, I.R. The Role of Endothelium in COVID-19. Int. J. Mol. Sci. 2021, 22, 11920. https://doi.org/10.3390/ijms222111920
Ionescu M, Stoian AP, Rizzo M, Serban D, Nuzzo D, Mazilu L, Suceveanu AI, Dascalu AM, Parepa IR. The Role of Endothelium in COVID-19. International Journal of Molecular Sciences. 2021; 22(21):11920. https://doi.org/10.3390/ijms222111920
Chicago/Turabian StyleIonescu, Mihaela, Anca Pantea Stoian, Manfredi Rizzo, Dragos Serban, Domenico Nuzzo, Laura Mazilu, Andra Iulia Suceveanu, Ana Maria Dascalu, and Irinel Raluca Parepa. 2021. "The Role of Endothelium in COVID-19" International Journal of Molecular Sciences 22, no. 21: 11920. https://doi.org/10.3390/ijms222111920
APA StyleIonescu, M., Stoian, A. P., Rizzo, M., Serban, D., Nuzzo, D., Mazilu, L., Suceveanu, A. I., Dascalu, A. M., & Parepa, I. R. (2021). The Role of Endothelium in COVID-19. International Journal of Molecular Sciences, 22(21), 11920. https://doi.org/10.3390/ijms222111920