
Multi-Run Concrete Autoencoder to Identify Prognostic lncRNAs for 12 Cancers

 ,
, 
Abstract
:1. Introduction
2. Results
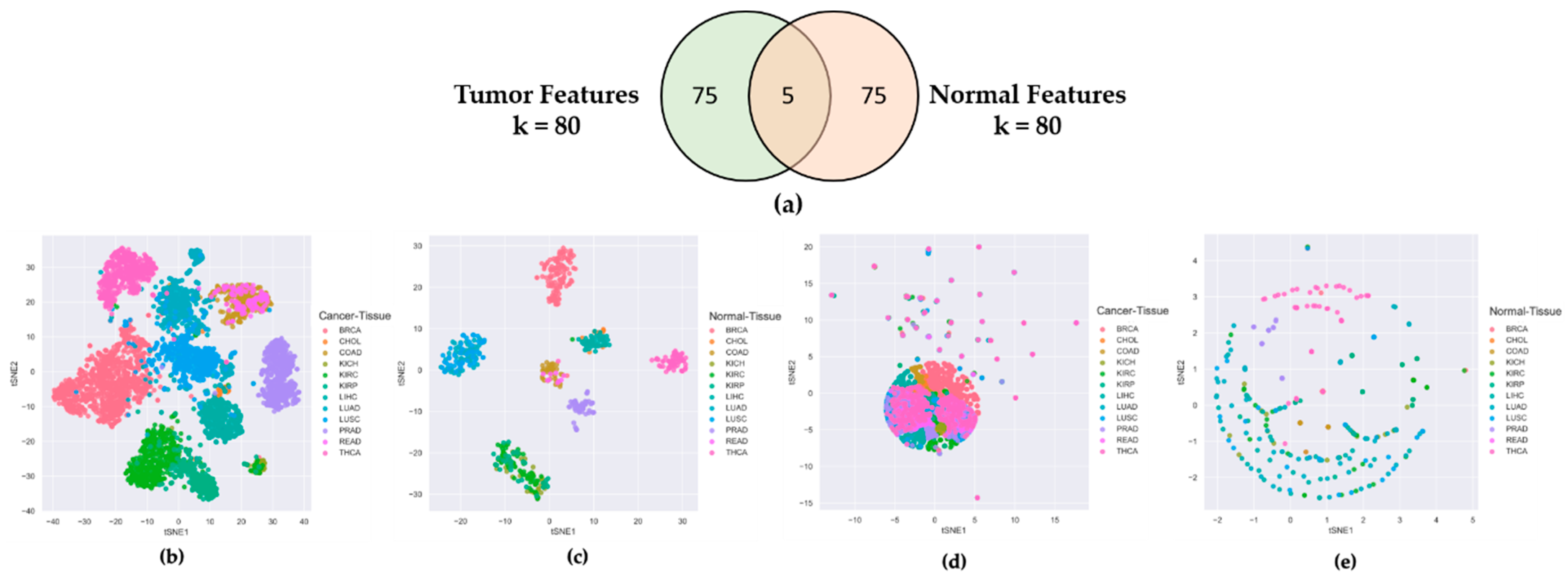
2.1. Features Selected from Tumor Tissues Are Cancer-Specific Not Organ-Specific
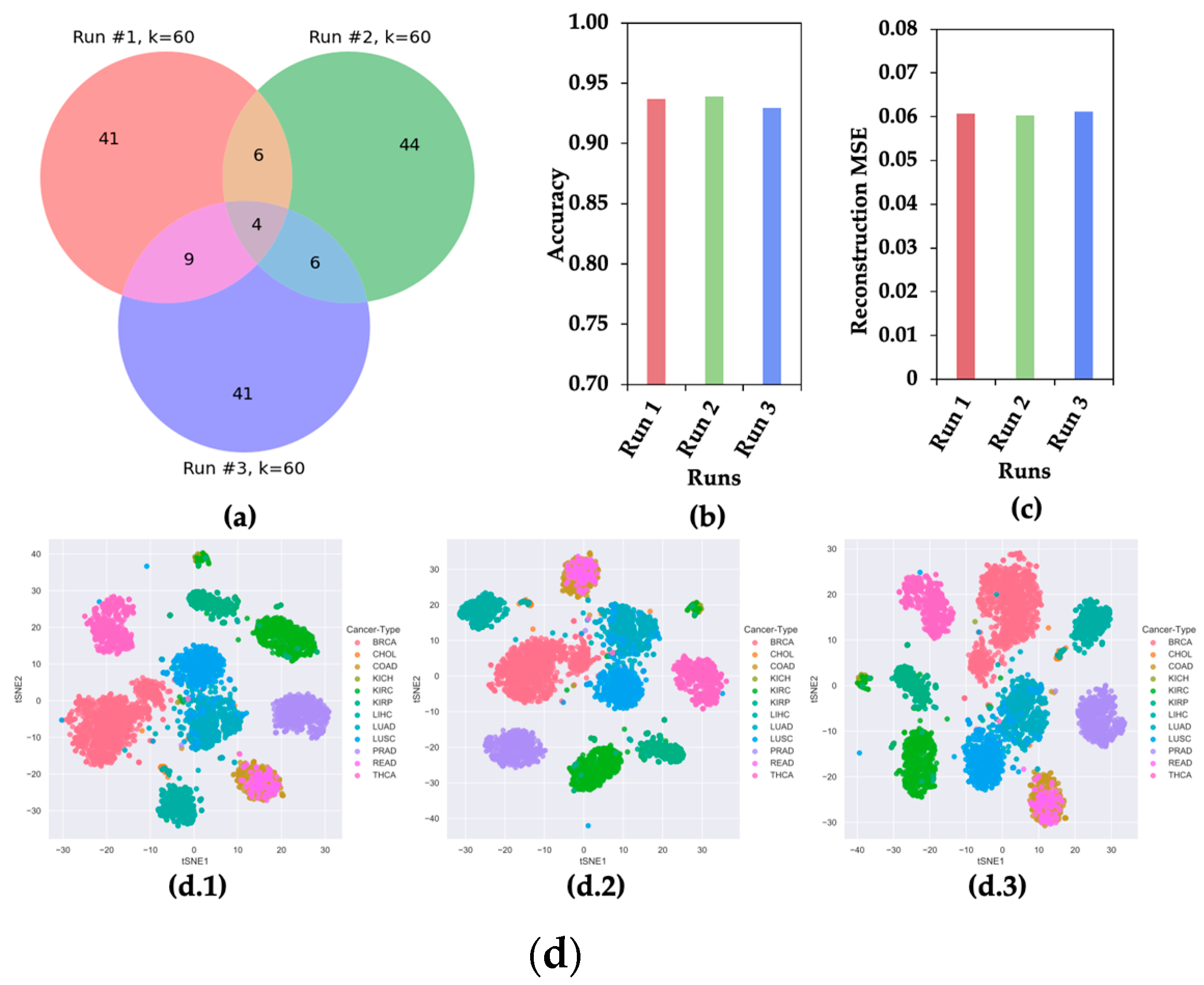
2.2. CAE Produces Different Sets of Significant Features in Different Runs
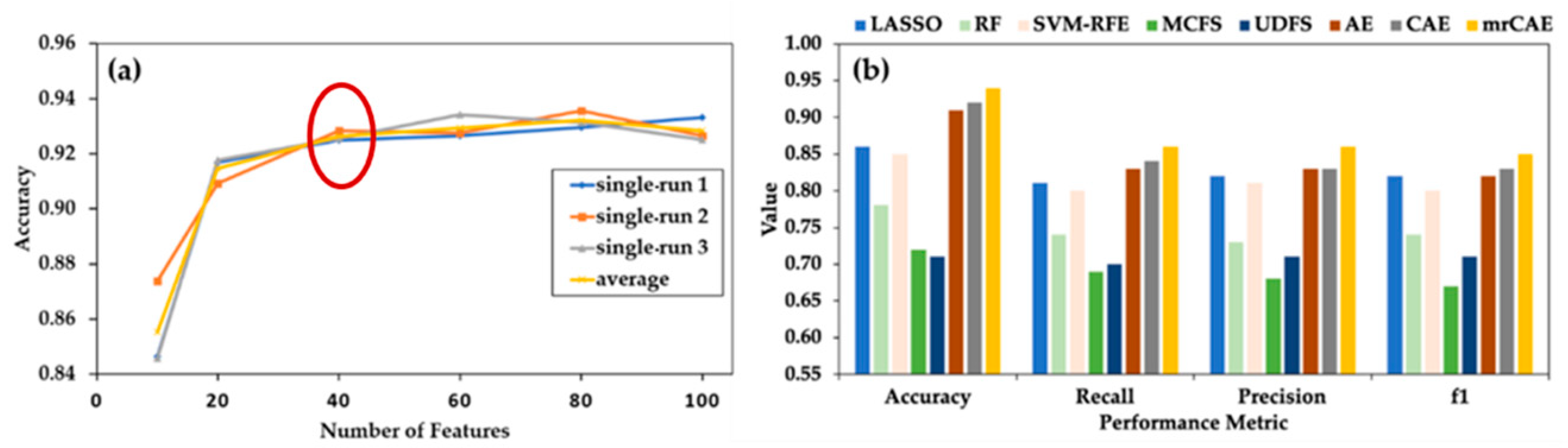
2.3. Comparison of mrCAE with Existing Feature Selection Approaches
2.4. mrCAE to Select a Stable Set of Features
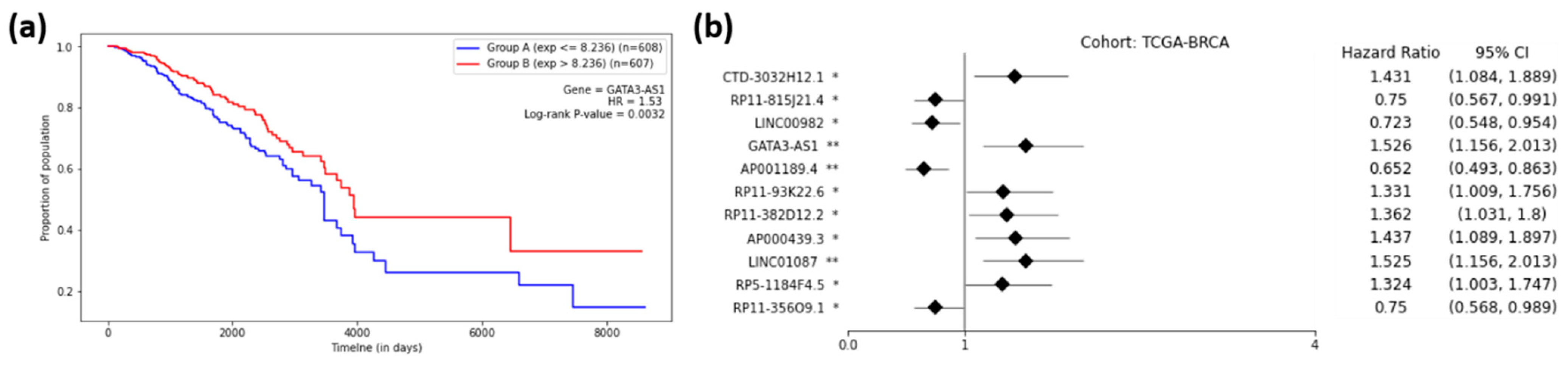
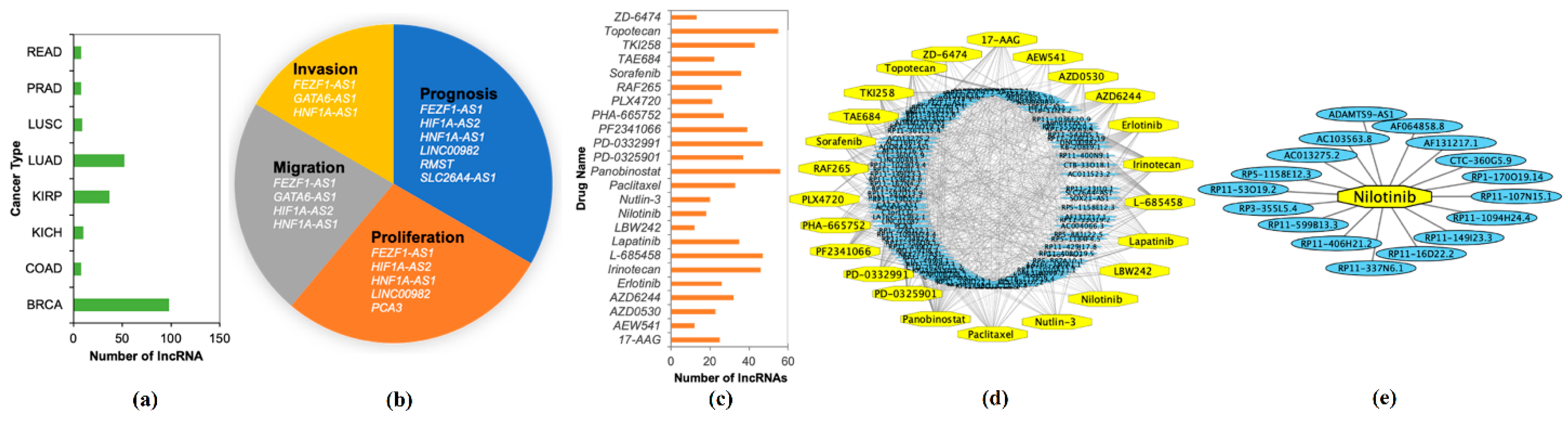
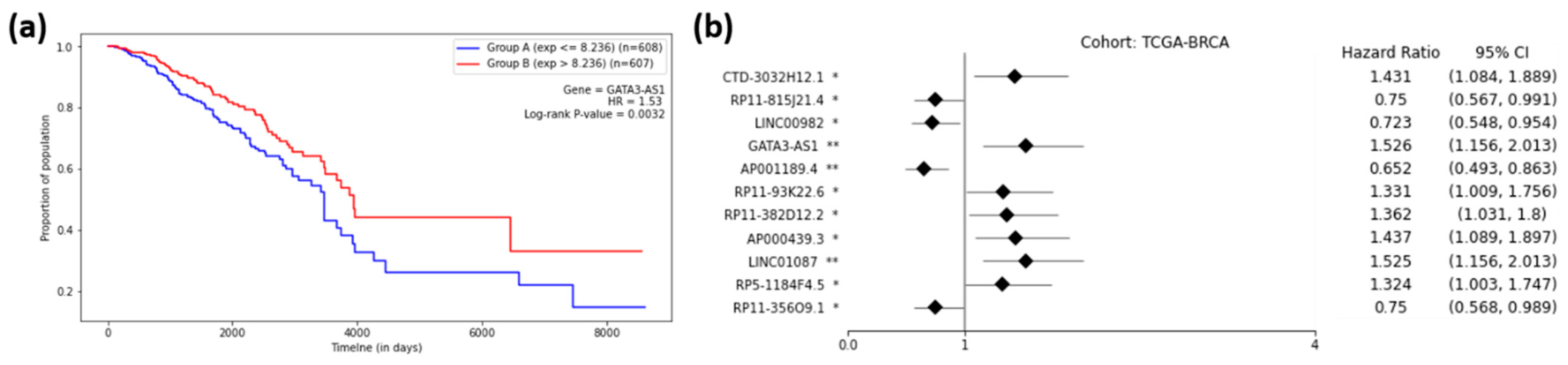
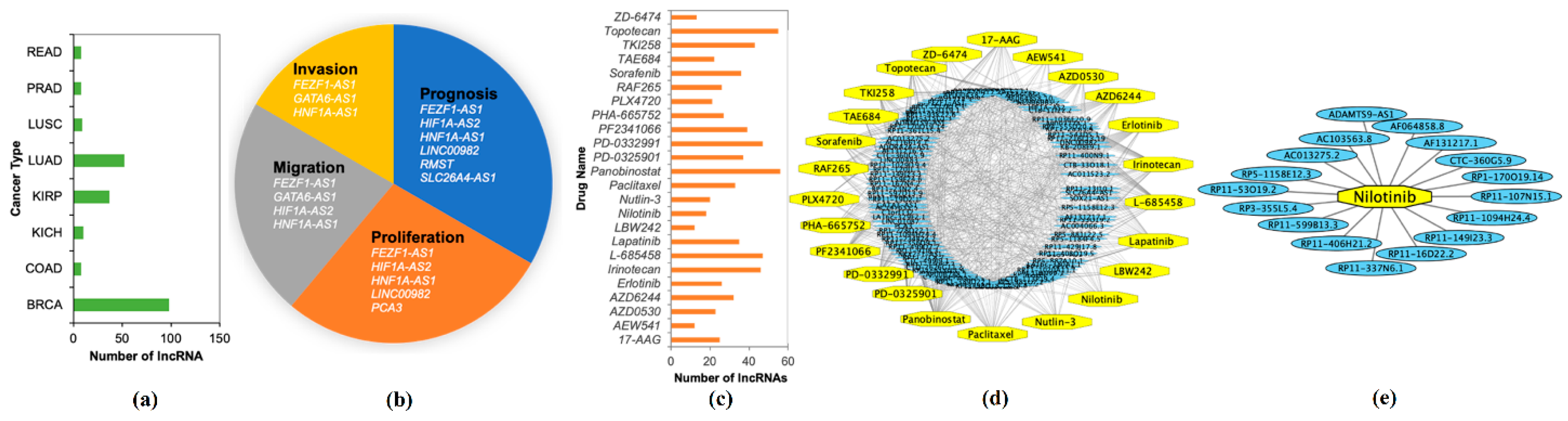
2.5. Prognostic Capability of Significant lncRNAs
2.6. Validations
3. Discussion
4. Materials and Methods
4.1. Data Preparation
4.2. Features Selection Using Multi-Run Concrete Autoencoder
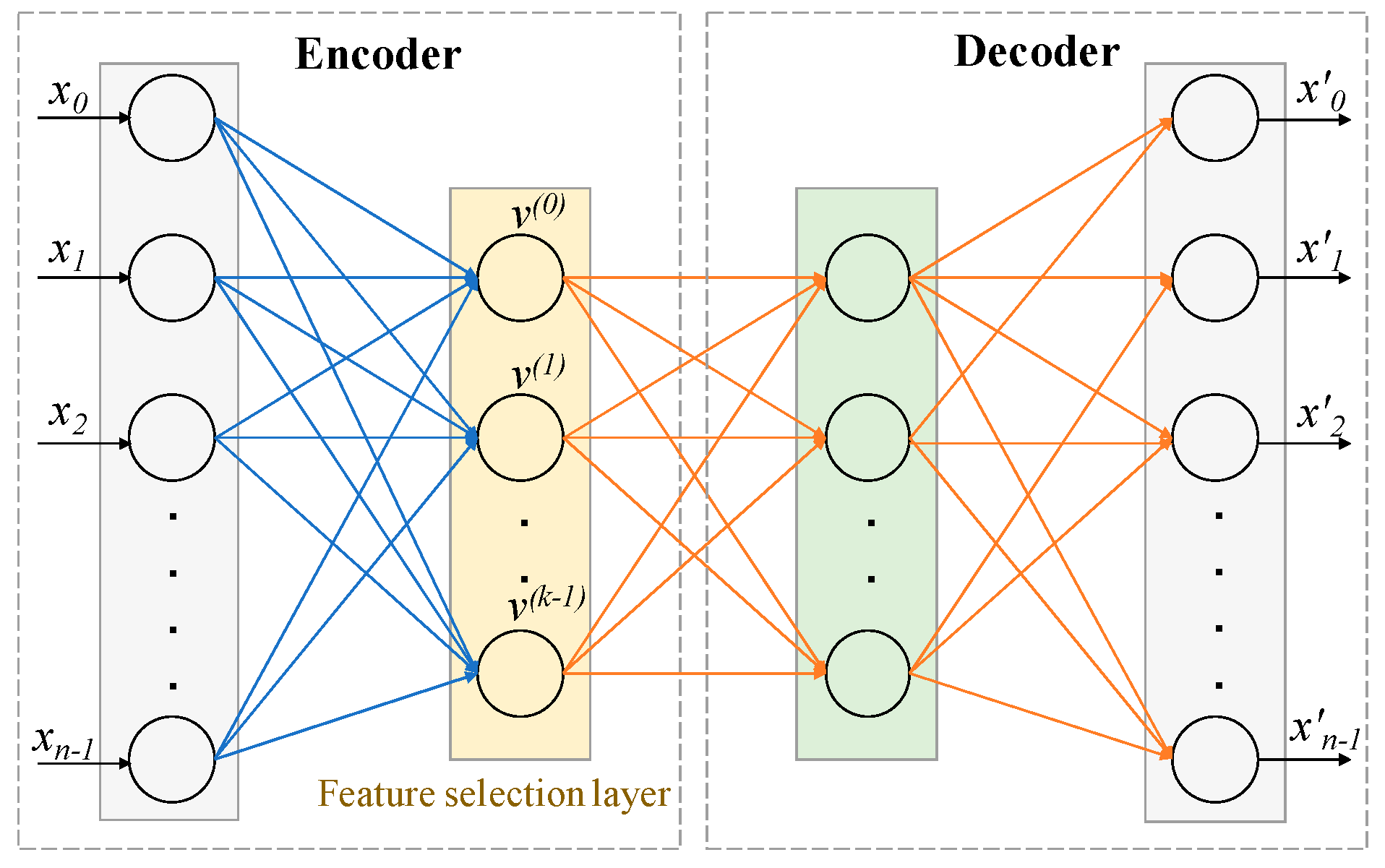
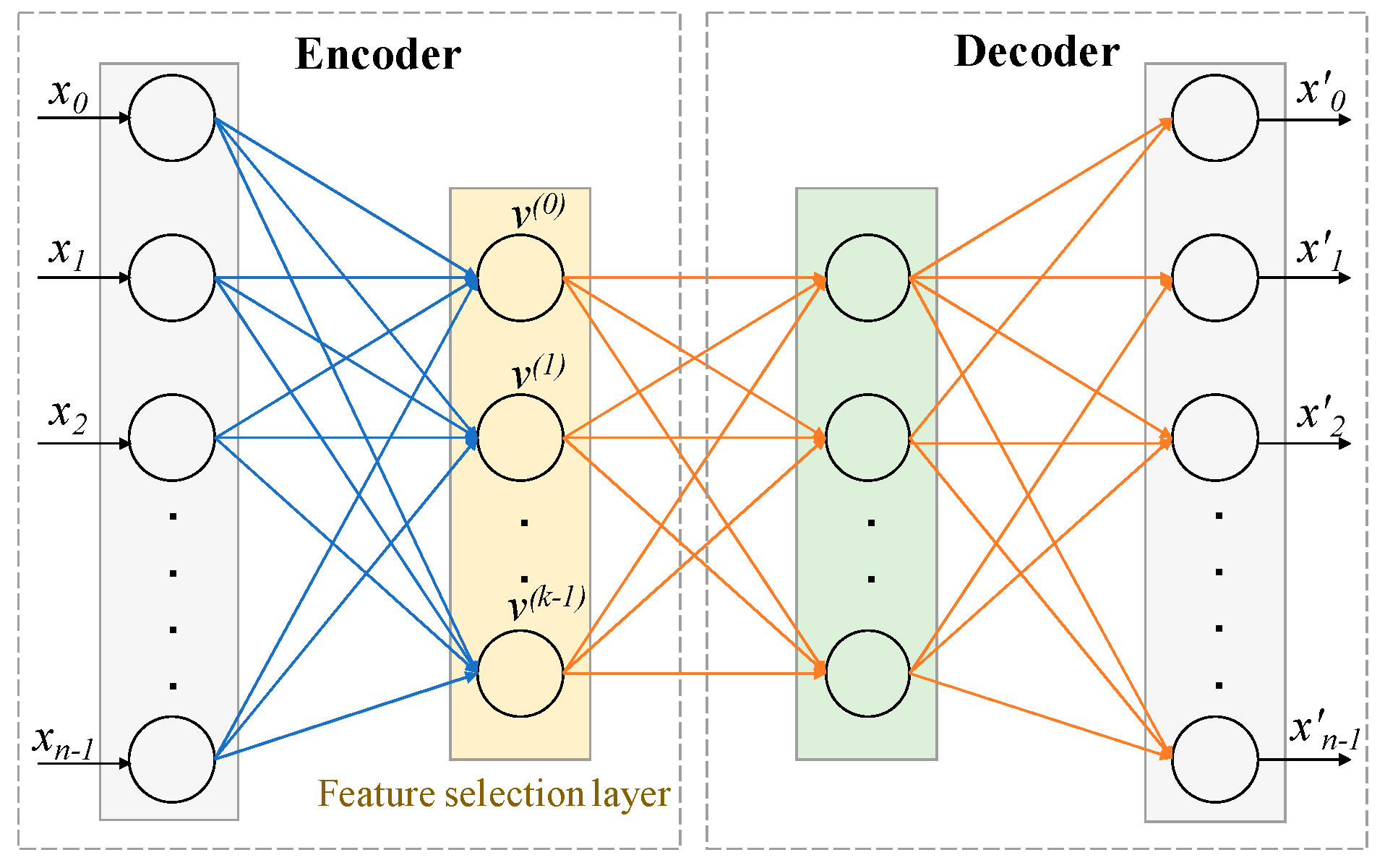
4.2.1. Architecture and Working Principle of CAE
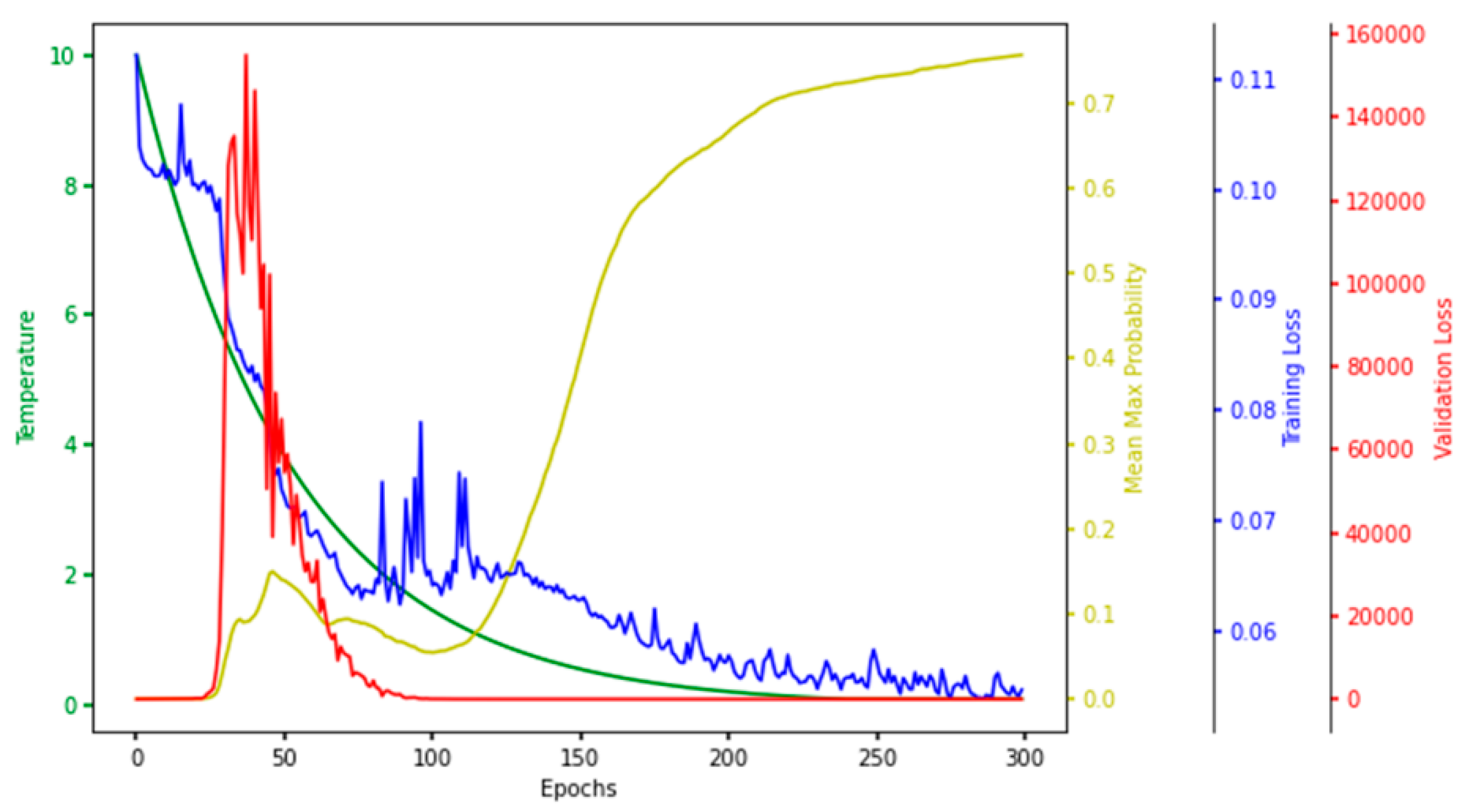
4.2.2. Hyperparameter Tuning for CAE
4.3. Comparing mrCAE with Other Feature Selection Approaches
4.4. Implementation of Feature Selection Algorithms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Yang, J.-J.; Zhou, X.; Deng, Z.-Y.; Shi, K.-H.; Li, J. Emerging role of long noncoding RNAs in lung cancer: Current status and future prospects. Respir. Med. 2016, 110, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, A.; Balin, M.F.; Zou, J. Concrete autoencoders: Differentiable feature selection and reconstruction. In Proceedings of the 36th International Conference on Machine Learning, PMLR, San Francisco, CA, USA, 27–30 June 2019; pp. 694–711. [Google Scholar]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417. [Google Scholar] [CrossRef]
- Hinton, G.E.; Salakhutdinov, R.R. Reducing the dimensionality of data with neural networks. Science 2006, 313, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, A.; Pourahmadi, V.; Soltani, M.; Sheikhzadeh, H. Deep feature selection using a teacher-student network. Neurocomputing 2020, 383, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Fan, Y.; Lv, J.; Noble, W.S. DeepPINK: Reproducible feature selection in deep neural networks. arXiv 2018, arXiv:1809.01185. [Google Scholar]
- Borisov, V.; Haug, J.; Kasneci, G. CancelOut: A Layer for Feature Selection in Deep Neural Networks. In Proceedings of the International Conference on Artificial Neural Networks, Munich, Germany, 17–19 September 2019; pp. 72–83. [Google Scholar]
- Al Mamun, A.; Duan, W.; Mondal, A.M. Pan-cancer Feature Selection and Classification Reveals Important Long Non-coding RNAs. In Proceedings of the 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM 2020), Seoul, Korea, 16–19 December 2020; pp. 2417–2424. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. Ournal R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Genuer, R.; Poggi, J.-M.; Tuleau-Malot, C. Variable selection using random forests. Pattern Recognit. Lett. 2010, 31, 2225–2236. [Google Scholar] [CrossRef] [Green Version]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, J.; Gao, Y.; Li, Y.; Feng, C.; Song, C.; Ning, Z.; Zhou, X.; Zhao, J.; Feng, M.; et al. LncSEA: A platform for long non-coding RNA related sets and enrichment analysis. Nucleic Acids Res. 2021, 49, D969–D980. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Wang, Z.; Pan, T.; Sahni, N.; Jin, X.; Wang, G.; Li, J.; Zheng, X.; Zhang, Y.; et al. LncMAP: Pan-cancer Atlas of long noncoding RNA-mediated transcriptional network perturbations. Nucleic Acids Res. 2018, 46, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Cui, T.; Zhang, L.; Huang, Y.; Yi, Y.; Tan, P.; Zhao, Y.; Hu, Y.; Xu, L.; Lin, Z.; Wang, D. MNDR v2.0: An updated resource of ncRNA-disease associations in mammals. Nucleic Acids Res. 2018, 46, D371–D374. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, S.; Zhang, J.; Wang, P.; Zhi, H.; Wang, J.; Liu, Y.; Gao, Y.; Guo, M.; Yue, M.; Wang, L.; et al. Lnc2Cancer: A manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016, 44, D980–D985. [Google Scholar] [CrossRef]
- Zhou, B.; Zhao, H.; Yu, J.; Guo, C.; Dou, X.; Song, F.; Hu, G.; Cao, Z.; Qu, Y.; Yang, Y.; et al. EVLncRNAs: A manually curated database for long non-coding RNAs validated by low-throughput experiments. Nucleic Acids Res. 2018, 46, D100–D105. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, X.; Chen, W.; Li, J.; Liu, C. CRlncRNA: A manually curated database of cancer-related long non-coding RNAs with experimental proof of functions on clinicopathological and molecular features. BMC Med. Genom. 2018, 11, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Craft, B.; Brooks, A.; Zhu, J.; Haussler, D. The UCSC Xena Platform for cancer genomics data visualization and interpretation. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Bergstra, J.; Bengio, Y. Random search for hyper-parameter optimization. J. Mach. Learn. Res. 2012, 13, 281–305. [Google Scholar]
- Cai, D.; Zhang, C.; He, X. Unsupervised feature selection for Multi-Cluster data. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Washington, DC, USA, 24–28 July 2010; pp. 333–342. [Google Scholar]
- Yang, Y.; Shen, H.T.; Ma, Z.; Huang, Z.; Zhou, X. L2, 1-Norm Regularized Discriminative Feature Selection for Unsupervised Learning. In Proceedings of the Twenty-Second International Joint Conference on Artificial Intelligence, Barcelona, Spain, 16–22 July 2011. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mrCAE | Total LncRNAs | Min Frequency | Max Frequency |
|---|---|---|---|
| 10-run mrCAE | 223 | 1 | 10 |
| 20-run mrCAE | 313 | 1 | 20 |
| 40-run mrCAE | 400 | 1 | 40 |
| 60-run mrCAE | 464 | 1 | 60 |
| 80-run mrCAE | 499 | 1 | 80 |
| 100-run mrCAE | 534 | 1 | 98 |
| 120-run mrCAE | 575 | 1 | 117 |
| Ranges of Frequency | ||||||
|---|---|---|---|---|---|---|
| mrCAE | Top-10 | Top-20 | Top-40 | Top-60 | Top-80 | Top-100 |
| 10-run mrCAE | (10–10) | (9–10) | (6–10) | (4–10) | (3–10) | (2–10) |
| 20-run mrCAE | (19–20) | (15–20) | (11–20) | (8–20) | (5–20) | (4–20) |
| 40-run mrCAE | (36–40) | (29–40) | (22–40) | (15–40) | (11–40) | (8—40) |
| 60-run mrCAE | (53–60) | (44–60) | (31–60) | (21–60) | (16–60) | (13–60) |
| 80-run mrCAE | (69–80) | (60–80) | (42–80) | (28–80) | (22—80) | (17–80) |
| 100-run mrCAE | (84–98) | (74–98) | (53–98) | (35–98) | (27–98) | (21–98) |
| 120-run mrCAE | (99–117) | (85–117) | (62–117) | (44–117) | (34–117) | (25–117) |
| BRCA | CHOL | COAD | KICH | KIRC | KIRP | LIHC | LUAD | LUSC | PRAD | READ | THCA | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11 | 0 | 3 | 3 | 31 | 15 | 1 | 22 | 18 | 4 | 4 | 10 | 76 |
| BRCA | CHOL | COAD | KICH | KIRC | KIRP | LIHC | LUAD | LUSC | PRAD | READ | THCA | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | 113 | 9 | 41 | 23 | 72 | 32 | 50 | 57 | 49 | 52 | 9 | 58 |
| Cancer | 1088 | 36 | 301 | 65 | 527 | 286 | 369 | 510 | 498 | 493 | 94 | 501 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Mamun, A.; Tanvir, R.B.; Sobhan, M.; Mathee, K.; Narasimhan, G.; Holt, G.E.; Mondal, A.M. Multi-Run Concrete Autoencoder to Identify Prognostic lncRNAs for 12 Cancers. Int. J. Mol. Sci. 2021, 22, 11919. https://doi.org/10.3390/ijms222111919
Al Mamun A, Tanvir RB, Sobhan M, Mathee K, Narasimhan G, Holt GE, Mondal AM. Multi-Run Concrete Autoencoder to Identify Prognostic lncRNAs for 12 Cancers. International Journal of Molecular Sciences. 2021; 22(21):11919. https://doi.org/10.3390/ijms222111919
Chicago/Turabian StyleAl Mamun, Abdullah, Raihanul Bari Tanvir, Masrur Sobhan, Kalai Mathee, Giri Narasimhan, Gregory E. Holt, and Ananda Mohan Mondal. 2021. "Multi-Run Concrete Autoencoder to Identify Prognostic lncRNAs for 12 Cancers" International Journal of Molecular Sciences 22, no. 21: 11919. https://doi.org/10.3390/ijms222111919
APA StyleAl Mamun, A., Tanvir, R. B., Sobhan, M., Mathee, K., Narasimhan, G., Holt, G. E., & Mondal, A. M. (2021). Multi-Run Concrete Autoencoder to Identify Prognostic lncRNAs for 12 Cancers. International Journal of Molecular Sciences, 22(21), 11919. https://doi.org/10.3390/ijms222111919