
Regulation of Endoplasmic Reticulum–Mitochondria Tethering and Ca2+ Fluxes by TDP-43 via GSK3β

 , , , ,
, , , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
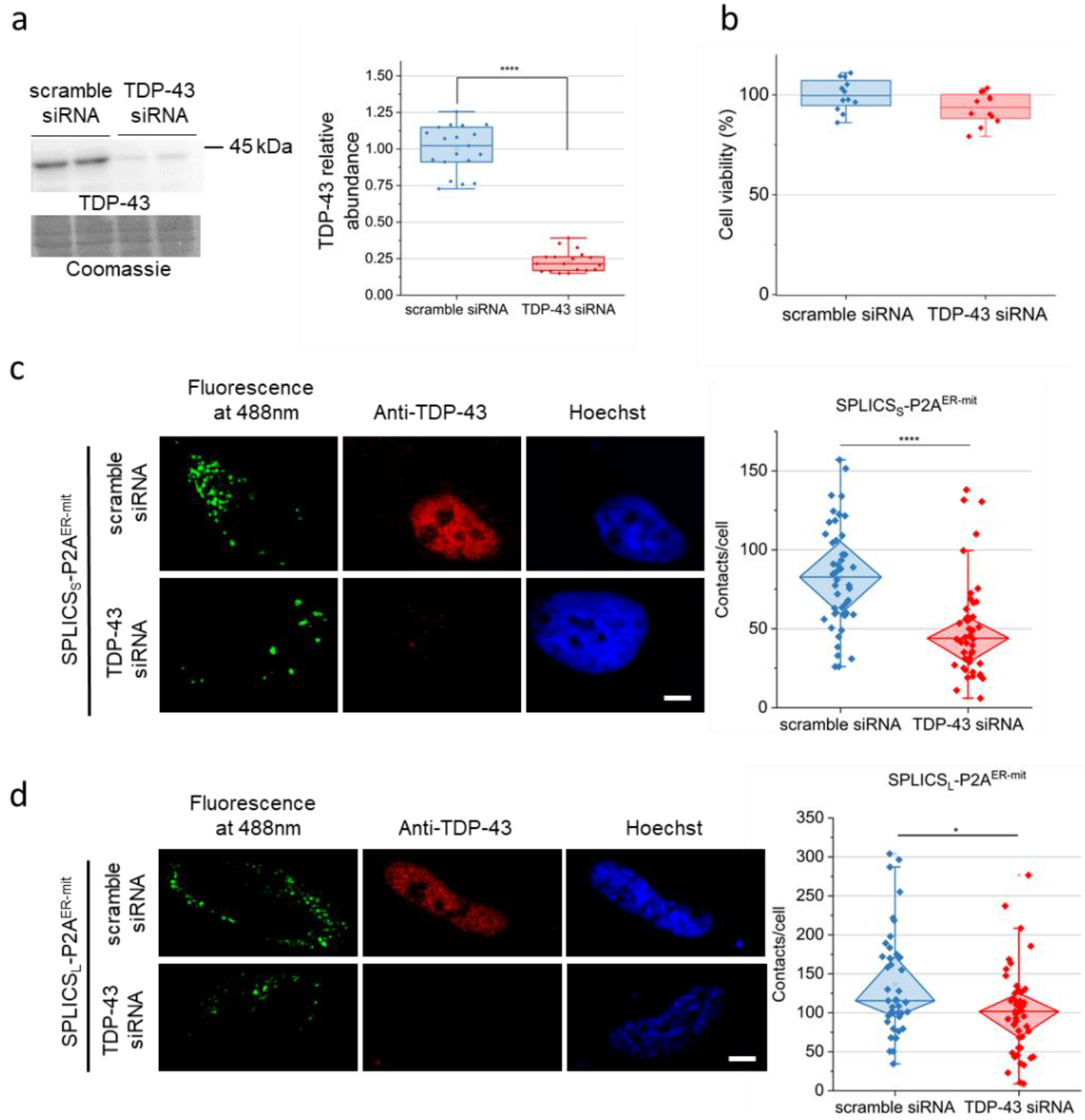
2.1. TDP-43 down-Regulation Affects ER–Mitochondria Tethering
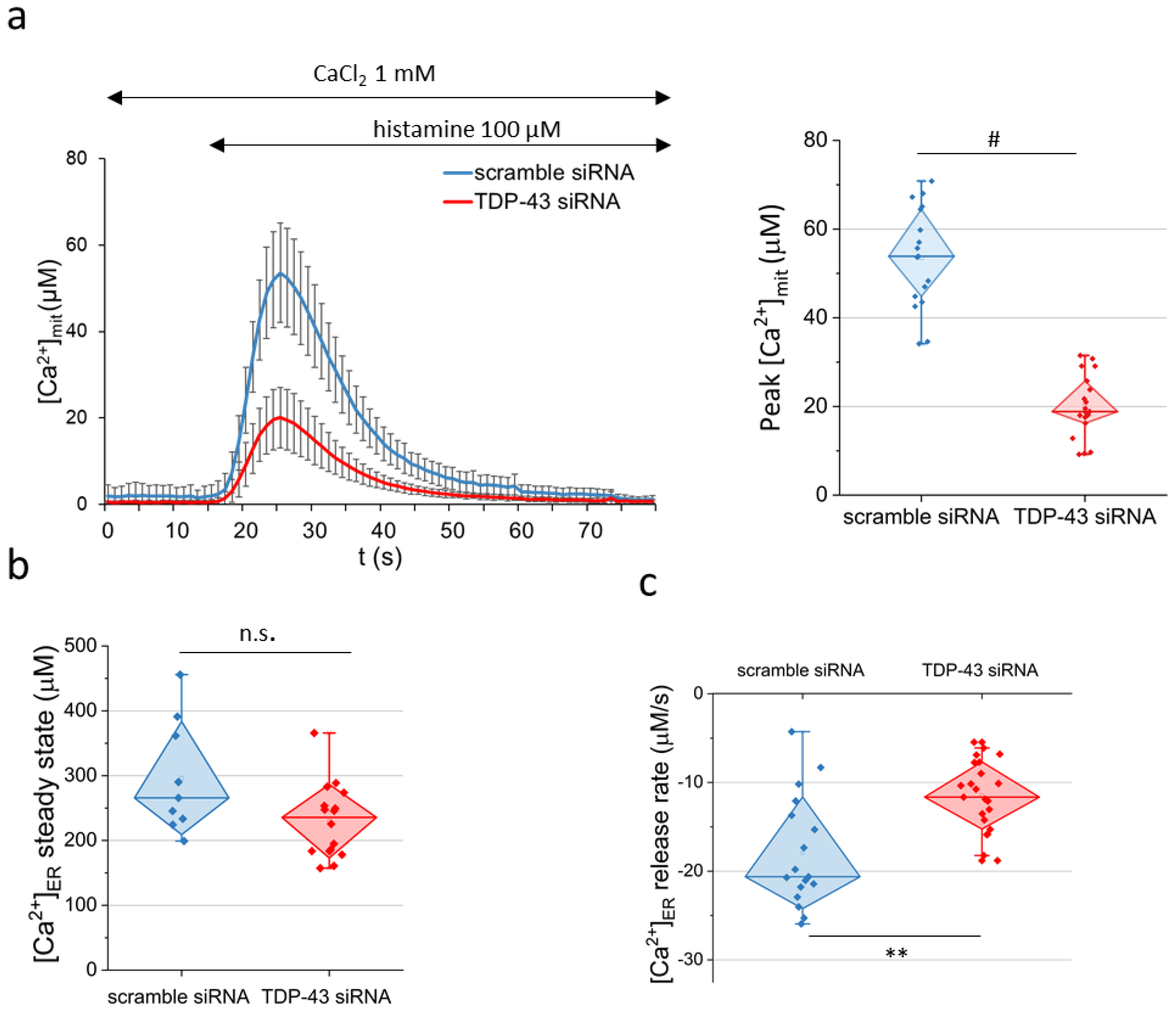
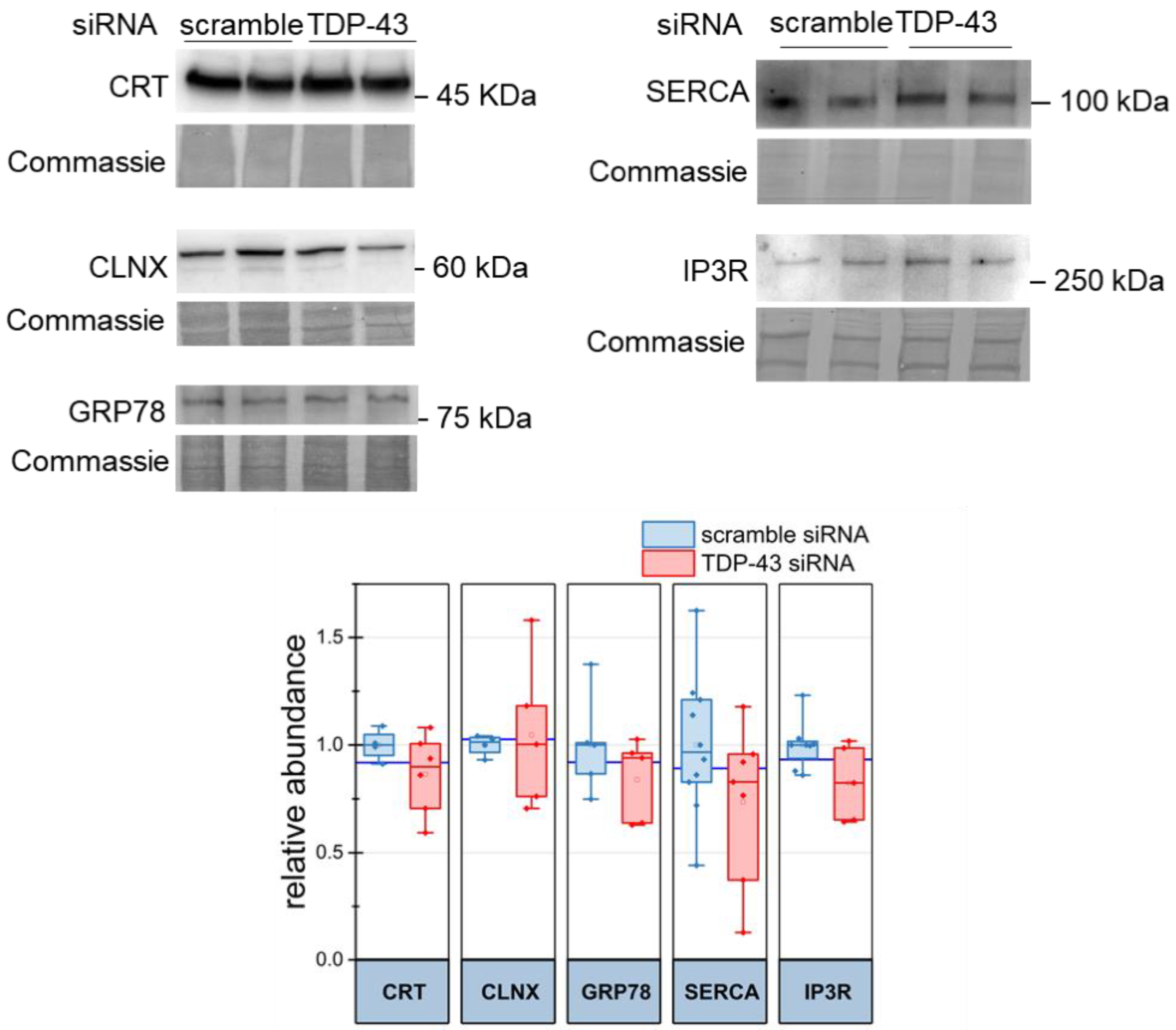
2.2. Dysregulation of Mitochondria Ca2+ Uptake and ER Ca2+ Discharge in TDP-43 Knock-Down HeLa Cells
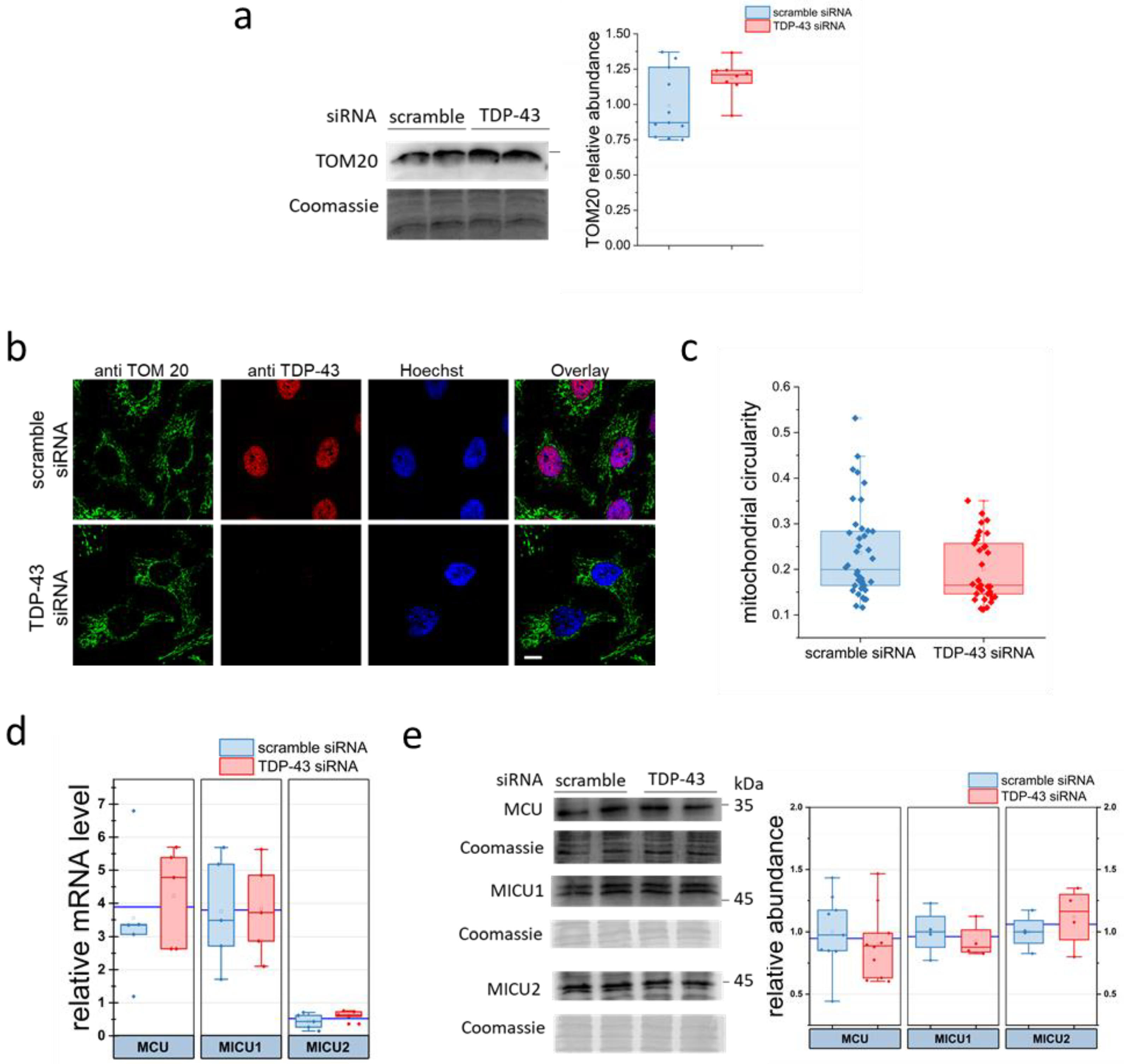
2.3. Evaluation of Mitochondrial Parameters in TDP-43 Knock-Down HeLa Cells
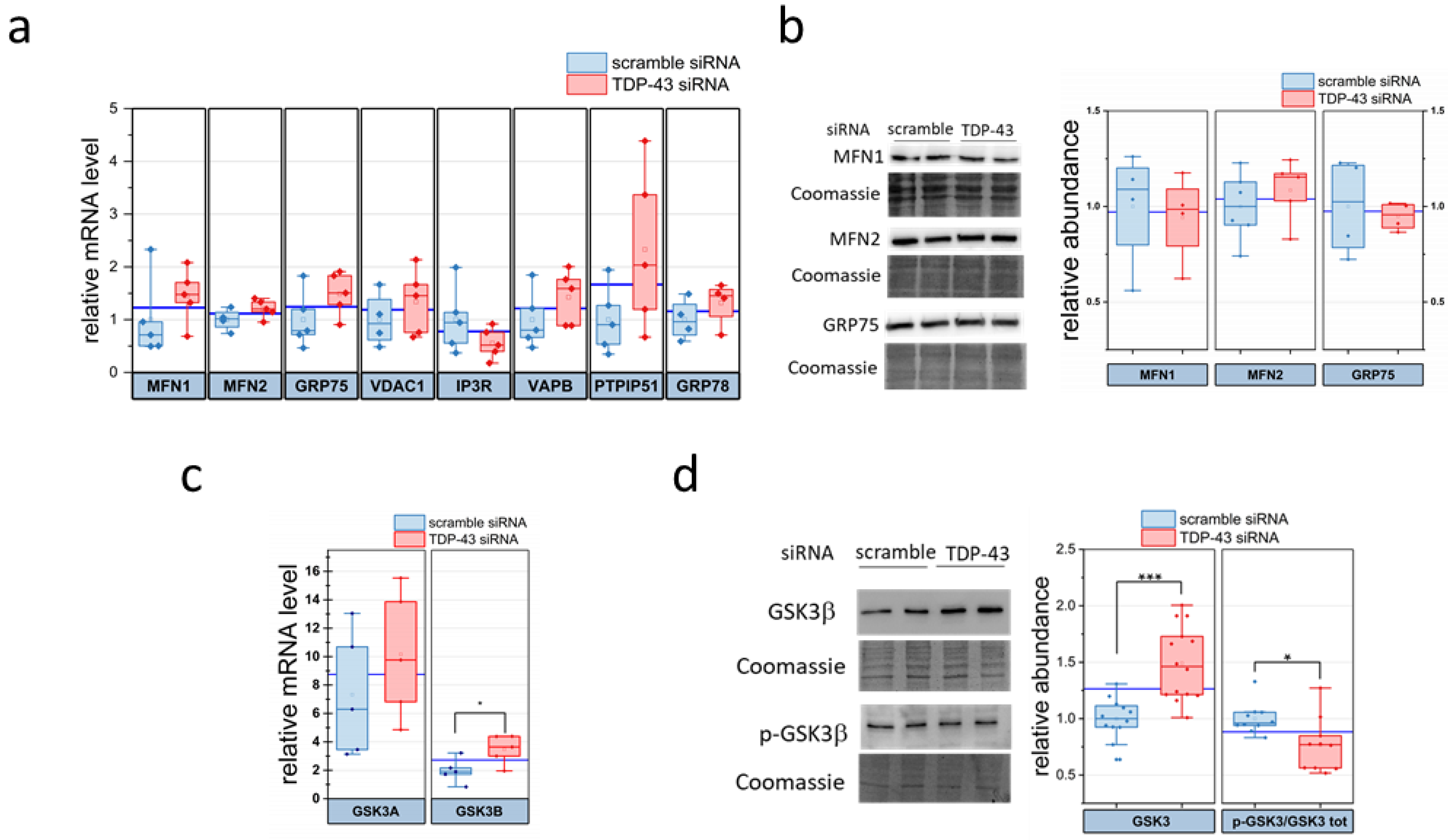
2.4. TDP-43 Alters the Abundance of the ER–Mitochondria Contacts Regulator GSK3β
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Cultures, siRNA-Based TDP-43 Silencing and Cell Viability Assay
4.3. Transfection with Plasmids Coding for SPLIC Probes
4.4. Immunocytochemistry
4.5. Confocal Microscopy, Image Acquisition and Processing
4.6. AEQ-Based [Ca2+] Measurements
4.7. WB Analysis
4.8. RNA Isolation and Real-Time RT-PCR
4.9. Statistical Analysis and Graphics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.R.; Dyer, R.F.; Skalko, R.G. Continuities between mitochondria and endoplasmic reticulum in the mammalian ovary. Z. Zellforsch Mikrosk. Anat. 1969, 97, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. Chapter Four—The Role of Mitochondria-Associated Membranes in Cellular Homeostasis and Diseases. In Biology of the Endoplasmic Reticulum; Kepp, O., Galluzzi, L.B.T.-I.R.C., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 350, pp. 119–196. ISBN 1937-6448. [Google Scholar]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- English, A.M.; Schuler, M.-H.; Xiao, T.; Kornmann, B.; Shaw, J.M.; Hughes, A.L. ER-mitochondria contacts promote mitochondrial-derived compartment biogenesis. J. Cell Biol. 2020, 219, e202002144. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Vance, J.E. Phosphatidylserine Synthase-1 and -2 Are Localized to Mitochondria-associated Membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Bittremieux, M.; Missiroli, S.; Morganti, C.; Patergnani, S.; Sbano, L.; Rimessi, A.; Kerkhofs, M.; Parys, J.B.; Bultynck, G.; et al. Endoplasmic Reticulum-Mitochondria Communication 1 Through Ca(2+) S. Adv. Exp. Med. Biol. 2017, 997, 49–67. [Google Scholar]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER–mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.L.; Metzakopian, E. ER-mitochondria contact sites in neurodegeneration: Genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 2021, 28, 1804–1821. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, X.; Tong, C. Endoplasmic Reticulum–Mitochondria Contact Sites and Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 428. [Google Scholar] [CrossRef]
- Manfredi, G.; Kawamata, H. Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 90, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 1789. [Google Scholar] [CrossRef]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- Bernard-Marissal, N.; Chrast, R.; Schneider, B.L. Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: A broken relationship? Cell Death Dis. 2018, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.R.; Walters, R.; Waldron, F.M.; Pal, S.; Selvaraj, B.T.; Macleod, M.R.; Hardingham, G.E.; Chandran, S.; Gregory, J.M. Targeting mitochondrial dysfunction in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Brain Commun. 2019, 1, fcz009. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.-P. Mitochondria in Amyotrophic Lateral Sclerosis: A Trigger and a Target. Neurodegener. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.J.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Misteli, T.; Baralle, F.E. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. USA. 2008, 105, 3785–3789. [Google Scholar] [CrossRef] [Green Version]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Chen, X.; Fan, Z.; McGee, W.; Chen, M.; Kong, R.; Wen, P.; Xiao, T.; Chen, X.; Liu, J.; Zhu, L.; et al. TDP-43 regulates cancer-associated microRNAs. Protein Cell 2018, 9, 848–866. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Bennett, C.F.; Cleveland, D.W.; Yeo, G.W. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baralle, F.E.; Buratti, E. RNA and splicing regulation in neurodegeneration. Mol. Cell. Neurosci. 2013, 56, 404–405. [Google Scholar] [CrossRef]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 2012, 287, 15635–15647. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.B.; Lee, V.M.Y.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.S. Does a loss of TDP-43 function cause neurodegeneration? Mol. Neurodegener. 2012, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prpar Mihevc, S.; Baralle, M.; Buratti, E.; Rogelj, B. TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009, 583, 1586–1592. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Klima, R.; Feiguin, F. TDP-43 prevents retrotransposon activation in the Drosophila motor system through regulation of Dicer-2 activity. BMC Biol. 2020, 18, 82. [Google Scholar] [CrossRef]
- Giamogante, F.; Barazzuol, L.; Brini, M.; Calì, T. ER–Mitochondria Contact Sites Reporters: Strengths and Weaknesses of the Available Approaches. Int. J. Mol. Sci. 2020, 21, 8157. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Brini, M. Split Green Fluorescent Protein-Based Contact Site Sensor (SPLICS ) for Heterotypic Organelle Juxtaposition as Applied to ER -Mitochondria Proximities. Methods Mol. Biol. 2021, 2275, 363–378. [Google Scholar] [PubMed]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018, 25, 1131–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3b to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Lau, D.H.W.; Hartopp, N.; Welsh, N.J.; Mueller, S.; Glennon, E.B.; Mórotz, G.M.; Annibali, A.; Gomez-Suaga, P.; Stoica, R.; Paillusson, S.; et al. Disruption of ER−mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis. 2018, 9, 327. [Google Scholar] [CrossRef] [Green Version]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef] [Green Version]
- Vallese, F.; Catoni, C.; Cieri, D.; Barazzuol, L.; Ramirez, O.; Calore, V.; Bonora, M.; Giamogante, F.; Pinton, P.; Brini, M.; et al. An expanded palette of improved SPLICS reporters detects multiple organelle contacts in vitro and in vivo. Nat. Commun. 2020, 11, 6069. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Massimino, M.L.; Stella, R.; Bortolotto, R.; Agostini, J.; Maldi, A.; Sartori, G.; Tonello, F.; Bertoli, A.; Lopreiato, R. Nucleolin Rescues TDP-43 Toxicity in Yeast and Human Cell Models. Front. Cell. Neurosci. 2021, 15, 115. [Google Scholar] [CrossRef]
- Perry, A.J.; Rimmer, K.A.; Mertens, H.D.T.; Waller, R.F.; Mulhern, T.D.; Lithgow, T.; Gooley, P.R. Structure, topology and function of the translocase of the outer membrane of mitochondria. Plant Physiol. Biochem. 2008, 46, 265–274. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Alevriadou, B.R.; Patel, A.; Noble, M.; Ghosh, S.; Gohil, V.M.; Stathopulos, P.B.; Madesh, M. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol.—Cell Physiol. 2021, 320, C465–C482. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-Associated Membranes: Composition, Molecular Mechanisms, and Physiopathological Implications. Antioxidants Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca2+ transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta—Bioenerg. 2009, 1787, 1342–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, H.; Peixoto, P.; Konrad, C.; Palomo, G.; Bredvik, K.; Gerges, M.; Valsecchi, F.; Petrucelli, L.; Ravits, J.M.; Starkov, A.; et al. Mutant TDP-43 does not impair mitochondrial bioenergetics in vitro and in vivo. Mol. Neurodegener. 2017, 12, 1–15. [Google Scholar] [CrossRef]
- Andres-Benito, P.; Gelpi, E.; Povedano, M.; Santpere, G.; Ferrer, I. Gene expression profile in frontal cortex in sporadic frontotemporal lobar degeneration-TDP. J. Neuropathol. Exp. Neurol. 2018, 77, 608–627. [Google Scholar] [CrossRef]
- Briese, M.; Saal-Bauernschubert, L.; Lüningschrör, P.; Moradi, M.; Dombert, B.; Surrey, V.; Appenzeller, S.; Deng, C.; Jablonka, S.; Sendtner, M. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 2020, 8, 116. [Google Scholar] [CrossRef]
- Vanden Broeck, L.; Callaerts, P.; Dermaut, B. TDP-43-mediated neurodegeneration: Towards a loss-of-function hypothesis? Trends Mol. Med. 2014, 20, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Sephton, C.F.; Cenik, B.; Cenik, B.K.; Herz, J.; Yu, G. TDP-43 in central nervous system development and function: Clues to TDP-43-associated neurodegeneration. Biol. Chem. 2012, 393, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godena, V.K.; Romano, G.; Romano, M.; Appocher, C.; Klima, R.; Buratti, E.; Baralle, F.E.; Feiguin, F. TDP-43 regulates drosophila neuromuscular junctions growth by modulating futsch/MAP1B levels and synaptic microtubules organization. PLoS ONE 2011, 6, e17808. [Google Scholar] [CrossRef] [Green Version]
- Arnold, E.S.; Ling, S.C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018, 146, 7–20. [Google Scholar] [CrossRef]
- Sasaki, S.; Takeda, T.; Shibata, N.; Kobayashi, M. Alterations in subcellular localization of TDP-43 immunoreactivity in the anterior horns in sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 2010, 478, 72–76. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.E18. [Google Scholar] [CrossRef] [PubMed]
- Izumikawa, K.; Nobe, Y.; Yoshikawa, H.; Ishikawa, H.; Miura, Y.; Nakayama, H.; Nonaka, T.; Hasegawa, M.; Egawa, N.; Inoue, H.; et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luty, A.A.; Kwok, J.B.J.; Dobson-Stone, C.; Loy, C.T.; Coupland, K.G.; Karlström, H.; Sobow, T.; Tchorzewska, J.; Maruszak, A.; Barcikowska, M.; et al. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann. Neurol. 2010, 68, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; Médard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T. The Sigma Receptor: Evolution of the Concept in Neuropsychopharmacology. Curr. Neuropharmacol. 2005, 3, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Haass, C. Fused in sarcoma (FUS): An oncogene goes awry in neurodegeneration. Mol. Cell. Neurosci. 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langou, K.; Moumen, A.; Pellegrino, C.; Aebischer, J.; Medina, I.; Aebischer, P.; Raoul, C. AAV-mediated expression of wild-type and ALS-linked mutant VAPB selectively triggers death of motoneurons through a Ca2+-dependent ER-associated pathway. J. Neurochem. 2010, 114, 795–809. [Google Scholar] [CrossRef]
- Mórotz, G.M.; De Vos, K.J.; Vagnoni, A.; Ackerley, S.; Shaw, C.E.; Miller, C.C.J. Amyotrophic lateral sclerosis-associated mutant VAPBP56s perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 2012, 21, 1979–1988. [Google Scholar] [CrossRef] [Green Version]
- Cadoni, M.P.L.; Biggio, M.L.; Arru, G.; Secchi, G.; Orrù, N.; Clemente, M.G.; Sechi, G.; Yamoah, A.; Tripathi, P.; Orrù, S.; et al. VAPB ER-Aggregates, A Possible New Biomarker in ALS Pathology. Cells 2020, 9, 164. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wen, Y.; Dong, J.; Cao, C.; Yuan, S. Systematic In-Depth Proteomic Analysis of Mitochondria-Associated Endoplasmic Reticulum Membranes in Mouse and Human Testes. Proteomics 2018, 18, e1700478. [Google Scholar] [CrossRef] [PubMed]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 2013, 79, 219–230. [Google Scholar] [CrossRef]
- Kwak, C.; Shin, S.; Park, J.S.; Jung, M.; My Nhung, T.T.; Kang, M.G.; Lee, C.; Kwon, T.H.; Park, S.K.; Mun, J.Y.; et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl. Acad. Sci. USA 2020, 117, 12109–12120. [Google Scholar] [CrossRef]
- Kaytor, M.D.; Orr, H.T. The GSK3β signaling cascade and neurodegenerative disease. Curr. Opin. Neurobiol. 2002, 12, 275–278. [Google Scholar] [CrossRef]
- Choi, H.J.; Cha, S.J.; Lee, J.W.; Kim, H.J.; Kim, K. Recent advances on the role of gsk3β in the pathogenesis of amyotrophic lateral sclerosis. Brain Sci. 2020, 10, 675. [Google Scholar] [CrossRef]
- D’mello, S.R. When good kinases go rogue: Gsk3, p38 mapk and cdks as therapeutic targets for alzheimer’s and huntington’s disease. Int. J. Mol. Sci. 2021, 22, 5911. [Google Scholar] [CrossRef]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef]
- Yang, W.; Leystra-Lantz, C.; Strong, M.J. Upregulation of GSK3β expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci). Brain Res. 2008, 1196, 131–139. [Google Scholar] [CrossRef]
- Manabe, Y.; Warita, H.; Murakami, T.; Shiote, M.; Hayashi, T.; Nagano, I.; Shoji, M.; Abe, K. Early decrease of redox factor-1 in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Brain Res. 2001, 915, 104–107. [Google Scholar] [CrossRef]
- Sreedharan, J.; Neukomm, L.J.; Brown, R.H.; Freeman, M.R. Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr. Biol. 2015, 25, 2130–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombrita, C.; Onesto, E.; Buratti, E.; de la Grange, P.; Gumina, V.; Baralle, F.E.; Silani, V.; Ratti, A. From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models. Biochim. Biophys. Acta 2015, 1849, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Coba, M.P.; Komiyama, N.H.; Nithianantharajah, J.; Kopanitsa, M.V.; Indersmitten, T.; Skene, N.G.; Tuck, E.J.; Fricker, D.G.; Elsegood, K.A.; Stanford, L.E.; et al. TNiK is required for postsynaptic and nuclear signaling pathways and cognitive function. J. Neurosci. 2012, 32, 13987–13999. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Miuzzo, M.; Pierobon, N.; Negro, A.; Sorgato, M.C. The prion protein and its paralogue doppel affect calcium signaling in Chinese hamster ovary cells. Mol. Biol. Cell 2005, 16, 2799–2808. [Google Scholar] [CrossRef] [Green Version]
- Lazzari, C.; Peggion, C.; Stella, R.; Massimino, M.L.; Lim, D.; Bertoli, A.; Sorgato, M.C. Cellular prion protein is implicated in the regulation of local Ca2+ movements in cerebellar granule neurons. J. Neurochem. 2011, 116, 881–890. [Google Scholar] [CrossRef]
- De Mario, A.; Castellani, A.; Peggion, C.; Massimino, M.L.; Lim, D.; Hill, A.F.; Sorgato, M.C.; Bertoli, A. The prion protein constitutively controls neuronal store-operated Ca(2+) entry through Fyn kinase. Front. Cell. Neurosci. 2015, 9, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquet, A.; Cottet-Rousselle, C.; Arnaud, J.; Julien Saint Amand, K.; Ben Messaoud, R.; Lenon, M.; Demeilliers, C.; Moulis, J.-M. Mitochondrial Morphology and Function of the Pancreatic beta-Cells INS-1 Model upon Chronic Exposure to Sub-Lethal Cadmium Doses. Toxics 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Brini, M.; Pizzo, P.; Murgia, M.; Pozzan, T. Chimeric green fluorescent protein as atool for visualizing subcellular organelles in living cells. Curr. Biol. 1995, 5, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Marsault, R.; Bastianutto, C.; Alvarez, J.; Pozzan, T.; Rizzuto, R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. J. Biol. Chem. 1995, 270, 9896–9903. [Google Scholar] [CrossRef] [Green Version]
- Colella, A.D.; Chegenii, N.; Tea, M.N.; Gibbins, I.L.; Williams, K.A.; Chataway, T.K. Comparison of Stain-Free gels with traditional immunoblot loading control methodology. Anal. Biochem. 2012, 430, 108–110. [Google Scholar] [CrossRef]
- Alessio, E.; Buson, L.; Chemello, F.; Peggion, C.; Grespi, F.; Martini, P.; Massimino, M.L.; Pacchioni, B.; Millino, C.; Romualdi, C.; et al. Single cell analysis reveals the involvement of the long non-coding RNA Pvt1 in the modulation of muscle atrophy and mitochondrial network. Nucleic Acids Res. 2019, 47, 1653–1670. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peggion, C.; Massimino, M.L.; Bonadio, R.S.; Lia, F.; Lopreiato, R.; Cagnin, S.; Calì, T.; Bertoli, A. Regulation of Endoplasmic Reticulum–Mitochondria Tethering and Ca2+ Fluxes by TDP-43 via GSK3β. Int. J. Mol. Sci. 2021, 22, 11853. https://doi.org/10.3390/ijms222111853
Peggion C, Massimino ML, Bonadio RS, Lia F, Lopreiato R, Cagnin S, Calì T, Bertoli A. Regulation of Endoplasmic Reticulum–Mitochondria Tethering and Ca2+ Fluxes by TDP-43 via GSK3β. International Journal of Molecular Sciences. 2021; 22(21):11853. https://doi.org/10.3390/ijms222111853
Chicago/Turabian StylePeggion, Caterina, Maria Lina Massimino, Raphael Severino Bonadio, Federica Lia, Raffaele Lopreiato, Stefano Cagnin, Tito Calì, and Alessandro Bertoli. 2021. "Regulation of Endoplasmic Reticulum–Mitochondria Tethering and Ca2+ Fluxes by TDP-43 via GSK3β" International Journal of Molecular Sciences 22, no. 21: 11853. https://doi.org/10.3390/ijms222111853
APA StylePeggion, C., Massimino, M. L., Bonadio, R. S., Lia, F., Lopreiato, R., Cagnin, S., Calì, T., & Bertoli, A. (2021). Regulation of Endoplasmic Reticulum–Mitochondria Tethering and Ca2+ Fluxes by TDP-43 via GSK3β. International Journal of Molecular Sciences, 22(21), 11853. https://doi.org/10.3390/ijms222111853