
Early-Life Origins of Metabolic Syndrome: Mechanisms and Preventive Aspects





Abstract
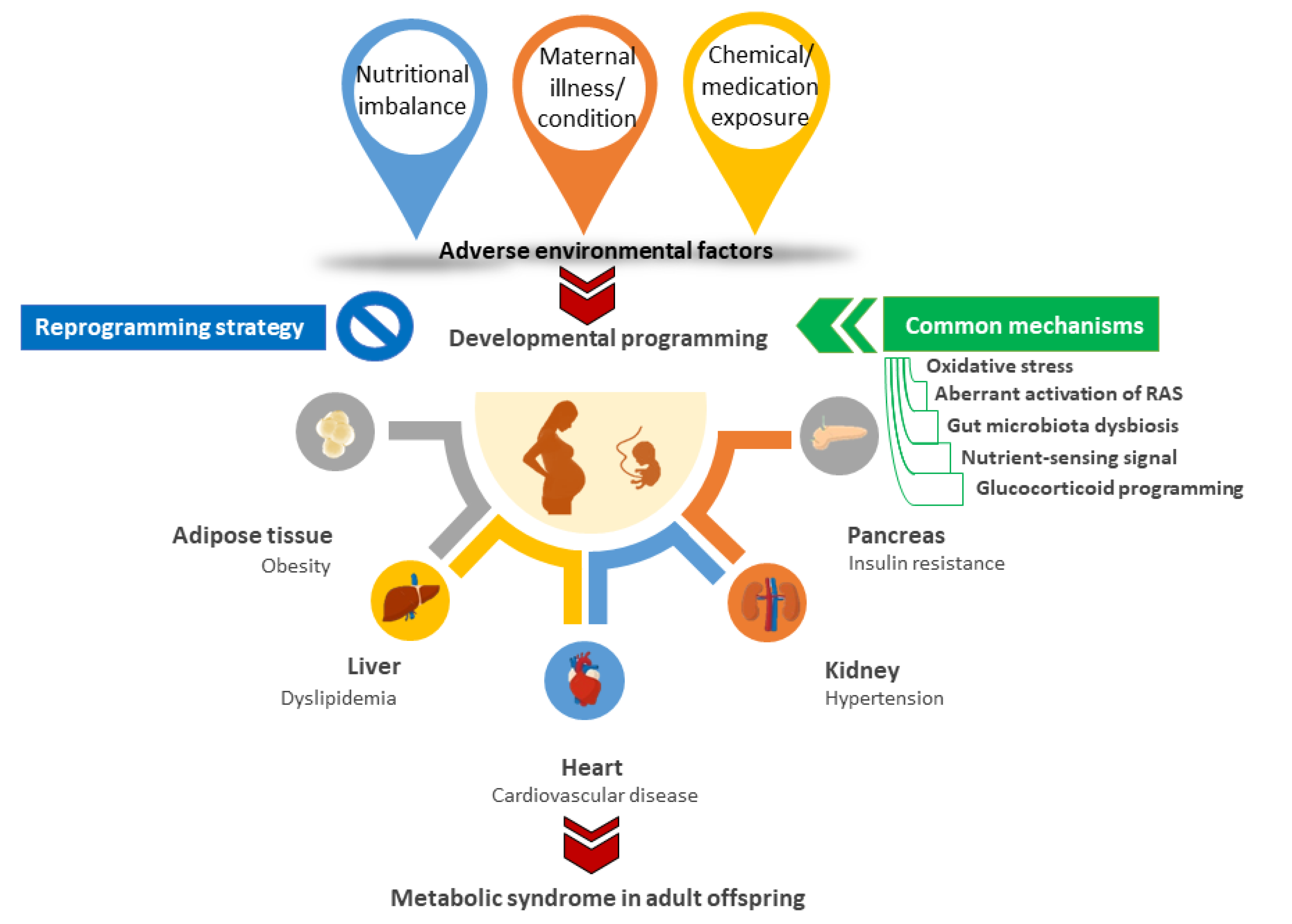
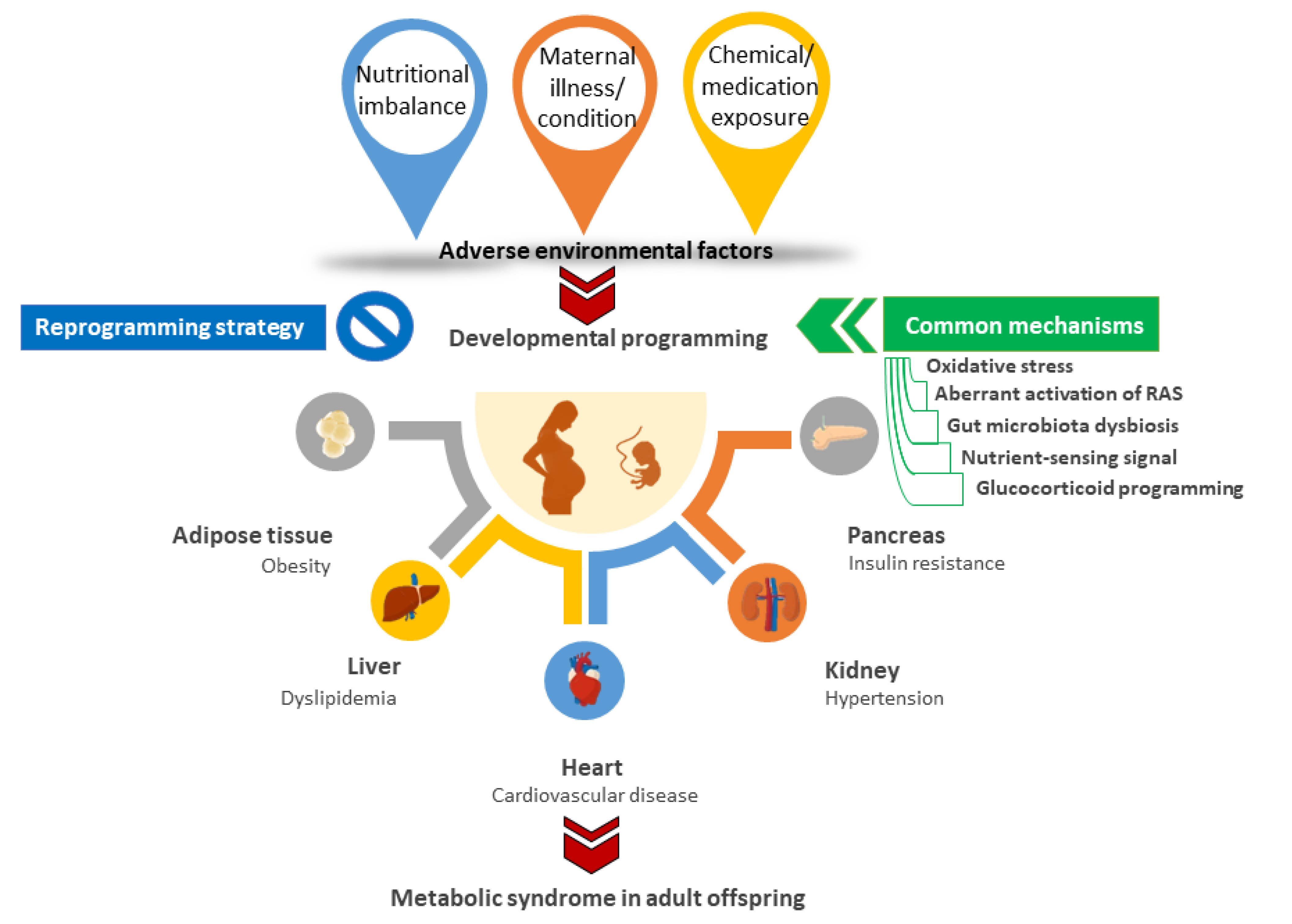
:1. Introduction
2. Epidemiological Evidence Linked Early-Life Insults with Offspring MetS
3. Animal Models for Developmental Origins of MetS
3.1. Nutritional Imbalance
3.2. Maternal Illnesses and Conditions
3.3. Chemical and Medication Exposures
4. Common Mechanisms behind Metabolic Syndrome of Developmental Origins
4.1. Oxidative Stress
4.2. Aberrant Activation of RAS
4.3. Gut Microbiota Dysbiosis
4.4. Dysregulated Nitrient-Sensing Signal
4.5. Glucocorticoid Programming
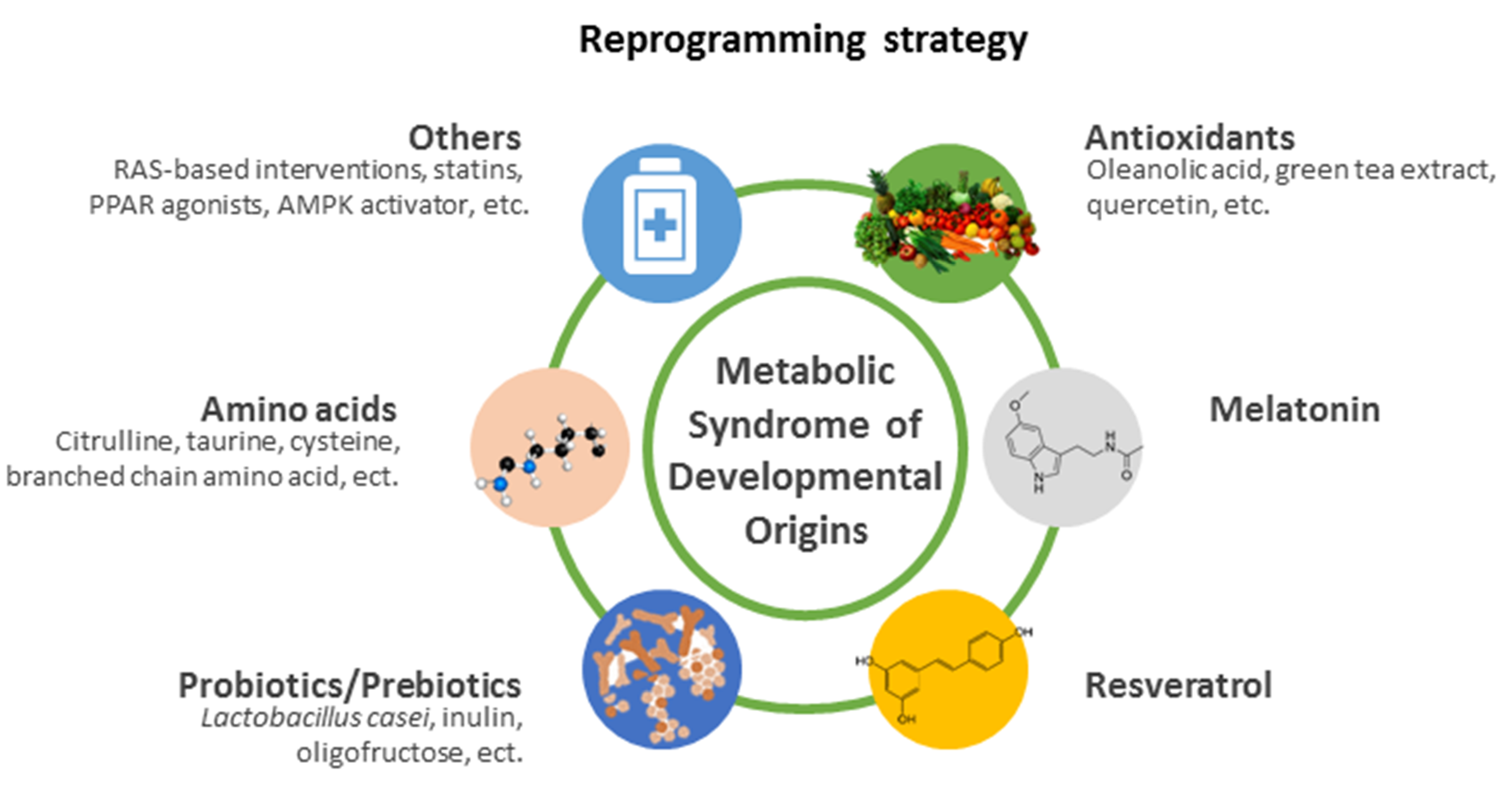
5. Reprogramming Strategy
5.1. Antioxidants
5.2. Melatonin
5.3. Resveratrol
5.4. Probiotics/Prebiotics
5.5. Amino Acids
5.6. Others
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Zarocostas, J. Need to increase focus on non-communicable diseases in global health, says WHO. Br. Med. J. 2010, 341, c7065. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M. The birth and future health of DOHaD. J. Dev. Orig. Health Dis. 2015, 6, 434–437. [Google Scholar] [CrossRef]
- Hanson, M.; Gluckman, P. Developmental origins of noncommunicable disease: Population and public health implications. Am. J. Clin. Nutr. 2011, 94, 1754S–1758S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A. The developmental origins of the metabolic syndrome. Trends Endocrinol. Metab. 2004, 15, 183–187. [Google Scholar] [CrossRef]
- Armitage, J.A.; Khan, I.Y.; Taylor, P.D.; Nathanielsz, P.W.; Poston, L. Developmental programming of the metabolic syndrome by maternal nutritional imbalance: How strong is the evidence from experimental models in mammals? J. Physiol. 2004, 561, 355–377. [Google Scholar] [CrossRef] [PubMed]
- de Gusmão Correia, M.L.; Volpato, A.M.; Águila, M.B.; Mandarim-de-Lacerda, C.A. Developmental origins of health and disease: Experimental and human evidence of fetal programming for metabolic syndrome. J. Hum. Hypertens. 2012, 26, 405–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, N.; Hardy, D.B. The Fetal Origins of the Metabolic Syndrome: Can We Intervene? J. Pregnancy 2012, 2012, 482690. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2015, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Lumey, L.H. Reproductive outcomes in women prenatally exposed to undernutrition: A review of findings from the Dutch famine birth cohort. Proc. Nutr. Soc. 1998, 57, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Schulz, L.C. The Dutch Hunger Winter and the Developmental Origins of Health and Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef] [Green Version]
- Stanner, S.A.; Yudkin, J.S. Fetal programming and the Leningrad Siege study. Twin Res. 2001, 4, 287–292. [Google Scholar] [CrossRef]
- Hult, M.; Tornhammar, P.; Ueda, P.; Chima, C.; Bonamy, A.K.; Ozumba, B.; Norman, M. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS ONE 2010, 5, e13582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Chen, Z.; Bartell, T.; Wang, X. Early Life Origins of Metabolic Syndrome: The Role of Environmental Toxicants. Curr. Environ. Health Rep. 2014, 1, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Bo, S.; Cavallo-Perin, P.; Ciccone, G.; Scaglione, L.; Pagano, G. The metabolic syndrome in twins: A consequence of low birth weight or of being a twin? Exp. Clin. Endocrinol. Diabetes 2001, 109, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Vaag, A.; Poulsen, P. Twins in metabolic and diabetes research: What do they tell us? Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 591–596. [Google Scholar] [CrossRef]
- Kelishadi, R.; Haghdoost, A.A.; Jamshidi, F.; Aliramezany, M.; Moosazadeh, M. Low birthweight or rapid catch-up growth: Which is more associated with cardiovascular disease and its risk factors in later life? A systematic review and cryptanalysis. Paediatr. Int. Child Health 2015, 35, 110–123. [Google Scholar] [CrossRef]
- Hrudey, E.J.; Reynolds, R.M.; Oostvogels, A.J.; Brouwer, I.A.; Vrijkotte, T.G. The association between maternal 25-hydroxyvitamin D concentration during gestation and early childhood cardio-metabolic outcomes: Is there interaction with pre-pregnancy BMI? PLoS ONE 2015, 10, e0133313. [Google Scholar] [CrossRef]
- Boney, C.M.; Verma, A.; Tucker, R.; Vohr, B.R. Metabolic syndrome in childhood: Association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005, 115, e290–e296. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.H.; Ma, R.C.W.; Ozaki, R.; Li, A.M.; Chan, M.H.M.; Yuen, L.Y.; Lao, T.T.H.; Yang, X.; Ho, C.S.; Tutino, G.E.; et al. In utero exposure to maternal hyperglycemia increases childhood cardiometabolic risk in offspring. Diabetes Care 2017, 40, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, A.; Tilling, K.; Macdonald-Wallis, C.; Sattar, N.; Brion, M.J.; Benfield, L.; Ness, A.; Deanfield, J.; Hingorani, A.; Nelson, S.M.; et al. Association of maternal weight gain in pregnancy with offspring obesity and metabolic and vascular traits in childhood. Circulation 2010, 121, 2557–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubry, A. Epigenetics as a Driver of Developmental Origins of Health and Disease: Did We Forget the Fathers? Bioessays 2018, 40, 1700113. [Google Scholar] [CrossRef]
- Eberle, C.; Kirchner, M.F.; Herden, R.; Stichling, S. Paternal metabolic and cardiovascular programming of their offspring: A systematic scoping review. PLoS ONE 2020, 15, e0244826. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease. Int. J. Mol. Sci. 2017, 18, 841. [Google Scholar] [CrossRef]
- Chong, E.; Yosypiv, I.V. Developmental programming of hypertension and kidney disease. Int. J. Nephrol. 2012, 2012, 760580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Tain, Y.L. Early Origins of Hypertension: Should Prevention Start Before Birth Using Natural Antioxidants? Antioxidants 2020, 9, 1034. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Animal Models for DOHaD Research: Focus on Hypertension of Developmental Origins. Biomedicines 2021, 9, 623. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal L-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar] [CrossRef]
- Franco Mdo, C.; Ponzio, B.F.; Gomes, G.N.; Gil, F.Z.; Tostes, R.; Carvalho, M.H.; Fortes, Z.B. Micronutrient prenatal supplementation prevents the development of hypertension and vascular endothelial damage induced by intrauterine malnutrition. Life Sci. 2009, 85, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Holemans, K.; Verhaeghe, J.; Dequeker, J.; Van Assche, F.A. Insulin sensitivity in adult female rats subjected to malnutrition during the perinatal period. J. Soc. Gynecol. Investig. 1996, 3, 71–77. [Google Scholar] [CrossRef]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Lê, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef] [PubMed]
- Ozanne, S.E.; Smith, G.D.; Tikerpae, J.; Hales, C.N. Altered regulation of hepatic glucose output in the male offspring of protein-malnourished rat dams. Am. J. Physiol. 1996, 270, E559–E564. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal Garlic Oil Supplementation Prevents High-Fat Diet-Induced Hypertension in Adult Rat Offspring: Implications of H2S-Generating Pathway in the Gut and Kidneys. Mol. Nutr. Food Res. 2021, 65, e2001116. [Google Scholar] [CrossRef]
- Tsai, T.A.; Tsai, C.K.; Huang, L.T.; Sheen, J.M.; Tiao, M.M.; Tain, Y.L.; Chen, C.C.; Lin, I.C.; Lai, Y.J.; Tsai, C.C.; et al. Maternal Resveratrol Treatment Re-Programs and Maternal High-Fat Diet-Induced Retroperitoneal Adiposity in Male Offspring. Int. J. Environ. Res. Public Health 2020, 17, 2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheen, J.M.; Yu, H.R.; Tain, Y.L.; Tsai, W.L.; Tiao, M.M.; Lin, I.C.; Tsai, C.C.; Lin, Y.J.; Huang, L.T. Combined maternal and postnatal high-fat diet leads to metabolic syndrome and is effectively reversed by resveratrol: A multiple-organ study. Sci. Rep. 2018, 8, 5607. [Google Scholar] [CrossRef]
- Wu, Z.; Zhao, J.; Xu, H.; Lyv, Y.; Feng, X.; Fang, Y.; Xu, Y. Maternal quercetin administration during gestation and lactation decrease endoplasmic reticulum stress and related inflammation in the adult offspring of obese female rats. Eur. J. Nutr. 2014, 53, 1669–1683. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Hou, C.Y.; Tain, Y.L. Maternal Administration of Probiotic or Prebiotic Prevents Male Adult Rat Offspring against Developmental Programming of Hypertension Induced by High Fructose Consumption in Pregnancy and Lactation. Nutrients 2018, 10, 1229. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.M.; Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Developmental programming of the metabolic syndrome: Next-generation sequencing analysis of transcriptome expression in a rat model of maternal high fructose intake. Sheng Li Xue Bao 2016, 68, 557–567. [Google Scholar]
- Saad, A.F.; Dickerson, J.; Kechichian, T.B.; Yin, H.; Gamble, P.; Salazar, A.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Costantine, M.M. High-fructose diet in pregnancy leads to fetal programming of hypertension, insulin resistance, and obesity in adult offspring. Am. J. Obstet. Gynecol. 2016, 215, 378.e1–378.e6. [Google Scholar] [CrossRef]
- Yamada-Obara, N.; Yamagishi, S.I.; Taguchi, K.; Kaida, Y.; Yokoro, M.; Nakayama, Y.; Ando, R.; Asanuma, K.; Matsui, T.; Ueda, S.; et al. Maternal exposure to high-fat and high-fructose diet evokes hypoadiponectinemia and kidney injury in rat offspring. Clin. Exp. Nephrol. 2016, 20, 853–886. [Google Scholar] [CrossRef]
- Li, M.; Reynolds, C.M.; Gray, C.; Patel, R.; Sloboda, D.M.; Vickers, M.H. Long-term effects of a maternal high-fat: High-fructose diet on offspring growth and metabolism and impact of maternal taurine supplementation. J. Dev. Orig. Health Dis. 2020, 11, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Bergel, E.; Belizán, J.M. A deficient maternal calcium intake during pregnancy increases blood pressure of the offspring in adult rats. BJOG 2002, 109, 540–545. [Google Scholar] [CrossRef]
- Park, S.; Kang, S.; Kim, D.S. Severe calcium deficiency increased visceral fat accumulation, down-regulating genes associated with fat oxidation, and increased insulin resistance while elevating serum parathyroid hormone in estrogen-deficient rats. Nutr. Res. 2020, 73, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Tomat, A.; Elesgaray, R.; Zago, V.; Fasoli, H.; Fellet, A.; Balaszczuk, A.M.; Schreier, L.; Costa, M.A.; Arranz, C. Exposure to zinc deficiency in fetal and postnatal life determines nitric oxide system activity and arterial blood pressure levels in adult rats. Br. J. Nutr. 2010, 104, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jou, M.Y.; Philipps, A.F.; Lönnerdal, B. Maternal zinc deficiency in rats affects growth and glucose metabolism in the offspring by inducing insulin resistance postnatally. J. Nutr. 2010, 140, 1621–1627. [Google Scholar] [CrossRef]
- Tare, M.; Emmett, S.J.; Coleman, H.A.; Skordilis, C.; Eyles, D.W.; Morley, R.; Parkington, H.C. Vitamin D insufficiency is associated with impaired vascular endothelial and smooth muscle function and hypertension in young rats. J. Physiol. 2011, 589, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chu, X.; Huang, Y.; Li, G.; Wang, Y.; Li, Y.; Sun, C. Maternal vitamin D deficiency during pregnancy results in insulin resistance in rat offspring, which is associated with inflammation and Iκbα methylation. Diabetologia 2014, 57, 2165–2172. [Google Scholar] [CrossRef]
- Wlodek, M.E.; Westcott, K.; Siebel, A.L.; Owens, J.A.; Moritz, K.M. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008, 74, 187–195. [Google Scholar] [CrossRef]
- Nüsken, K.D.; Dötsch, J.; Rauh, M.; Rascher, W.; Schneider, H. Uteroplacental insufficiency after bilateral uterine artery ligation in the rat: Impact on postnatal glucose and lipid metabolism and evidence for metabolic programming of the offspring by sham operation. Endocrinology 2008, 149, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.B.; Sarsour, N.; Salehi, M.; Schroering, A.; Mell, B.; Joe, B.; Hill, J.W. Prenatal androgen exposure causes hypertension and gut microbiota dysbiosis. Gut Microbes 2018, 9, 400–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuchowski, Y.; Dalmasso, C.; Shawky, N.M.; Reckelhoff, J.F. Cardiometabolic consequences of maternal hyperandrogenemia in male offspring. Physiol. Rep. 2021, 9, e14941. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, W.; Ciriello, J. Effect of maternal chronic intermittent hypoxia during gestation on offspring growth in the rat. Am. J. Obstet. Gynecol. 2013, 209, 564.e1–564.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, N.; Deng, Y.; Wei, Y.; Huang, Y.; Pu, X.; Li, L.; Zheng, Y.; Guo, J.; Yu, J.; et al. Ascorbic Acid Protects against Hypertension through Downregulation of ACE1 Gene Expression Mediated by Histone Deacetylation in Prenatal Inflammation-Induced Offspring. Sci. Rep. 2016, 6, 39469. [Google Scholar] [CrossRef]
- Tsosura, T.V.S.; Chiba, F.Y.; Mattera, M.S.L.C.; Pereira, R.F.; Cintra, L.T.A.; Conti, L.C.; Santos, R.M.D.; Mateus, J.H.P.; Garbin, C.A.S.; Sumida, D.H. Maternal apical periodontitis is associated with insulin resistance in adult offspring. Int. Endod. J. 2019, 52, 1040–1050. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar]
- Oliveira, A.C.; Andreotti, S.; Chimin, P.; Sertié, R.A.; Farias Tda, S.; Torres-Leal, F.L.; de Proença, A.R.; Campaña, A.B.; D’Avila, L.S.; Oliveira, K.A.; et al. Neonatal streptozotocin-induced diabetes in mothers promotes metabolic programming of adipose tissue in male rat offspring. Life Sci. 2015, 136, 151–156. [Google Scholar] [CrossRef]
- Thaeomor, A.; Teangphuck, P.; Chaisakul, J.; Seanthaweesuk, S.; Somparn, N.; Roysommuti, S. Perinatal Taurine Supplementation Prevents Metabolic and Cardiovascular Effects of Maternal Diabetes in Adult Rat Offspring. Adv. Exp. Med. Biol. 2017, 975, 295–305. [Google Scholar]
- Tain, Y.L.; Lin, Y.J.; Chan, J.Y.H.; Lee, C.T.; Hsu, C.N. Maternal melatonin or agomelatine therapy prevents programmed hypertension in male offspring of mother exposed to continuous light. Biol. Reprod. 2017, 97, 636–643. [Google Scholar] [CrossRef]
- Ferreira, D.S.; Amaral, F.G.; Mesquita, C.C.; Barbosa, A.P.; Lellis-Santos, C.; Turati, A.O.; Santos, L.R.; Sollon, C.S.; Gomes, P.R.; Faria, J.A.; et al. Maternal melatonin programs the daily pattern of energy metabolism in adult offspring. PLoS ONE 2012, 7, e38795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Song, L.; Wei, J.; Chen, T.; Chen, J.; Lin, Y.; Xia, W.; Xu, B.; Li, X.; Chen, X.; et al. Maternal exposure to di-(2-ethylhexyl)phthalate alters kidney development through the renin-angiotensin system in offspring. Toxicol. Lett. 2012, 212, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, G.; Bhaskaran, R.S.; Karundevi, B. Maternal di-(2-ethylhexyl) phthalate exposure alters hepatic insulin signal transduction and glucoregulatory events in rat F1 male offspring. J. Appl. Toxicol. 2019, 39, 751–763. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Tain, Y.L. Maternal exposure to bisphenol A combined with high-fat diet-induced programmed hypertension in adult male rat offspring: Effects of resveratrol. Int. J. Mol. Sci. 2019, 20, 4382. [Google Scholar] [CrossRef] [Green Version]
- Galyon, K.D.; Farshidi, F.; Han, G.; Ross, M.G.; Desai, M.; Jellyman, J.K. Maternal bisphenol A exposure alters rat offspring hepatic and skeletal muscle insulin signaling protein abundance. Am. J. Obstet. Gynecol. 2017, 216, 290.e1–290.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, S.P.; Denton, K.M.; Cullen-McEwen, L.; Bertram, J.F.; Moritz, K.M. Prenatal exposure to alcohol reduces nephron number and raises blood pressure in progeny. J. Am. Soc. Nephrol. 2010, 21, 1891–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.M.T.; Steane, S.E.; Moritz, K.M.; Akison, L.K. Prenatal alcohol exposure programmes offspring disease: Insulin resistance in adult males in a rat model of acute exposure. J. Physiol. 2019, 597, 5619–5637. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Huang, X.; Li, Y.; Dasgupta, C.; Wang, L.; Zhang, L. Antenatal Antioxidant Prevents Nicotine-Mediated Hypertensive Response in Rat Adult Offspring. Biol. Reprod. 2015, 93, 66. [Google Scholar] [CrossRef] [Green Version]
- Holloway, A.C.; Lim, G.E.; Petrik, J.J.; Foster, W.G.; Morrison, K.M.; Gerstein, H.C. Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia 2005, 48, 2661–2666. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef]
- Chang, H.Y.; Tain, Y.L. Postnatal dexamethasone-induced programmed hypertension is related to the regulation of melatonin and its receptors. Steroids 2016, 108, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nyirenda, M.J.; Lindsay, R.S.; Kenyon, C.J.; Burchell, A.; Seckl, J.R. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J. Clin. Investig. 1998, 101, 2174–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullen, S.; Mostyn, A. Animal models for the study of the developmental origins of health and disease. Proc. Nutr. Soc. 2009, 68, 306–320. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar]
- Hsu, C.N.; Tain, Y.L. The Double-Edged Sword Effects of Maternal Nutrition in the Developmental Programming of Hypertension. Nutrients 2018, 10, 1917. [Google Scholar] [CrossRef] [Green Version]
- Langley-Evans, S.C. Critical differences between two low protein diet protocols in the programming of hypertension in the rat. Int. J. Food Sci. Nutr. 2000, 51, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Schölmerich, J.; Bollheimer, L.C. High-fat diets: Modeling the metabolic disorders of human obesity in rodents. Obesity 2007, 15, 798–808. [Google Scholar] [CrossRef]
- Williams, L.; Seki, Y.; Vuguin, P.M.; Charron, M.J. Animal models of in utero exposure to a high fat diet: A review. Biochim. Biophys. Acta 2014, 1842, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.H.; Sheen, J.M.; Lin, I.C.; Yu, H.R.; Tiao, M.M.; Tain, Y.L.; Huang, L.T. Effects of Maternal Resveratrol on Maternal High-Fat Diet/Obesity with or without Postnatal High-Fat Diet. Int. J. Mol. Sci. 2020, 21, 3428. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lin, Y.J.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Tsai, C.C.; Huang, L.T.; Hsu, C.N. High Fat Diets Sex-Specifically Affect the Renal Transcriptome and Program Obesity, Kidney Injury, and Hypertension in the Offspring. Nutrients 2017, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Wu, K.L.H.; Leu, S.; Tain, Y.L. Translational insights on developmental origins of metabolic syndrome: Focus on fructose consumption. Biomed. J. 2018, 41, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J.B.; Yzydorczyk, C.; Siddeek, B.; Peyter, A.C.; Simeoni, U. Intrauterine growth restriction: Clinical consequences on health and disease at adulthood. Reprod. Toxicol. 2021, 99, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Bahri Khomami, M.; Joham, A.E.; Boyle, J.A.; Piltonen, T.; Silagy, M.; Arora, C.; Misso, M.L.; Teede, H.J.; Moran, L.J. Increased maternal pregnancy complications in polycystic ovary syndrome appear to be independent of obesity-A systematic review, meta-analysis, and meta-regression. Obes. Rev. 2019, 20, 659–674. [Google Scholar] [CrossRef]
- Kalagiri, R.R.; Carder, T.; Choudhury, S.; Vora, N.; Ballard, A.R.; Govande, V.; Drever, N.; Beeram, M.R.; Uddin, M.N. Inflammation in Complicated Pregnancy and Its Outcome. Am. J. Perinatol. 2016, 33, 1337–1356. [Google Scholar] [CrossRef]
- Saravanan, P.; Diabetes in Pregnancy Working Group; Maternal Medicine Clinical Study Group; Royal College of Obstetricians and Gynaecologists, UK. Gestational diabetes: Opportunities for improving maternal and child health. Lancet Diabetes Endocrinol. 2020, 8, 793–800. [Google Scholar] [CrossRef]
- Zimmet, P.; Alberti, K.G.M.M.; Stern, N.; Bilu, C.; El-Osta, A.; Einat, H.; Kronfeld-Schor, N. The Circadian Syndrome: Is the Metabolic Syndrome and much more! J. Intern. Med. 2019, 286, 181–191. [Google Scholar] [CrossRef]
- Lian, Y.; Yuan, Q.; Wang, G.; Tang, F. Association between sleep quality and metabolic syndrome: A systematic review and meta-analysis. Psychiatry Res. 2019, 274, 66–74. [Google Scholar] [CrossRef]
- Scheer, F.A.; Hilton, M.F.; Mantzoros, C.S.; Shea, S.A. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl. Acad. Sci. USA 2009, 106, 4453–4458. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Sumová, A.; Bendová, Z.; Sládek, M.; El-Hennamy, R.; Laurinová, K.; Jindráková, Z.; Illnerová, H. Setting the biological time in central and peripheral clocks during ontogenesis. FEBS Lett. 2006, 580, 2836–2842. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Tan, D.X.; Korkmaz, A.; Rosales-Corral, S.A. Melatonin and stable circadian rhythms optimize maternal, placental and fetal physiology. Hum. Reprod. Update 2014, 20, 293–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatani, Y.; Okamoto, K.; Hayashi, K.; Wakatsuki, A.; Tamura, S.; Sagara, Y. Maternal-fetal transfer of melatonin in pregnant women near term. J. Pineal Res. 1998, 25, 129–134. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Light and Circadian Signaling Pathway in Pregnancy: Programming of Adult Health and Disease. Int. J. Mol. Sci. 2020, 21, 2232. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Adverse Impact of Environmental Chemicals on Developmental Origins of Kidney Disease and Hypertension. Front. Endocrinol. 2021, 12, 745716. [Google Scholar] [CrossRef]
- Kirkley, A.G.; Sargis, R.M. Environmental endocrine disruption of energy metabolism and cardiovascular risk. Curr. Diabetes Rep. 2014, 14, 494. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, T.A. Cholinergic systems in brain development and disruption by neurotoxicants: Nicotine, environmental tobacco smoke, organophosphates. Toxicol. Appl. Pharmacol. 2004, 198, 132–151. [Google Scholar] [CrossRef] [PubMed]
- Slabiak-Blaz, N.; Adamczak, M.; Gut, N.; Grajoszek, A.; Nyengaard, J.R.; Ritz, E.; Wiecek, A. Administration of cyclosporine a in pregnant rats—The effect on blood pressure and on the glomerular number in their offspring. Kidney Blood Press. Res. 2015, 40, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Chan, J.Y.H.; Wu, K.L.H.; Yu, H.R.; Lee, W.C.; Hou, C.Y.; Tain, Y.L. Altered Gut Microbiota and Its Metabolites in Hypertension of Developmental Origins: Exploring Differences between Fructose and Antibiotics Exposure. Int. J. Mol. Sci. 2021, 22, 2674. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.J.; Sinclair, K.D. Paternal low protein diet affects adult offspring cardiovascular and metabolic function in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1444–H1452. [Google Scholar] [CrossRef] [Green Version]
- Masuyama, H.; Mitsui, T.; Eguchi, T.; Tamada, S.; Hiramatsu, Y. The effects of paternal high-fat diet exposure on offspring metabolism with epigenetic changes in the mouse adiponectin and leptin gene promoters. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E236–E245. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shi, X.; Hou, Y.; Cao, X.; Gong, L.; Wang, H.; Li, J.; Li, J.; Wu, C.; Xiao, D.; et al. Paternal hyperglycemia induces transgenerational inheritance of susceptibility to hepatic steatosis in rats involving altered methylation on Pparα promoter. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 147–160. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef]
- Bogdarina, I.; Welham, S.; King, P.J.; Burns, S.P.; Clark, A.J. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 2007, 100, 520–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Tain, Y.L. Targeting the Renin-Angiotensin-Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef]
- Jašarević, E.; Bale, T.L. Prenatal and postnatal contributions of the maternal microbiome on offspring programming. Front. Neuroendocrinol. 2019, 55, 100797. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Hou, C.Y.; Hsu, W.H.; Tain, Y.L. Cardiovascular Diseases of Developmental Origins: Preventive Aspects of Gut Microbiota-Targeted Therapy. Nutrients 2021, 13, 2290. [Google Scholar] [CrossRef]
- Cottrell, E.C.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Dennery, P.A. Oxidative stress in development: Nature or nurture? Free Radic. Biol. Med. 2010, 49, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Hanson, M.A. The developmental origins, mechanisms, and implications of metabolic syndrome. J. Nutr. 2010, 140, 648–652. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Chan, J.Y.; Hsu, C.N. Maternal fructose intake affects transcriptome changes and programmed hypertension in offspring in later life. Nutrients 2016, 8, 757. [Google Scholar] [CrossRef] [Green Version]
- Ching, R.H.; Yeung, L.O.; Tse, I.M.; Sit, W.H.; Li, E.T. Supplementation of bitter melon to rats fed a high-fructose diet during gestation and lactation ameliorates fructose-induced dyslipidemia and hepatic oxidative stress in male offspring. J. Nutr. 2011, 141, 1664e72. [Google Scholar] [CrossRef] [Green Version]
- Rodrıguez, L.; Otero, P.; Panadero, M.I.; Rodrigo, S.; Alvarez-Millan, J.J.; Bocos, C. Maternal fructose intake induces insulin resistance and oxidative stress in male, but not female, offspring. J. Nutr. Metab. 2015, 2015, 158091. [Google Scholar] [CrossRef] [PubMed]
- Litvinova, L.; Atochin, D.N.; Fattakhov, N.; Vasilenko, M.; Zatolokin, P.; Kirienkova, E. Nitric oxide and mitochondria in metabolic syndrome. Front. Physiol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Hsieh, C.S.; Chang, K.A.; Tain, Y.L. Roles of nitric oxide and asymmetric dimethylarginine in pregnancy and fetal programming. Int. J. Mol. Sci. 2012, 13, 14606–14622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Hsu, C.N. Toxic Dimethylarginines: Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Fan, E.; Zhang, L.; Jiang, S.; Bai, Y. Beneficial effects of resveratrol on atherosclerosis. J. Med. Food 2008, 11, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Vega, C.C.; Reyes-Castro, L.A.; Rodríguez-González, G.L.; Bautista, C.J.; Vázquez-Martínez, M.; Larrea, F.; Chamorro-Cevallos, G.A.; Nathanielsz, P.W.; Zambrano, E. Resveratrol partially prevents oxidative stress and metabolic dysfunction in pregnant rats fed a low protein diet and their offspring. J. Physiol. 2016, 594, 1483–1499. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Huang, L.T.; Tain, Y.L. Perinatal Use of Melatonin for Offspring Health: Focus on Cardiovascular and Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 5681. [Google Scholar] [CrossRef] [Green Version]
- Putnam, K.; Shoemaker, R.; Yiannikouris, F.; Cassis, L.A. The renin-angiotensin system: A target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1219–H1230. [Google Scholar] [CrossRef] [Green Version]
- Sahajpal, V.; Ashton, N. Increased glomerular angiotensin II binding in rats exposed to a maternal low protein diet in utero. J. Physiol. 2005, 563, 193–201. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Chan, J.Y.H.; Lee, C.T.; Tain, Y.L. Hypertension Programmed by Perinatal High-Fat Diet: Effect of Maternal Gut Microbiota-Targeted Therapy. Nutrients 2019, 11, 2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y.; Tain, Y.L. Aliskiren Administration during Early Postnatal Life Sex-Specifically Alleviates Hypertension Programmed by Maternal High Fructose Consumption. Front. Physiol. 2016, 7, 299. [Google Scholar] [CrossRef] [PubMed]
- Grigore, D.; Ojeda, N.B.; Robertson, E.B.; Dawson, A.S.; Huffman, C.A.; Bourassa, E.A.; Speth, R.C.; Brosnihan, K.B.; Alexander, B.T. Placental insufficiency results in temporal alterations in the renin angiotensin system in male hypertensive growth restricted offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R804–R811. [Google Scholar] [CrossRef] [Green Version]
- Walton, S.L.; Bielefeldt-Ohmann, H.; Singh, R.R.; Li, J.; Paravicini, T.M.; Little, M.H.; Moritz, K.M. Prenatal hypoxia leads to hypertension, renal renin-angiotensin system activation and exacerbates salt-induced pathology in a sex-specific manner. Sci. Rep. 2017, 7, 8241. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.Q.; Zhang, H.G.; Yuan, Z.B.; Yang, D.L.; Hao, L.Y.; Li, X.H. Prenatal exposure to lipopolysaccharide alters the intrarenal renin-angiotensin system and renal damage in offspring rats. Hypertens. Res. 2010, 33, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Chenier, I.; Tran, S.; Scotcher, M.; Chang, S.Y.; Zhang, S.L. Maternal diabetes programs hypertension and kidney injury in offspring. Pediatr. Nephrol. 2010, 25, 1319–1329. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Bessa, A.S.M.; Jesus, É.F.; Nunes, A.D.C.; Pontes, C.N.R.; Lacerda, I.S.; Costa, J.M.; Souza, E.J.; Lino-Júnior, R.S.; Biancardi, M.F.; Dos Santos, F.C.A.; et al. Stimulation of the ACE2/Ang-(1-7)/Mas axis in hypertensive pregnant rats attenuates cardiovascular dysfunction in adult male offspring. Hypertens. Res. 2019, 42, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early life: Implications for health outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef] [Green Version]
- Guimarães, K.S.L.; Braga, V.A.; Noronha, S.I.S.R.; Costa, W.K.A.D.; Makki, K.; Cruz, J.C.; Brandão, L.R.; Chianca Junior, D.A.; Meugnier, E.; Leulier, F.; et al. Lactiplantibacillus plantarum WJL administration during pregnancy and lactation improves lipid profile, insulin sensitivity and gut microbiota diversity in dyslipidemic dams and protects male offspring against cardiovascular dysfunction in later life. Food Funct. 2020, 11, 8939–8950. [Google Scholar] [CrossRef]
- De Oliveira, Y.; Cavalcante, R.G.S.; Cavalcanti Neto, M.P.; Magnani, M.; Braga, V.A.; de Souza, E.L.; de Brito Alves, J.L. Oral administration of Lactobacillus fermentum post-weaning improves the lipid profile and autonomic dysfunction in rat offspring exposed to maternal dyslipidemia. Food Funct. 2020, 11, 5581–5594. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on gut microbial metabolite trimethylamine-N-Oxide and short-chain fatty acid to prevent maternal high-fructose-diet-induced developmental programming of hypertension in adult male offspring. Mol. Nutr. Food Res. 2019, 63, e1900073. [Google Scholar] [CrossRef] [PubMed]
- Khodor, S.A.; Reichert, B.; Shatat, I.F. The microbiome and blood pressure: Can microbes regulate our blood pressure? Front. Pediatr. 2017, 5, 138. [Google Scholar] [CrossRef]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Developmental programming and reprogramming of hypertension and kidney disease: Impact of tryptophan metabolism. Int. J. Mol. Sci. 2020, 21, 8705. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatarek, P.; Kaluzna-Czaplinska, J. Trimethylamine N-oxide (TMAO) in human health. EXCLI J. 2021, 20, 301–319. [Google Scholar]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, H.A.; Collins, K.H.; Nicolucci, A.C.; Urbanski, S.J.; Hart, D.A.; Vogel, H.J.; Reimer, R.A. Maternal prebiotic supplementation reduces fatty liver development in offspring through altered microbial and metabolomic profiles in rats. FASEB J. 2019, 33, 5153–5167. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, T.; Powell, T.L. Role of placental nutrient sensing in developmental programming. Clin. Obstet. Gynecol. 2013, 56, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck, B.N.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 2007, 115, 2540–2548. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y. PPARs Link Early Life Nutritional insults to later programmed hypertension and metabolic syndrome. Int. J. Mol. Sci. 2015, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.S.; Cantó, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Preventive Aspects of Early Resveratrol Supplementation in Cardiovascular and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 4210. [Google Scholar] [CrossRef]
- Shah, A.; Reyes, L.M.; Morton, J.S.; Fung, D.; Schneider, J.; Davidge, S.T. Effect of resveratrol on metabolic and cardiovascular function in male and female adult offspring exposed to prenatal hypoxia and a high-fat diet. J. Physiol. 2016, 594, 1465–1482. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.H.; Leu, S.; Chan, J.Y.H. Resveratrol Prevents the Development of Hypertension Programmed by Maternal Plus Post-Weaning High-Fructose Consumption Through Modulation of Oxidative Stress, Nutrient-Sensing Signals, and Gut Microbiota. Mol. Nutr. Food Res. 2018, 62, e1800066. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lin, Y.J.; Sheen, J.M.; Lin, I.C.; Yu, H.R.; Huang, L.T.; Hsu, C.N. Resveratrol prevents the combined maternal plus post weaning high-fat-diets-induced hypertension in male offspring. J. Nutr. Biochem. 2017, 48, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Moisiadis, V.G.; Matthews, S.G. Glucocorticoids and fetal programming part 1: Outcomes. Nat. Rev. Endocrinol. 2014, 10, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.L.; Murphy, B.E. The maternal-fetal cortisol gradient during pregnancy and at delivery. J. Clin. Endocrinol. Metab. 1977, 45, 435–440. [Google Scholar] [CrossRef]
- Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Chang, H.Y.; Tain, Y.L. Prenatal dexamethasone-induced programmed hypertension and renal programming. Life Sci. 2015, 132, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Sferruzzi-Perri, A.N.; Camm, E.J. The Programming Power of the Placenta. Front. Physiol. 2016, 7, 33. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Amino Acids and Developmental Origins of Hypertension. Nutrients 2020, 12, 1763. [Google Scholar] [CrossRef]
- Wu, F.; Tian, F.J.; Lin, Y. Oxidative Stress in Placenta: Health and Diseases. BioMed Res. Int. 2015, 2015, 293271. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Goffau, M.C.; Lager, S.; Sovio, U.; Gaccioli, F.; Cook, E.; Peacock, S.J.; Parkhill, J.; Charnock-Jones, D.S.; Smith, G.C.S. Human placenta has no microbiome but can contain potential pathogens. Nature 2019, 572, 329–334. [Google Scholar] [CrossRef]
- Olaniyi, K.S.; Moodley, J.; Mahabeer, Y.; Mackraj, I. Placental Microbial Colonization and Its Association with Pre-eclampsia. Front. Cell. Infect. Microbiol. 2020, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, K.G.; Adeshina, K.A.; Bello, M.B.; Malami, I.; Abubakar, B.; Abubakar, M.B.; Imam, M.U. Prophylactic Use of Natural Products against Developmentally Programmed Metabolic Syndrome. Planta Med. 2021. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. The First Thousand Days: Kidney Health and Beyond. Healthcare 2021, 9, 1332. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Nimse, S.B.; Palb, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC. Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Matumba, M.G.; Ayeleso, A.O.; Nyakudya, T.; Erlwanger, K.; Chegou, N.N.; Mukwevho, E. Long-term impact of neonatal intake of oleanolic acid on the expression of AMP-activated protein kinase, adiponectin and inflammatory cytokines in rats fed with a high fructose diet. Nutrients 2019, 11, 226. [Google Scholar] [CrossRef] [Green Version]
- Nyakudya, T.T.; Mukwevho, E.; Erlwanger, K.H. The protective effect of neonatal oral administration of oleanolic acid against the subsequent development of fructose-induced metabolic dysfunction in male and female rats. Nutr. Metab. 2019, 15, 82. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tse, I.M.Y.; Li, E.T.S. Maternal green tea extract supplementation to rats fed a high-fat diet ameliorates insulin resistance in adult male offspring. J. Nutr. Biochem. 2012, 23, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiao, X.; Zhang, Q.; Zheng, J.; Deng, M. Maternal genistein intake mitigates the deleterious effects of high-fat diet on glucose and lipid metabolism and modulates gut microbiota in adult life of male mice. Front. Physiol. 2019, 10, 985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, H.; Nakamura, Y.; Terron, M.P.; Flores, L.J.; Manchester, L.C.; Tan, D.X.; Sugino, N.; Reiter, R.J. Melatonin and pregnancy in the human. Reprod. Toxicol. 2008, 25, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N.; Lee, C.T. Melatonin therapy prevents programmed hypertension and nitric oxide deficiency in offspring exposed to maternal caloric restriction. Oxid. Med. Cell. Longev. 2014, 2014, 283180. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N(G)-Nitro-L-arginine-methyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Lin, Y.J.; Yu, H.R.; Sheen, J.M.; Tain, Y.L.; Huang, L.T.; Tiao, M.M. Melatonin alleviates liver steatosis induced by prenatal dexamethasone exposure and postnatal high-fat diet. Exp. Ther. Med. 2018, 16, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N. Developmental Programming of Adult Disease: Reprogramming by Melatonin? Int. J. Mol. Sci. 2017, 18, 426. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.P.; Gögenur, I.; Rosenberg, J.; Reiter, R.J. The Safety of Melatonin in Humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef]
- Chen, Y.C.; Tain, Y.L.; Sheen, J.M.; Huang, L.T. Melatonin utility in neonates and children. J. Formos. Med. Assoc. 2012, 111, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Gerevini, G.T.; Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Developmental Programming of the Metabolic Syndrome: Can We Reprogram with Resveratrol? Int. J. Mol. Sci. 2018, 19, 2584. [Google Scholar] [CrossRef] [Green Version]
- Zou, T.; Chen, D.; Yang, Q.; Wang, B.; Zhu, M.J.; Nathanielsz, P.W.; Du, M. Resveratrol supplementation of high-fat diet-fed pregnant mice promotes brown and beige adipocyte development and prevents obesity in male offspring. J. Physiol. 2017, 595, 1547–1562. [Google Scholar] [CrossRef]
- Ros, P.; Díaz, F.; Freire-Regatillo, A.; Argente-Arizón, P.; Barrios, V.; Argente, J.; Chowen, J.A. Resveratrol Intake during Pregnancy and Lactation Modulates the Early Metabolic Effects of Maternal Nutrition Differently in Male and Female Offspring. Endocrinology 2018, 159, 810–825. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Rueda-Clausen, C.F.; Morton, J.S.; Davidge, S.T.; Dyck, J.R. Continued postnatal administration of resveratrol prevents diet-induced metabolic syndrome in rat offspring born growth restricted. Diabetes 2011, 60, 2274–2284. [Google Scholar] [CrossRef] [Green Version]
- Tenorio-Jiménez, C.; Martínez-Ramírez, M.J.; Gil, Á.; Gómez-Llorente, C. Effects of Probiotics on Metabolic Syndrome: A Systematic Review of Randomized Clinical Trials. Nutrients 2020, 12, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonson, M.; Boirie, Y.; Guillet, C. Protein, amino acids and obesity treatment. Rev. Endocr. Metab. Disord. 2020, 21, 341–353. [Google Scholar] [CrossRef]
- Koeners, M.P.; Racasan, S.; Koomans, H.A.; Joles, J.A.; Braam, B. Nitric oxide, superoxide and renal blood flow autoregulation in SHR after perinatal L-arginine and antioxidants. Acta Physiol. 2007, 190, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Roysommuti, S.; Lerdweeraphon, W.; Malila, P.; Jirakulsomchok, D.; Wyss, J.M. Perinatal taurine alters arterial pressure control and renal function in adult offspring. Adv. Exp. Med. Biol. 2009, 643, 145–156. [Google Scholar] [PubMed] [Green Version]
- Tain, Y.L.; Huang, L.T.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal citrulline supplementation prevents prenatal NG-nitro-l-arginine-methyl ester (L-NAME)-induced programmed hypertension in rats. Biol. Reprod. 2015, 92, 7. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Early supplementation of D-cysteine or L-cysteine prevents hypertension and kidney damage in spontaneously hypertensive rats exposed to high-salt intake. Mol. Nutr. Food Res. 2018, 62, 2. [Google Scholar] [CrossRef]
- Fujii, T.; Yura, S.; Tatsumi, K.; Kondoh, E.; Mogami, H.; Fujita, K.; Kakui, K.; Aoe, S.; Itoh, H.; Sagawa, N.; et al. Branched-chain amino acid supplemented diet during maternal food restriction prevents developmental hypertension in adult rat offspring. J. Dev. Orig. Health Dis. 2011, 2, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Cynober, L.; Moinard, C.; De Bandt, J.P. The 2009 ESPEN Sir David Cuthbertson. Citrulline: A new major signaling molecule or just another player in the pharmaconutrition game? Clin. Nutr. 2010, 29, 545–551. [Google Scholar] [CrossRef]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Sarfati, G.; Nubret, E.; Kapel, N.; Waligora-Dupriet, A.J.; Bergheim, I.; Cynober, L.; De-Bandt, J.P. Effect of specific amino acids on hepatic lipid metabolism in fructose-induced non-alcoholic fatty liver disease. Clin. Nutr. 2016, 35, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Dias, S.C.; Obonye, N.; Johnson, R.; Louw, J.; Nkambule, B.B. A Systematic Review on the Protective Effect of N-Acetyl Cysteine against Diabetes-Associated Cardiovascular Complications. Am. J. Cardiovasc. Drugs 2018, 18, 283–298. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rask Larsen, J.; Dima, L.; Correll, C.U.; Manu, P. The pharmacological management of metabolic syndrome. Expert Rev. Clin. Pharmacol. 2018, 11, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Elahi, M.M.; Cagampang, F.R.; Ohri, S.K.; Hanson, M.A. Long-term statin administration to dams on high-fat diet protects not only them but also their offspring from cardiovascular risk. Ann. Nutr. Metab. 2013, 62, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Elahi, M.M.; Cagampang, F.R.; Anthony, F.W.; Curzen, N.; Ohri, S.K.; Hanson, M.A. Statin treatment in hypercholesterolemic pregnant mice reduces cardiovascular risk factors in their offspring. Hypertension 2008, 51, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhl, E.S.; Jessen, N.; Pold, R.; Ledet, T.; Flyvbjerg, A.; Pedersen, S.B.; Pedersen, O.; Schmitz, O.; Lund, S. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes 2002, 51, 2199–2206. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.L.; Hsu, C.N.; Tain, Y.L. Whether AICAR in Pregnancy or Lactation Prevents Hypertension Programmed by High Saturated Fat Diet: A Pilot Study. Nutrients 2020, 12, 448. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Animal Models | Timing | Experimental Animal | Offspring Outcomes Relating to MetS |
|---|---|---|---|
| Nutritional imbalance | |||
| Caloric restriction | Pregnancy and lactation | SD rats [30]/Wistar rats [31,32] | ↑BP 12-16 wk [30,31], ↓insulin levels and exhibit insulin resistance 14 wk [32] |
| Protein restriction | Pregnancy | Wistar rats | ↑BP 12 wk [33], exhibit insulin resistance 12 wk [34] |
| High-fat diet | Pregnancy and lactation | SD rats | ↑BP 16 wk [35], ↑adiposity 16 wk [36], dyslipidemia 16 wk [37], ↑BW, exhibit dyslipidemia and hyperinsulinemia 100 day [38] |
| High-fructose diet | Pregnancy and lactation | SD rats/C57BL6J mice | ↑BP [39], exhibit insulin resistance and dyslipidemia 12 wk [40], ↑BP, exhibit insulin resistance and obesity 1 year [41] |
| High-fructose diet plus high-fat diet | Pregnancy and lactation | SD rats [42]/Wistar rats [43] | ↑BP 16 wk [42], ↑BW and adiposity 150 days [43] |
| Calcium-deficient diet | Pregnancy | WKY rats [44]/SD rats [45] | ↑BP 1 year [44], ↑adiposity, exhibit insulin resistance and dyslipidemia [45] |
| Zinc-deficient diet | Pregnancy and lactation | Wistar rats [46]/SD rats [47] | ↑BP 12 wk [46], ↑BW, exhibit insulin resistance 15 wk [47] |
| Vitamin D restricted diet | Pregnancy and lactation | SD rats | ↑BP 8 wk [48], exhibit insulin resistance 16 wk [49] |
| Maternal illness/condition | |||
| Uteroplacental insufficiency | Pregnancy | WKY rats [50]/Wistar rats [51] | ↑BP 22 wk [50], exhibit dyslipidemia and insulin resistance 30 wk [51] |
| Polycystic ovary syndrome | Pregnancy | Wistar rat [52], SD rats [53] | ↑BP 120 days [52], exhibit dyslipidemia 16 wk [53] |
| Maternal hypoxia | Pregnancy | Wistar rats [52]/SD rats [54] | ↑BP 4 mo [52], ↑BW and adiposity, exhibit insulin resistance 12 wk [54] |
| Maternal inflammation | Pregnancy | SD rats [55]/Wistar rats [56] | ↑BP 12 wk [55], insulin resistance 75 days [56] |
| Diabetes | Pregnancy | SD rats [57]/Wistar rats [58,59] | ↑BP 12 wk [57], ↑BW and adiposity 12 wk [58], exhibit insulin resistance and dyslipidemia 16 wk [59] |
| Chronodisruption | Pregnancy and lactation | SD rats [60]/Wistar rats [61] | ↑BP 12 wk [60], exhibit insulin resistance 18 wk [61] |
| Chemical/medication exposure | |||
| DEHP | Pregnancy | Wistar rats | ↑BP 21 wk [62], exhibit insulin resistance 80 day [63] |
| BPA | Pregnancy and lactation | SD rats | ↑BP 16 wk [64], exhibit insulin resistance 6 mo [65] |
| Alcohol | Pregnancy | SD rats | ↑BP 6 mo [66], exhibit insulin resistance 6 mo [67] |
| Nicotine | Pregnancy | Wistar rats | ↑BP 8 mo [68], ↑BW and adiposity, exhibit insulin resistance [69] |
| Glucocorticoid | Pregnancy and postnatal days1-3 | SD rats [70,71]/Wistar rats [72] | ↑BP 12 mo [70,71], exhibit insulin resistance 6 mo [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-N.; Hou, C.-Y.; Hsu, W.-H.; Tain, Y.-L. Early-Life Origins of Metabolic Syndrome: Mechanisms and Preventive Aspects. Int. J. Mol. Sci. 2021, 22, 11872. https://doi.org/10.3390/ijms222111872
Hsu C-N, Hou C-Y, Hsu W-H, Tain Y-L. Early-Life Origins of Metabolic Syndrome: Mechanisms and Preventive Aspects. International Journal of Molecular Sciences. 2021; 22(21):11872. https://doi.org/10.3390/ijms222111872
Chicago/Turabian StyleHsu, Chien-Ning, Chih-Yao Hou, Wei-Hsuan Hsu, and You-Lin Tain. 2021. "Early-Life Origins of Metabolic Syndrome: Mechanisms and Preventive Aspects" International Journal of Molecular Sciences 22, no. 21: 11872. https://doi.org/10.3390/ijms222111872
APA StyleHsu, C.-N., Hou, C.-Y., Hsu, W.-H., & Tain, Y.-L. (2021). Early-Life Origins of Metabolic Syndrome: Mechanisms and Preventive Aspects. International Journal of Molecular Sciences, 22(21), 11872. https://doi.org/10.3390/ijms222111872