
Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress


Abstract
1. Introduction
2. Normal Aging
3. Mitochondria in the Brain
3.1. Mitochondrial Respiratory Chain and ROS Production
3.2. Mitochondria and Cellular Calcium Homeostasis
- -
- Bap31 (B cell receptor-associated protein 31), which interacts with the OMM protein Fis1 [82];
- -
- VAPB (Vesicle-associated membrane protein-associated protein B), which interacts with the OMM protein tyrosine phosphatase-interacting protein 51 [83];
- -
- -
- Protein kinase-like endoplasmic reticulum kinase (PERK), which, when activated reduces protein synthesis until the accumulated unfolded protein is cleared [86].
3.3. Mitochondrial Dynamics
3.4. Autophagy
- -
- Macroautophagy, in which the autophagosome, a double-membraned vesicle, forms and fuses with lysosomes after which their content is degraded by the acidic hydrolases of the lysosomes;
- -
- Microautophagy, a process during which lysosomes wrap around various cytosolic compounds which are degraded after the involution of the membrane [109];
- -
- Chaperone-mediated autophagy, a process during which chaperones bind to damaged proteins and to receptors on the lysosomal membrane, followed by translocation of the protein into the lysosome for degradation [110].
4. The Brain and Oxidative Stress
4.1. Vulnerability of the Brain to Oxidative Stress
- -
- Action potentials cause calcium influx and raise the intracellular calcium concentration from approximately 0.001 μm to roughly 100 μm [163]. High intracellular Ca2+ activates nNOS (neuronal nitric oxide synthase) and leads to NO (nitric oxide) formation [164], which binds to cytochrome c oxidase and inhibits mitochondrial respiration [165]. Mitochondria attempt to buffer intracellular calcium, but the subsequent calcium overload causes prolonged opening of the MPTP and inhibits ATP generation, inducing apoptosis [166].
- -
- -
- -
- -
- -
- -
- The brain is enriched in redox active transition metals, such as Cu+ or Fe2+ [175]. Iron is a catalyzer in the hydroxyl radical-generating Fenton reaction, and also catalyzes peroxyl and alkoxyl radical generation, thereby contributing to ferroptosis, a form of cell death dependent on lipid peroxidation and Fe2+ [176]. Cu+ is a co-factor for Cu/ZnSOD and is important for cell signaling [177,178] but enhances copper-catalyzed Fenton reaction [175].
- -
- The brain is particularly rich in cholesterol, which may undergo auto-oxidation [179] and brain cells have a higher membrane surface/cytoplasmic volume ratio, cellular membranes being rich in polyunsaturated fatty acids (PUFA), which are highly susceptible to peroxidation through free radical attack [153].
- -
4.2. Sources of Free Radicals
4.2.1. Mitochondria and Oxidative Stress
4.2.2. NADPH Oxidase as a Source of ROS
4.2.3. Monoamine Oxidase as a Source of ROS
4.2.4. Peroxisomes and ROS Production
4.2.5. Exogenous Sources of ROS
4.3. Targets of ROS
4.3.1. Proteins and ROS
4.3.2. Lipids and ROS
4.3.3. DNA
4.3.4. RNA and Oxidative Damage
5. Selective Neuron Vulnerability in Neurodegenerative Diseases
5.1. Selective Neuronal Vulnerability in Alzheimer’s Disease
5.2. Selective Neuronal Vulnerability in Parkinson’s Disease
5.3. Motor Neuron Vulnerability in Amyotrophic Lateral Sclerosis
6. Oxidative Stress in Neurodegenerative Diseases
6.1. Oxidative Stress in Alzheimer’s Disease
6.2. Oxidative Stress in Parkinson’s Disease
6.3. Oxidative Stress in Amyotrophic Lateral Sclerosis
7. Translating Theoretical Knowledge into Therapy
7.1. Targeting Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease
7.2. Targeting Oxidative Stress and Mitochondrial Dysfunction in Parkinson’s Disease
7.3. Targeting Oxidative Stress and Mitochondria in ALS
8. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Albers, D.S.; Flint Beal, M. Mitochondrial Dysfunction and Oxidative Stress in Aging and Neurodegenerative Disease. In Advances in Dementia Research; Jellinger, K., Schmidt, R., Windisch, M., Eds.; Springer: Vienna, Austria, 2000. [Google Scholar]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 6. [Google Scholar] [CrossRef]
- Huffman, K.J. The developing, aging neocortex: How genetics and epigenetics influence early developmental patterning and age-related change. Front. Genet. 2012, 3, 212. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Allison, D.B.; Weindruch, R. Caloric intake and aging: Mechanisms in rodents and a study in nonhuman primates. Toxicol. Sci. 1999, 52, 35–40. [Google Scholar] [CrossRef]
- Błaszczyk, J.W. Energy metabolism decline in the aging brain–pathogenesis of neurodegenerative disorders. Metabolites 2020, 10, 450. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Kmiec, Z. Central regulation of food intake in ageing. J. Physiol. Pharmacol. 2006, 57, 7–16. [Google Scholar] [PubMed]
- Zhang, X.Y.; Yu, L.; Zhuang, Q.X.; Zhu, J.N.; Wang, J.J. Central functions of the orexinergic system. Neurosci. Bull. 2013, 29, 355–365. [Google Scholar] [CrossRef]
- Waterson, M.J.; Horvath, T.L. Neuronal regulation of energy homeostasis: Beyond the hypothalamus and feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Rolls, A. Hypothalamic control of sleep in aging. Neuromol. Med. 2012, 14, 139–153. [Google Scholar] [CrossRef]
- Cahine, L.M.; Amara, A.W.; Videnovic, A. A systematic review of the literature on disorder of sleep and wakefulness in Parkinson’s disease from 2005 to 2015. Sleep Med. Rev. 2016, 35, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Brzecka, A.; Leszek, J.; Ashraf, G.M.; Ejma, M.; Ávila-Rodriguez, M.F.; Yarla, N.S.; Tarasov, V.V.; Chubarev, V.N.; Samsonova, A.N.; Barreto, G.E.; et al. Sleep disorders associated with Alzheimer’s disease: A perspective. Front. Neurosci. 2018, 12, 330. [Google Scholar] [CrossRef]
- Aalling, N.N.; Nedergaard, M.; DiNuzzo, M. Cerebral metabolic changes during sleep. Curr. Neurol. Neurosci. Rep. 2018, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, G.J.; Swiatek, K.R.; Chao, K.L. Lactate, 3-hydroxybutyrate, and glucose as substrates for the early postnatal rat brain. Neurochem. Res. 1989, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Leithner, C.; Royl, G. The oxygen paradox of neurovascular coupling. J. Cerebr. Blood Flow Metab. 2014, 34, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Liang, J.; Zhou, B. Glucose metabolic dysfunction in neurodegenerative diseases–new mechanistic insights and the potential of hypoxia as a prospective therapy targeting metabolic reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef]
- Romano, A.H.; Conway, T. Evolution of carbohydrate metabolic pathways. Res. Microbiol. 1996, 147, 448–455. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. Br. J. Pharmacol. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell 2019, 176, 73–84. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Cini, M.; Moretti, A. Studies on lipid peroxidation and protein oxidation in the aging brain. Neurobiol. Aging 1995, 16, 53–57. [Google Scholar] [CrossRef]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in nortmal aging and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Stahon, K.E.; Bastian, C.; Griffith, S.; KJidd, G.J.; Brunet, S.; Baltan, S. Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. J. Neurosci. 2016, 36, 9990–10001. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Pollard, A.K.; Craig, E.L.; Chakrabarti, L. Mitochondrial complex I activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS ONE 2016, 11, e0157405. [Google Scholar] [CrossRef]
- Leslie, S.W.; Chandler, L.J.; Barr, E.M.; Farrar, R.P. Reduced calcium uptake by rat brain mitochondria and synaptosomes in response to aging. Brain Res. 1985, 329, 177–183. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in aging: Molecular mechanisms and translational implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Kudo, N.; Thelen, J.N.; Ito, A.; Yoshida, M.; Denu, J.M. Kinetic and structural basis for acyl-group selectivity and NAD+ dependence in sirtuin-catalyzed deacylation. Biochemistry 2015, 54, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.-I.; Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S. Emerging therapeutic roles for NAD+ metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016, 16, 1–11. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-I. Sirt 1 extends life span and delays aging in mice through the regulation of Nk2 homebox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Satoh, A.; Imai, S.-I. Systemic regulation of mammalian ageing and longevity by brain sirtuins. Nat. Commun. 2014, 5, 4211. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef]
- Fischer, H.G.; Morawski, M.; Bruckner, M.K.; Mittag, A.; Tarnok, A.; Arendt, T. Changes in neuronal DNA content variation in the human brain during aging. Aging Cell 2012, 11, 628–633. [Google Scholar] [CrossRef]
- Mosch, B.; Morawski, M.; Mittag, A.; Lenz, D.; Tarnok, A.; Arendt, T. Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J. Neurosci. 2007, 27, 6859–6967. [Google Scholar] [CrossRef]
- Ceafalan, L.C.; Popescu, B.O. Juxtacerebral tissue regeneration potential: Telocytes contribution. Adv. Exp. Med. Biol. 2016, 913, 397–402. [Google Scholar]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, A. Emerging role of NF-κB signalling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Baldwin, A.S., Jr. The NF-κB and IκB proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–681. [Google Scholar] [CrossRef]
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive oxygen species: Stuck in the middle of neurodegeneration. J. Alzheime. Dis. 2010, 20, S357–S367. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef]
- Snow, W.M.; Stoesz, B.M.; Kelly, D.M.; Albensi, B.C. Roles for NF-κB and gene targets of NF-κB in synaptic plasticity, memory, and navigation. Mol. Neurobiol. 2014, 49, 757–770. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Sastie, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Sazanova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Sazanova, M.D.; Kirichenko, T.V.; Khotina, V.A.; Khasanova, Z.B.; Doroschuk, N.A.; Karagodin, V.P.; Orekhov, A.N.; et al. Some molecular and cellular stress mechanisms associated with neurodegenerative diseases and atherosclerosis. Int. J. Mol. Sci. 2021, 22, 699. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef]
- Kwon, S.K.; Sando, R., 3rd; Lewis, T.L.; Hirabayashi, Y.; Maximov, A.; Polleux, F. LKB1 regulates mitochondria-dependent presynaptic calcium clearance and neurotransmitter release properties at excitatory synapses along cortical axons. PLoS Biol. 2016, 14, e1002516. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Rugarli, E.I.; Langer, T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012, 31, 1336–1349. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Benard, G.; Karbowski, M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol. 2009, 20, 365–374. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation, and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Rad. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Horigane, S.-I.; Ozawa, Y.; Yamada, H.; Takemoto-Kimura, S. Calcium signalling: A key regulator of neuronal migration. J. Biochem. 2019, 165, 401–409. [Google Scholar] [CrossRef]
- Verkhratsky, A. Endoplasmic reticulum calcium signaling in nerve cells. Biol. Res. 2004, 37, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Karagas, N.E.; Venkatachalam, K. Roles for the endoplasmic reticulum in regulation of neuronal calcium homeostasis. Cells 2019, 8, 1232. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2010, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Primeau, J.O.; Armanious, G.P.; Fisher, M.E.; Young, H.S. The SarcoEndoplasmic Reticulum Calcium ATPase. Subcell. Biochem. 2018, 87, 229–258. [Google Scholar]
- McCormack, J.G.; Denton, R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993, 21, 793–799. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharm. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2009, 107, 436–441. [Google Scholar] [CrossRef]
- Paß, T.; Wiesner, R.J.; Pla-Martin, D. Selective neuron vulnerability in common and rare diseases–mitochondria in the focus. Front. Mol. Biosci. 2021, 8, 676187. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Manella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell. Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular dysfunctions of mitochondria-associated membranes (MAMs) in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef]
- Rizzuto, P.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hing, C.-H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef]
- Youker, R.T.; Shinde, U.; Day, R.; Thomas, G. At the crossroads of homeostasis and disease: Roles of the PACS proteins in membrane traficking and apoptosis. Biochem. J. 2009, 421, 1–15. [Google Scholar] [CrossRef]
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 569–610. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Pawlak, K.J.; Burak, W.E.; Perry, E.E.; Marshall, B.; Whittal, R.M.; Bose, H.S. Mitochondrial metabolic regulation by GRP78. Sci. Adv. 2017, 3, e1602038. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Minczuk, M. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 30, 2953–2960. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [PubMed]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Cho, B.; Cho, H.M.; Jo, Y.; Kim, H.D.; Song, M.; Moon, C.; Kim, H.; Kim, K.; Sesaki, H.; Rhyu, I.J.; et al. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat. Commun. 2017, 8, 15754. [Google Scholar] [CrossRef] [PubMed]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; Van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease-the long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Park, Y.Y.; Nguyen, O.T.; Kang, H.; Cho, H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014, 5, e1172. [Google Scholar] [CrossRef]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, M.; Kenny, S.J.; Liu, D.; Maeda, M.; Saito, K.; Mathur, A.; Xu, K.; Schekman, R. Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep. 2017, 18, 1586–1603. [Google Scholar] [CrossRef]
- Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the brain. Int. J. Mol. Sci. 2020, 21, 9661. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Young, L.N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of human Atg12-Atg5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Le Grand, J.N.; Chakrama, F.Z.; Seguin-Py, S.; Fraichard, A.; Delage-Mourroux, R.; Jouvenot, M.; Boyer-Guittaut, M. GABARAPL1 (GEC1): Original or copycat? Autophagy 2011, 7, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M. ULK1, mammalian target of rapamycin, and mitochondria: Linking nutrient availability and autophagy. Antioxid. Redox Signal. 2011, 14, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Beertsen, W.; Eskelinen, E.L. LAMP-2: A control step for phagosome and autophagosome maturation. Autophagy 2008, 4, 510–512. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against α-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; De Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2012, 107, 378–383. [Google Scholar] [CrossRef]
- Jin, M.S.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson’s disease pathogenesis. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Fung, G.; Deng, H.; Zhang, J.; Fiesel, F.C.; Springer, W.; Li, X.; Luo, H. NBR1 is dispensable for PARK2-mediated mitophagy regardless of the presence or absence of SQSTM1. Cell Death Dis. 2015, 6, e1943. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Davis, C.H.; Marsh-Armstrong, N. Discovery and implications of transcellular mitophagy. Autophagy 2014, 10, 2383–2384. [Google Scholar] [CrossRef]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [PubMed]
- Hass, D.T.; Barnstable, C.J. Mitochondrial uncoupling protein 2 knock-out promotes mitophagy to decrease retinal ganglion cell death in a mouse model of glaucoma. J. Neurosci. 2019, 39, 3582–3596. [Google Scholar] [PubMed]
- Zorov, D.B.; Kinally, K.W.; Tedeschi, H. Voltage activation of heart inner mitochondrial membrane channels. J. Bioenerg. Biomembr. 1992, 24, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Tinari, A.; Garofalo, T.; Sorice, M.; Degli Esposti, M.; Malorni, W. Mitoptosis: Different pathways for mitochondrial execution. Autophagy 2007, 3, 282–284. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, C.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Dames, S.A.; Mulet, J.M.; Rathgeb-Szabo, K.; Hall, M.N.; Grzesiek, S. The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J. Biol. Chem. 2005, 280, 20558–20564. [Google Scholar] [CrossRef]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.D.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell. 2011, 43, 19–32. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yus, Z. Distinct regulation opf autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rubinsztein, S.C. Inositol and IP3 levels regulate autophagy: Biology and therapeutic speculations. Autophagy 2006, 2, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T.R. DAPK-1 binding to a linear peptide motif in MAP18 stimulates autophagy and membrane blebbing. J.Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Yamamoto, M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol. 2002, 348, 182–190. [Google Scholar] [PubMed]
- Fujita, K.; Maeda, D.; Xiao, Q.; Srinivasuta, S.M. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 1427–1432. [Google Scholar] [CrossRef]
- Li, P.F.; Dietz, R.; Von Harsdorf, R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J. 1999, 18, 6027–6036. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Carrington, S.J.; Tantillo, D.J.; Harrison, J.G.; Ignarro, L.J.; Freeman, B.A.; Chen, A.; Wink, D.A. Small molecule signaling agents: The integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012, 25, 769–793. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamenta theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Cobley, N.J.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–513. [Google Scholar] [CrossRef]
- Massad, C.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Peltier, J.; Store, D.; Schaffer, D.V.; Chang, C.J. Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 2011, 7, 106–112. [Google Scholar] [CrossRef]
- Gauron, C.; Meda, F.; Dupont, E.; Albadri, S.; Quenech’Du, N.; Ipendey, E.; Volovitch, M.; Del Bene, F.; Joliot, A.; Rampon, C.; et al. Hydrogen peroxide (H2O2) controls axon pathfinding during zebrafish development. Dev. Biol. 2016, 414, 133–141. [Google Scholar] [CrossRef]
- Pasterkamp, R.J. Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 2012, 13, 605–618. [Google Scholar] [CrossRef]
- Bailey, D.M.; Bärtsch, P.; Knauth, M.; Baumgartner, R.W. Emerging concepts in acute mountain sickness and high-altitude cerebral edema: From the molecular to the morphological. Cell. Moll. Life Sci. 2009, 66, 3583–3594. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell. Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed]
- Simion, A.; Jurcau, A. The role of antioxidant treatment in acute ischemic stroke: Past, present and future. Neurol. Res. Surg. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Oxidative stress in the pathogenesis of Alzheimer’s disease and cerebrovascular disease with therapeutic implications. CNS Neurol. Disord. Drug Targets 2020, 19, 24–108. [Google Scholar] [CrossRef]
- Zucker, R.S. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol. 1999, 9, 305–313. [Google Scholar] [CrossRef]
- Lipton, S.A.; Choi, Y.-P.; Pan, Z.-H.; Lei, S.Z.; Chen, H.-S.V.; Sucher, N.J.; Loscalzo, J.; Singel, D.J.; Stamler, J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 1993, 364, 626–632. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide and mitochondrial respiration. Biochim. Biophys Acta-Bioenerg. 1999, 1411, 351–369. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2010, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar] [CrossRef]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anion by a CNS macrophage, the microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef]
- Edmondson, D. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: Biological implications. Curr. Pharm. Des. 2014, 20, 155–160. [Google Scholar] [CrossRef]
- Heikkila, R.E.; Cohen, G. 6-Hydroxydopamine: Evidence for superoxide radical as an oxidative intermediate. Science 1973, 181, 456–457. [Google Scholar] [CrossRef]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 39, 1517–1549. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of non-apoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Abreu, I.A.; Cabelli, D.E. Superoxide dismutases––A review of the metal-associated mechanistic variations. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 263–274. [Google Scholar] [CrossRef]
- Chang, C.J. Searching for harmony in transition-metal signaling. Nat. Chem. Biol. 2015, 11, 744–747. [Google Scholar] [CrossRef]
- Zerbinati, C.; Iuliano, L. Cholesterol and related sterols autoxidation. Free Radic. Biol. Med. 2017, 111, 151–155. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wolvetang, J.E.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef]
- Simms, C.L.; Zaher, H.S. Quality control of chemically damaged RNA. Cell. Mol. Life Sci. 2016, 73, 3639–3653. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Chock, P.B.; Stadtman, E.R. Oxidized messenger RNA induces translation errors. Proc. Natl. Acad. Sci. USA 2006, 104, 66–71. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2006, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and disease. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; He, W.; Liou, Y.-C. The redox language in neurodegenerative diseases: Oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 2021, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef]
- Sato, K. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef]
- Zoccarato, F. Succinate modulation of H2O2 release at NADH: Ubiquinone oxidoreductase (Complex I) in brain mitochondria. Biochem. J. 2007, 406, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.L.; Salter, J.D.; Brookes, P.S. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: Implications for hypoxic cell signaling. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, H101–H108. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Ghandi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar]
- Pradeep, H.; Diya, J.B.; Shashikumar, S.; Rajanikant, G.K. Oxidative stress- assassin behind the ischemic stroke. Folia Neuropathol. 2012, 50, 219–230. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target 2015, 23, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Jacobson, J.; Wientjes, F.; Hothersall, J.; Canevari, L.; Duchen, M.R. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J. Neurosci. 2005, 25, 9176–9184. [Google Scholar] [CrossRef]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.-I.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Binda, C.; Wang, J.; Upadhyay, A.K.; Mattevi, A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 2009, 48, 4220–4230. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem J. 1997, 324, 1–18. [Google Scholar] [CrossRef]
- Chevion, M.; Berenshtein, E.; Stadtman, E.R. Human studies related to protein oxidation: Protein carbonyl content as a marker of damage. Free Radic. Res. 2000, 33, S99–S108. [Google Scholar] [PubMed]
- Pryor, W.A.; Jin, X.; Squadrito, G.L. One- and two-electron oxidations of methionine by peroxynitrite. Proc. Natl. Acad. Sci. USA 1994, 91, 11173–11177. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Koppal, T.; Howard, B.; Subramaniam, R.; Hall, N.; Hensley, K.; Yatin, S.; Allen, K.; Aksenov, M.; Aksenova, M.; et al. Structural and functional changes in proteins induced by free radical-mediated oxidative stress and protective action of the antioxidants N-tert-butyl-alpha-phenylnitrone and vitamin E. Ann. N. Y. Acad. Sci. 1998, 854, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Sorg, O.; Horn, T.F.; Yu, N.; Gruol, D.; Bloom, F.E. Inhibition of astrocyte glutamate uptake by reactive oxygen species: Role of antioxidant enzymes. Mol. Med. 1997, 3, 431–440. [Google Scholar] [CrossRef]
- Grune, T.; Reinheckel, T.; Davies, K.J. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997, 11, 526–534. [Google Scholar] [CrossRef]
- Grune, D.; Merker, K.; Sandig, G.; Davies, K.J. Selective degradation of oxidatively modified protein substrates by proteasome. Biochem. Biophys. Res. Commun. 2003, 305, 709–718. [Google Scholar] [CrossRef]
- Varshavsky, A. The ubiquitin system. Trends Biochem. Sci. 1997, 22, 383–387. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T. Johansen, T.; Dikic. I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009, 5, 732–733. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Hajieva, P.; Kaya, M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Perluigi, M.; Coccia, R.; Butterfield, D.A. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: A toxic combination illuminated by redox proteomics studies. Antioxid. Redox Signal. 2012, 17, 1590–1609. [Google Scholar] [CrossRef]
- Erejuwa, O.O.; Sulaiman, S.A.; Ab Wahab, M.S. Evidence in support of potential applications of lipid peroxidation products in cancer treatment. Oxid. Med. Cell. Longev. 2013, 2013, 931251. [Google Scholar] [CrossRef]
- Bradley-Whitman, M.A.; Lovell, M.A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): An update. Arch. Toxicol. 2015, 89, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Petersen, D.R. An overview of the chemistry and biology of reactive aldehydes. Free Radic. Biol. Med. 2013, 59, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, F.J.; Cipollina, C.; Freeman, B.A. Formation and signaling actions of electrophilic lipids. Chem Rev. 2011, 111, 5997–6021. [Google Scholar] [CrossRef]
- Kansanen, E.; Jyrkkanen, H.K.; Levonen, A.L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kozak, K.R.; Marnett, L.J. IkappaB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J. Biol. Chem. 2001, 276, 18223–18228. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Allan Butterfield, D. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Laurora, S.; Tamagno, E.; Briatore, F.; Bardini, P.; Pizzimenti, S.; Toaldo, C.; Reffo, P.; Costelli, P.; Dianzani, M.U.; Danni, O.; et al. 4-Hydroxynonenal modulation of p53 family gene expression in the SK-N-BE neuroblastoma cell line. Free Radic. Biol. Med. 2005, 38, 215–225. [Google Scholar] [CrossRef]
- Li, J.; Sharma, R.; Patrick, B.; Sharma, A.; Jeyabal, P.V.; Reddy, P.M.; Saini, M.K.; Dwivedi, S.; Dhanani, S.; Ansari, N.-H.; et al. Regulation of CD95 (Fas) expression and Fas-mediated apoptotic signaling in HLE B-3 cells by 4-hydroxynonenal. Biochemistry 2006, 45, 12253–12264. [Google Scholar] [CrossRef]
- Parola, M.; Robino, G.; Marra, F.; Pinzani, M.; Bellomo, G.; Leonarduzzi, G.; Chiarugi, P.; Camandola, S.; Poli, G.; Waeg, G.; et al. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J. Clin. Investig. 1998, 102, 1942–1950. [Google Scholar] [CrossRef]
- Song, B.J.; Soh, Y.; Bae, M.; Pie, J.; Wan, J.; Jeong, K. Apoptosis of PC12 cells by 4-hydroxy-2-nonenal is mediated through selective activation of the c-Jun N-Terminal protein kinase pathway. Chem. Biol. Interact. 2001, 130–132, 943–954. [Google Scholar] [CrossRef]
- Shibata, N.; Kato, Y.; Inose, Y.; Hiroi, A.; Yamamoto, T.; Morikawa, S.; Sawada, M.; Kobayashi, M. 4-hydroxy-2-nonenal upregulates and phosphorylates cytosolic phospholipase A2 in cultured Ra2 microglial cells via MAPK pathways. Neuropathology 2011, 31, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, C.E.; Yun, M.R.; Seo, K.W.; Park, H.M.; Yun, J.W.; Shin, H.K.; Bae, S.S.; Kim, C.D. 4-hydroxynonenal enhances MMP-9 production in murine macrophages via 5-lipoxygenase-mediated activation of ERK and p38 MAPK. Toxicol. Appl. Pharmacol. 2010, 242, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Narciso, L.; Parlanti, E.; Racaniello, M.; Simonelli, V.; Cardinale, A.; Merlo, D.; Dogliotti, E. The response to oxidative DNA damage in neurons: Mechanisms and disease. Neural Plast. 2016, 2016, 3619274. [Google Scholar] [CrossRef] [PubMed]
- Colnaghi, L.; Rondelli, D.; Muzi-Falconi, M.; Sertic, S. Tau and DNA damage in neurodegeneration. Brain Sci. 2020, 10, 946. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell. Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef]
- Hegde, M.L.; Mantha, A.K.; Hazra, T.K.; Bhakat, K.K.; Mitra, S.; Szczesny, B. Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mech. Ageing Dev. 2012, 133, 157–168. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Qu, D.; Zhang, Y.; Venderova, K.; Haque, M.E.; Rousseaux, M.W.C.; Slack, R.S.; Woulfe, J.M.; Park, D.S. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat. Cell Biol. 2010, 12, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.-Y.; Kim, D.K.; Mook-Yung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef]
- Biswas, G.; Anandatheerthavarada, H.K.; Zaidi, M.; Avadhani, N.G. Mitochondria to nucleus stress signaling: A distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J. Cell Biol. 2003, 161, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.S.; Haigis, M.C. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res. Rev. 2009, 8, 173–188. [Google Scholar] [CrossRef]
- Fukae, J.; Takanashi, M.; Kubo, S.-I.; Nishioka, K.-I.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005, 109, 256–262. [Google Scholar] [CrossRef]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000, 855, 116–123. [Google Scholar] [CrossRef]
- Weissman, L.; Jo, D.-G.; Sørensen, M.M.; De Souza-Pinto, M.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chenm, X.; Lim, Z.; Yem, W.; Dingm, H.; Lim, P.; Aung, L.H.H. Role of RNA oxidation in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5022. [Google Scholar] [CrossRef]
- Hofer, T.; Badouard, C.; Bajak, E.; Ravanat, J.L.; Mattsson, A.; Cotgreave, I.A. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem. 2005, 386, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yeo, G.; Muotri, A.R.; Kuwabara, T.; Gage, F.H. Noncoding RNAs in the mammalian central nervous system. Annu. Rev. Neurosci. 2006, 29, 77–103. [Google Scholar] [CrossRef]
- Barciszewski, J.; Barciszewska, M.Z.; Siboska, G.; Rattan, S.; Clark, B.F. Some unusual nucleic acid bases are products of hydroxyl radical oxidation of DNA and RNA. Mol. Biol. Rep. 1999, 26, 231–238. [Google Scholar] [CrossRef]
- Nunomura, A.; Lee, H.-G.; Zhu, X.; Perry, G. Consequences of RNA oxidation on protein synthesis rate and fidelity: Implications for the pathophysiology of neuropsychiatric disorders. Biochem. Soc. Trans. 2017, 45, 1053–1066. [Google Scholar] [CrossRef]
- Jacobs, A.C.; Resendiz, M.J.E.; Greenberg, M.M. Direct strand scission from a nucleobase radical in RNA. J. Am. Chem. Soc. 2010, 132, 3668–3669. [Google Scholar] [CrossRef][Green Version]
- Ding, Q.; Dimayuga, E.; Bruce-Keller, A.J. Oxidative stress alters neuronal RNA- and protein-synthesis: Implications for neural viability. Free. Radic. Res. 2007, 41, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.L.; Zaher, H.S. How do cells cope with RNA damage and its consequences? J. Biol. Chem. 2019, 294, 15158–15171. [Google Scholar] [CrossRef]
- Khanam, H.; Ali, A.; Asif, M.; Shamsuzzaman. Neurodegenerative diseases linked to misfolded proteins and their therapeutic approaches: A review. Eur. J. Med. Chem. 2016, 124, 1121–1141. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F.; Wong-Riley, M.T. Entorhinal cortex of the human, monkey, and rat: Metabolic map as revealed by cytochrome oxidase. J. Comp. Neurol. 1992, 326, 451–469. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Hof, P.R.; Nimchinsky, E.A.; Celio, M.R.; Bouras, C.; Morrison, J.H. Calretinin-immunoreactive neocortical interneurons are unaffected in Alzheimer’s diseaase. Neurosci. Lett. 1993, 152, 145–148. [Google Scholar] [CrossRef]
- Roussarie, J.-P.; Yao, V.; Rodriguez-Rodriguez, P.; Oughtred, R.; Rust, J.; Plautz, Z.; Kasturia, S.; Albornoz, C.; Wang, W.; Schmidt, E.F.; et al. Selective neuronal vulnerability in Alzheimer’s disease: A network-based analysis. Neuron 2020, 107, 821–835. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Del Tredici, K.; Thomas, T.L.; Braak, H.; Diamond, M.I. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 2018, 136, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Diehl Rodriguez, R.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, J.F.; Baris, O.R.; Hess, S.; Moser, N.; Schroder, H.; Chinta, S.J.; Andersen, J.K.; Kloppenburg, P.; Wiesner, R.J. Catecholamine metabolism drives generation of mitochondrial DNA deletions in dopaminergic neurons. Brain 2014, 137, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; Tsairis, P. Anatomy and innervation ratios in motor units of cat gastrocnemius. J. Physiol. 1973, 234, 749–765. [Google Scholar] [CrossRef]
- Enoka, R.M. Morphological features and activation patterns of motor units. J. Clin. Neurophysiol. 1995, 12, 538–559. [Google Scholar] [CrossRef]
- Zimmermann, L.; Morado-Díaz, C.J.; Davis-López de Carrizosa, M.A.; De la Cruz, R.R.; May, P.J.; Streicher, J.; Pastor, A.M.; Blumer, R. Axons giving rise to the palisade endings of feline extraocular muscle display motor features. J. Neurosci. 2013, 33, 2784–2793. [Google Scholar] [CrossRef] [PubMed]
- Alexianu, M.E.; Ho, B.-K.; Mohamed, A.H.; La Bella, V.; Smith, R.G.; Appel, S.H. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 1994, 36, 846–858. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis in intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J.Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell. Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. On the pathogenesis of Alzheimer’s disease: The MAM hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell. Calcium 2020, 86, 102150. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Morales, J.-C.; Arranz-Tagarro, J.-A.; Calvo-Gallardo, E.; Maroto, M.; Padín, J.-F.; García, A.G. Stabilizers of neuronal and mitochondrial calcium cycling as a strategy for developing a medicine for Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 873–883. [Google Scholar] [CrossRef]
- Magi, S.; Castaldo, P.; Macrí, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular calcium dysregulation: Implications for Alzheimer’s disease. Biomed. Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Ramachandran, S.; Azimov, R.; Kagan, B.L.; Lal, R. Ion channel formation bt Tau protein: Implications for Alzheimer’s disease and tauopathies. Biochemistry 2015, 50, 7320–7325. [Google Scholar] [CrossRef]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prá, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons–therapeutic relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2013, 10, 1634–1652. [Google Scholar]
- Jové, M.; Mota-Martorell, N.; Torres, P.; Ayala, V.; Portero-Otin, M.; Ferrer, I.; Pamplona, R. The causal role of lipoxidative damage in mitochondrial bioenergetic dysfunction linked to Alzheimer’s disease pathology. Life 2021, 11, 388. [Google Scholar] [CrossRef]
- Terni, B.; Boada, J.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol. 2010, 20, 222–233. [Google Scholar] [CrossRef]
- Jové, M.; Pradas, I.; Dominguez-Gonzalez, M.; Ferrer, I.; Pamplona, R. Lipids and lipoxidation in human brain aging. Mitochondrial ATP-synthase as a key lipoxidation target. Redox Biol. 2019, 23, 101083. [Google Scholar] [CrossRef] [PubMed]
- Tobore, T.O. On the central role of mitochondria dysfunction and oxidative stress in Alzheimer’s disease. Neurol. Sci. 2019, 40, 1527–1540. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Etheridge, N.; Dodd, P.R.; Nouwens, A.S. Targeted quantitative analysis of synaptic proteins in Alzheimer’s disease brain. Neurochem. Int. 2014, 75, 66–75. [Google Scholar] [CrossRef]
- Reed, T.T.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009, 1274, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteom. Clin. Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxynonenal (HNE) in the pathogenesis of Alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Pamplona, R.; Dalfó, E.; Ayala, V.; Bellmunt, M.J.; Prat, J.; Ferrer, I.; Portero-Otín, M. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 2005, 280, 21522–21530. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Xi, Y.; Gao, M.; Li, Z.; Xu, C.; Fan, S.; He, W. Gene expression profiles of entorhinal cortex in Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Demen. 2014, 29, 526–532. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paulo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J.Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef]
- Fabelo, N.; Martín, V.; Marín, R.; Moreno, D.; Ferrer, I.; Díaz, M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Reddy, P.H.; Williams, J.; Smith, F.; Bhatti, J.S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Wang, R.; Manczak, M.; et al. MicroRNAs, aging, cellular senescence, and Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 127–171. [Google Scholar]
- Crespo-Biel, N.; Theunis, C.; Van Leuven, F. Protein tau: Prime cause of synaptic and neuronal degeneration in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2012, 2012, 251426. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Tammineni, P. Alterations in mitochondrial quality control in Alzheimer’s disease. Front. Cell. Neurosci. 2016, 10, 24. [Google Scholar] [CrossRef]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.-H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife 2017, e21776. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.M.; Ke, M.; Tan, Y.; Huang, Z.; Zhang, K.; Ai, N.; Ge, W.; Qin, D.; Lu, J.H.; Su, H. Presenilin 1 deficiency suppresses autophagy in human neural stem cells through reducing γ-secretase-independent ERK/CREB signaling. Cell Death Dis. 2018, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Augusto-Oliveira, M.; Arrifano, G.P.; Lopes-Araújo, A.; Santos-Sacramento, L.; Takeda, P.Y.; Anthony, D.C.; Malva, J.O.; Crespo-Lopez, M.E. What do microglia really do in healthy adult brain? Cells 2019, 8, 1293. [Google Scholar] [CrossRef] [PubMed]
- Loughlin, A.J.; Woodroofe, M.N.; Cuzner, M.L. Regulation of Fc receptor and major histocompatibility complex antigen expression on isolated rat microglia by tumour necrosis factor, interleukin-1 and lipopolysaccharide: Effects on interferon-gamma induced activation. Immunology 1992, 75, 170–175. [Google Scholar] [PubMed]
- Song, X.; Shapiro, S.; Goldman, D.L.; Casadevall, A.; Scharff, M.; Lee, S.C. Fcgamma receptor I- and III-mediated macrophage inflammatory protein 1alpha induction in primary human and murine microglia. Infect. Immun. 2002, 70, 5177–5184. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; El Khoury, J.; Medeiros, L.A.; Terada, K.; Geula, C.; Luster, A.D.; Freeman, M.W. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J. Biol. Chem. 2002, 277, 47373–47379. [Google Scholar] [CrossRef]
- Anderson, K.V. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 2000, 12, 13–19. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; García-Rodríguez, C.; Villalobos, C.; Núñez, L. Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front. Immunol. 2020, 11, 1588. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenmayre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Mann, V.M.; Cooper, J.M.; Daniel, S.E.; Srai, K.; Jenner, P.; Marsden, C.D.; Schapira, A.H. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol. 1994, 36, 876–881. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Quing, H.; Zhang, Y.; Deng, Y.; McGeer, E.G.; McGeer, P.L. Lrrk2 intercation with α-synucleinin diffuse Lewy body disease. Biochem. Biophys. Res. Commun. 2009, 390, 1229–1234. [Google Scholar] [CrossRef]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; Van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef]
- Dalfo, E.; Ferrer, I. Early α-synuclein lipoxidation in neocortex in Lewy body diseases. Neurobiol. Aging 2008, 29, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef]
- Floor, E.; Wetzel, M.G. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal coretx measured with an improved dinitrophenylhydrazine assay. J. Neurochem. 1998, 70, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Chakravarthy, V.S. Influence of energy deficiency on the subcellular processes of substantia nigra pars compacta cell for understanding parkinsonian neurodegeneration. Sci. Rep. 2021, 11, 1754. [Google Scholar] [CrossRef]
- Pavlin, M.; Repič, M.; Vianello, R.; Mavri, J. The chemistry of neurodegeneration: Kinetic data and their implications. Mol. Neurobiol. 2016, 53, 3400–3415. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Yasuda, T. Iron accumulation in Parkinson’s disease. J. Neural Transm. 2012, 119, 1511–1514. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef]
- Stafa, K.; Tsika, E.; Moser, R.; Musso, A.; Glauser, L.; Jones, A.; Biskup, S.; Xiong, Y.; Bandopadhyay, R.; Dawson, V.L.; et al. Functional interaction of Parkinson’s disease-associated LRRK2 with membres of the dynamin GTPase superfamily. Hum. Mol. Genet. 2014, 23, 2055–2077. [Google Scholar] [CrossRef]
- Zilocchi, M.; Finzi, G.; Lualdi, M.; Sessa, F.; Fasano, M.; Alberio, T. Mitochondrial alterations in Parkinson’s disease human samples and cellular models. Neurochem. Int. 2018, 118, 61–72. [Google Scholar] [CrossRef]
- Perez-Carrion, M.; Pischedda, F.; Biosa, A.; Russo, I.; Straniero, L.; Civiero, L.; Guida, M.; Gloeckner, C.J.; Ticozzi, N.; Tiloca, C.; et al. The LRRK2 variant E193K prevents mitochondrial fission upon MPP+ treatment by altering LRRK2 binding to DRP1. Front. Mol. Neurosci. 2018, 11, 64. [Google Scholar] [CrossRef]
- Devoto, P.V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marín-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. αSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson’s disease. Sci. Rep. 2017, 7, 5042. [Google Scholar] [CrossRef]
- Xie, W.; Chung, K.K.K. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J. Neurochem. 2012, 122, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Melkov, A.; Abdu, U. Regulation of long-distance transport of mitochondria along microtubules. Cell. Mol. Life Sci. 2018, 75, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Trimbuch, T.; Rosenmund, C. Differential pH dynamics in synaptic vesicles from intact glutamatergic and GABAergic synapses. Front. Synaptic Neurosci. 2018, 10, 44. [Google Scholar] [CrossRef]
- Umek, N.; Gerŝak, B.; Vintar, N.; Ŝoŝtarič, M.; Mavri, J. Dopamine autoxidation is controlled by acidic pH. Front. Mol. Neurosci. 2018, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.S.; Guarnieri, M.; Xu, Z.-S.; Wong, P.C.; Brown, R.H.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Nat. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Yang, L.; DiMauro, J.P.; Fuller, S.W.; Licata, S.C.; Beal, M.F. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 2006, 22, 599–610. [Google Scholar] [CrossRef]
- Hatazawa, J.; Brooks, R.A.; Dalakas, M.C.; Mansi, L.; Di Chiro, G. Cortical motor-sensory hypometabolism in amyotrophic lateral sclerosis: A PET study. J. Comput. Assist. Tomogr. 1988, 12, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Raman, R.; Allen, S.P.; Goodall, E.F.; Kramer, S.; Ponger, L.L.; Heath, P.R.; Milo, M.; Hollinger, H.C.; Walsh, T.; Highley, J.R.; et al. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 2015, 41, 201–226. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Neuronal glucose metabolism is impaired while astrocytic TCA cycling is unaffected at symptomatic stages in the HSOD1G93A mouse model of amyotrophic lateral sclerosis. J. Cereb. Blood Flow Metab. 2019, 39, 1710–1724. [Google Scholar] [CrossRef]
- Diaz-Garcia, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 2017, 26, 361–374. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.; Estrela, J.M. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 71, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Could sirtuin activities modify ALS onset and progression? Cell. Mol. Neurobiol. 2017, 37, 1147–1160. [Google Scholar] [CrossRef]
- Siklós, L.; Engelhardt, J.I.; Alexianu, M.E.; Gurney, M.E.; Siddique, T.; Appel, S.H. Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J. Neuropathol. Exp. Neurol. 1998, 57, 571–587. [Google Scholar] [CrossRef]
- Guatteo, E.; Carunchio, I.; Pieri, M.; Albo, F.; Canu, N.; Mercuri, N.B.; Zona, C. Altered calcium homeostasis in motor neurons following AMPA receptor but not voltage-dependent calcium channels’ activation in a genetic model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2007, 28, 90–100. [Google Scholar] [CrossRef]
- Van den Bosch, L.; Vandenberghe, W.; Klaassen, H.; Van Houtte, E.; Robberecht, W. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 2000, 180, 29–34. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Padman, B.S.; Nguyen, T.N.; Uoselis, L.; Skulsuppaisarn, M.; Nguyen, L.K.; Lazarou, M. LC3/GABARAPs drive ubiquitin-independent recruitment of optineurin and NDP52 to amplify mitophagy. Nat. Commun. 2019, 10, 408. [Google Scholar] [CrossRef]
- Feng, S.-M.; Che, C.-H.; Feng, S.-Y.; Liu, C.-Y.; Li, L.-Y.; Li, Y.-X.; Huang, H.-P.; Zou, Z.-Y. Novel mutation in optineurin causing aggressive ALS+/- frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 2377–2383. [Google Scholar] [CrossRef]
- Babbar, M.; Sheikh, M.S. Metabolic stress and disorders related to alterations in mitochondrial fission or fusion. Mol. Cell Pharmacol. 2013, 5, 109–133. [Google Scholar]
- Lyons, T.J.; Liu, H.; Goto, J.J.; Nersissian, A.; Roe, J.A.; Graden, J.A.; Café, C.; Ellerby, L.M.; Bredesen, D.E.; Gralla, E.B.; et al. Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc. Natl. Acad. Sci. USA 1996, 93, 12240–12244. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-ΚB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Putcha, G.V.; Harris, C.A.; Moulder, K.L.; Easton, R.M.; Thompson, C.B.; Johnson, E.M. Intrinsic and extrinsic pathway signaling during neuronal apoptosis: Lessons from the analysis of mutant mice. J. Cell Biol. 2002, 157, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. P53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar] [CrossRef]
- Kabashi, E.; Agar, J.N.; Taylor, D.M.; Minotti, S.; Durham, H.D. Focal dysfunction of the proteasome: A pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004, 89, 1325–1335. [Google Scholar] [CrossRef]
- Ranganathan, S.; Bowser, R. P53 and cell cycle proteins participate in spinal motor neuron cell death in ALS. Open Pathol. J. 2010, 4, 11–22. [Google Scholar] [CrossRef]
- Mishra, P.-S.; Dhull, D.K.; Nalini, A.; Vijayalakshmi, K.; Sathyaprabha, T.N.; Alladi, P.A.; Raju, T.R. Astroglia acquires a toxic neuroinflammatory role in response to the cerebrospinal fluid from amyotrophic lateral sclerosis patients. J. Neuroinflamm. 2016, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, J.A. Astrogliosis in amyotrophic lateral sclerosis: Role and therapeutic potential of astrocytes. Neurotherapeutics 2010, 7, 471–481. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-ΚB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-C.; Qi, X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 2013, 22, 4545–4561. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Castanedes Casey, M.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Tang, F.-L.; Liu, W.; Hu, J.-X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.-C. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St. Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell. 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and axonal transport misregulation in tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar]
- Shaltouki, A.; Hsieh, C.-H.; Kim, M.J.; Wang, X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in Parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3 by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Grenier, K.; McLelland, G.L.; Fon, E.A. Parkin- and PINK1-dependent mitophagy in neurons: Will the real pathway please stand up? Front. Neurol. 2013, 4, 100. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin preserves dopaminergic substantia nigra neurons following mitochondrial DNA mutagenic stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef]
- Amo, T.; Saiki, S.; Sawayama, T.; Sato, S.; Hattori, N. Detailed analysis of mitochondrial respiratory chain defects caused by loss of PINK1. Neurosci. Lett. 2014, 580, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Hanss, Z.; Larsen, S.B.; Antony, P.; Mencke, P.; Massart, F.; Jarazo, J.; Schwamborn, J.C.; Barbuti, P.A.; Mellick, G.D.; Krüger, R. Mitochondrial and clearance impairment in p.D620N VPS35 patient-derived neurons. Mov. Disord. 2021, 36, 704–715. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Nguyen, T.T.D.; Nguyen, T.K.O.; Vo, T.K.; Vo, V.G. Advances in developing therapeutic strategies in Alzheimer’s disease. Biomed. Pharmacother. 2021, 139, 111623. [Google Scholar] [CrossRef]
- Dunn, B.; Stein, P.; Cavazzoni, P. Approval of Aducanumab for Alzheimer’s disease–the FDA’s perspective. JAMA Intern. Med. 2021, 181, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar]
- Lin, Y.G.; Kunnumakkara, A.B.; Nair, A.; Merritt, W.M.; Han, L.Y.; Armaiz-Pena, G.M.; Kamat, A.A.; Spannuth, W.A.; Gershenson, D.M.; Lutgendorf, S.K. Curcumin inhibits tumor growth and angiogenesis in ovarian carcinoma by targeting the nuclear factor-κB pathway. Clin. Cancer Res. 2007, 13, 3423–3430. [Google Scholar] [CrossRef] [PubMed]
- Marchiani, A.; Rozzo, C.; Fadda, A.; Delogu, G.; Ruzza, P. Curcumin and curcumin-like molecules: From spice to drugs. Curr. Med. Chem. 2014, 21, 204–222. [Google Scholar] [CrossRef]
- Nuzzo, D.; Amato, A.; Picone, P.; Terzo, S.; Galizzi, G.; Bonina, F.B.; Mulé, F.; Di Carlo, M. A natural dietary supplement with a combination of nutrients prevents neurodegeneration induced by a high fat diet in mice. Nutrients 2018, 10, 1130. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beige, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef]
- Available online: https://www.clinicaltrials.gov (accessed on 3 October 2021).
- Hannan, M.A.; Dash, R.; Sohag, A.M.; Haque, N. Neuroprotection against oxidative stress: Phytochemicals targeting TrkB signaling and the Nrf2-ARE antioxidant system. Front. Mol. Neurosci. 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.L.; Yang, J.Y.; Dong, Y.X.; Wang, J.M.; Cui, Y.H.; Ikeshima, T.; Zhao, Y.Q.; Wu, C.F. Resveratrol inhibits nitric oxide and TNF-alpha production by lipopolysaccharide-activated microglia. Int. Immunopharmacol. 2005, 5, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.W.; Grossman, H.; Neugroschl, J.; Parker, S.; Burden, A.; Luo, X.; Sano, M. A randomized, double-blind, placebo-controlled trial of resveratrol with glucose and malate (RGM) to slow the progression of Alzheimer’s disease: A pilot study. Alzheimers Dement. 2018, 4, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective effects of quercetin in Alzheimer’s disease. Biomolecules 2020, 10, 59. [Google Scholar] [CrossRef]
- Costa, L.G.; Garrick, J.M.; Roque, P.J.; Pellacani, G. Mechanisms of neuroprotection by quercetin: Counteracting oxidative stress and more. Oxid. Med. Cell Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, R.; Zhan, Z.; Li, X.; Zhou, F.; Xing, A.; Jiang, C.; Chen, Y.; An, L. Beneficial effects of sulforaphane treatment in Alzheimer’s disease may be mediated through reduced HDAC1/3 and increased p75NTR expression. Front. Aging Neurosci. 2017, 9, 121. [Google Scholar] [CrossRef]
- Gleason, C.E.; Carlsson, C.M.; Barnet, J.H.; Meade, S.A.; Setchell, K.D.; Atwood, C.S.; Johnson, S.C.; Ries, M.L.; Asthana, S. A preliminary study of the safety, feasibility and cognitive efficacy of soy isoflavone supplements in older men and women. Age Ageing 2009, 38, 86–93. [Google Scholar] [CrossRef]
- Wu, P.-S.; Ding, H.-Y.; Yen, J.-H.; Chen, S.-F.; Lee, K.-H.; Wu, M.-J. Anti-inflammatory activity of 8-hydroxydaidzein in LPS-stimulated BV2 microglial cells via activation of the Nrf2-antioxidant and attenuation of Akt/NF-κB-inflammatory signaling pathways, as well as inhibition of COX-2 activity. J. Agric. Food Chem. 2018, 66, 5790–5801. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Jang, Y.J.; Hwang, M.K.; Kang, N.J.; Lee, K.W.; Lee, H.J. Attenuation of oxidative neuronal cell death by coffee phenolic phytochemicals. Mutat. Res. 2009, 661, 18–24. [Google Scholar] [CrossRef]
- Ishida, K.; Yamamoto, M.; Misawa, K.; Nishimura, H.; Misawa, K.; Ota, N.; Shimotoyodome, A. Coffee polyphenols prevent cognitive dysfunction and suppress amyloid β plaques in APP/PS2 transgenic mouse. Neurosci. Res. 2020, 154, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Hughes, D.A. Alpha-lipoic acid modulates NF-kappaB activity in human monocytic cells by direct interaction with DNA. Exp. Gerontol. 2002, 37, 401–410. [Google Scholar] [CrossRef]
- Packer, L.; Roy, S.; Sen, C.K. Alpha-lipoic acid: A metabolic antioxidant and potential redox modulator of transcription. Adv. Pharmacol. 1997, 38, 79–101. [Google Scholar]
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 38, 111–210. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Alzheimer’s Disease Cooperative Study. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of antioxidant supplement use and dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. J. Am. Med Assoc. 2014, 311, 33–44. [Google Scholar] [CrossRef]
- Bachurin, S.; Shevtsova, E.; Kireeva, E.; Oxenkrug, G.; Sablin, S. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann. N. Y. Acad. Sci. 2003, 993, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D.; Dimebon Investigators. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Jones, R.W. Dimebon disappointment. Alzheime. Res Ther. 2010, 2, 25. [Google Scholar] [CrossRef][Green Version]
- Chau, S.; Herrmann, N.; Ruthirakuhan, M.T.; Chen, J.J.; Lanctôt, K.L. Latrepirdine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, 4, CD009524. [Google Scholar] [CrossRef]
- Chang, K.-S.; Chen, C.-M. The role of oxidative stress in Parkinson’s disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Barzilai, A.; Melamed, E.; Shirvan, A. Is there a rationale for neuroprotection against dopamine toxicity in Parkinson’s disease? Cell. Mol. Neurobiol. 2001, 21, 215–235. [Google Scholar] [CrossRef]
- Stoker, T.B.; Barker, R.A. Recent developments in the treatment of Parkinson’s Disease. F1000Res. 2020, 9, F1000 Faculty Rev-862. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.D.; Broer, S. Creatine as a booster for human brain function. How might it work? Neurochem. Int. 2015, 89, 249–259. [Google Scholar] [CrossRef]
- Bender, A.; Koch, W.; Elstner, M.; Schombacher, Y.; Bender, J.; Moeschl, M.; Gekeler, F.; Muller-Myhsok, B.; Gasser, T.; Tatsch, K.; et al. Creatine supplementation in Parkinson disease: A placebo-controlled randomized pilot trial. Neurology 2006, 67, 1262–1264. [Google Scholar] [CrossRef] [PubMed]
- The NINDS-NET PD Investigators. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson’s disease. Neurology 2006, 67, 1262–1264. [Google Scholar]
- Kieburtz, K.; Tilley, B.C.; Elm, J.J.; Babcock, D.; Hauser, R.; Ross, G.W.; Augustine, A.H.; Augustine, E.U.; Aminoff, M.J.; Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET-PD) Investigators; et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: A randomized clinical trial. J. Am. Med Assoc. 2015, 313, 584–593. [Google Scholar] [CrossRef]
- De Rijk, M.C.; Breteler, M.M.; Den Breeijen, J.H.; Launer, L.J.; Grobbee, D.E.; Van der Meche, F.G.; Hofman, A. Dietary antioxidants and Parkinson disease. The Rotterdam Study. Arch. Neurol. 1997, 54, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Hernán, M.A.; Chen, H.; Spiegelman, D.; Willett, W.C.; Ascherio, A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology 2002, 59, 1161–1169. [Google Scholar] [CrossRef]
- Fahn, S. A pilot trial of high-dose alpha-tocopherol and ascorbate in early Parkinson’s disease. Ann. Neurol. 1992, 32, S128–S132. [Google Scholar] [CrossRef] [PubMed]
- The Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Parkinson Study Group QE3 Investigators; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [PubMed]
- Storch, A.; Jost, W.H.; Vieregge, P.; Spiegel, J.; Greulich, W.; Durner, J.; Muller, T.; Kupsch, A.; Henningsen, H.; Oertel, W.H.; et al. Randomized, double-blind, placebo-controlled trial on symptomatic effects of coenzyme Q10 in Parkinson disease. Arch. Neurol. 2007, 64, 938–944. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Protect Study. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170. [Google Scholar] [CrossRef]
- Hauser, R.A.; Lyons, K.E.; McClain, T.; Carter, S.; Perlmutter, D. Randomized, double-blind, pilot evaluation of intravenous glutathione in Parkinson’s disease. Mov. Disord. 2009, 24, 979–983. [Google Scholar] [CrossRef]
- Mischley, L.K.; Lau, R.C.; Shankland, E.G.; Wilbur, T.K.; Padowski, J.M. Phase IIb study of intranasal glutathione in Parkinson’s disease. J. Parkinsons Dis. 2017, 7, 289–299. [Google Scholar] [CrossRef]
- Jurcau, A. The role of natural antioxidants in the prevention of dementia–where do we stand and future perspectives. Nutrients 2021, 13, 282. [Google Scholar] [CrossRef]
- Jurcau, A.; Nunkoo, V.S. Clinical markers may identify patients at risk for early Parkinson’s disease dementia: A prospective study. Am. J. Alzheime. Dis. Other Demen. 2021, 36, 15333175211021369. [Google Scholar] [CrossRef]
- Medeiros, C.A.; Carvalhedo de Bruin, P.F.; Lopes, L.A.; Magalhaes, M.C.; De Lourdes Seabra, M.; De Bruin, V.M. Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson’s disease. A randomized, double blind, placebo-controlled study. J. Neurol. 2007, 254, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed]
- NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [Google Scholar] [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Cho, H.; Shukla, S. Role of edaravone as a treatment option for patients with amyotrophic lateral sclerosis. Pharmaceuticals 2020, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tanaka, M.; Yuki, S.; Hirai, M.; Yamamoto, Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 2018, 62, 20–38. [Google Scholar] [CrossRef]
- Ramis, M.R.; Esteban, S.; Miralles, A.; Tan, D.-X.; Reiter, R.J. Protective effects of melatonin and mitochondria-targeted antioxidants against oxidative stress: A review. Curr. Med. Chem. 2015, 22, 2690–2711. [Google Scholar] [CrossRef]
- Bald, E.M.; Nance, C.S.; Schultz, J.L. Melatonin may slow disease progression in amyotrophic lateral sclerosis: Findings from the pooled resource open-access ALS clinic trials database. Muscle Nerve 2021, 63, 572–576. [Google Scholar] [CrossRef]
- Pattee, G.L.; Post, G.R.; Gerber, R.E.; Bennett, J.P. Reduction of oxidative stress in amyotrophic lateral sclerosis following pramipexole treatment. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 90–95. [Google Scholar] [CrossRef]
- Izumi, Y.; Sawada, H.; Yamamoto, N.; Kume, T.; Katsuki, H.; Shimohama, S.; Akaike, A. Novel neuroprotective mechanisms of pramipexole, an anti-Parkinson drug, against endogenous dopamine-mediated excitotoxicity. Eur. J. Pharmacol. 2007, 557, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; Van den Berg, L.H.; Shefner, J.M.; Mitsumoto, H.; Mora, J.S.; Ludolph, A.; Hardiman, O.; Bozik, M.E.; Ingersoll, E.W.; Archibald, D.; et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): A randomised, double-blind, phase 3 trial. Lancet Neurol. 2013, 12, 1059–1067. [Google Scholar] [CrossRef]
- Cudkowicz, M.; Bozik, M.E.; Ingersoll, E.W.; Miller, R.; Mitsumoto, H.; Shefner, J.; Moore, D.H.; Schoenfeld, D.; Mather, J.L.; Archibald, D.; et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat. Med. 2011, 17, 1652–1656. [Google Scholar] [CrossRef] [PubMed]
- Macchi, Z.; Wang, Y.; Moore, D.; Katz, J.; Saperstein, D.; Walk, D.; Simpson, E.; Genge, A.; Bertorini, T.; Fernandes, J.A.; et al. Western ALS (WALS) Rasagiline Study Group. A multi-center screening trial of rasagiline in patients with amyotrophic lateral sclerosis: Possible mitochondrial biomarker target engagement. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 345–352. [Google Scholar] [CrossRef]
- Statland, J.M.; Moore, D.; Wang, Y.; Walsh, M.; Mozaffar, T.; Elman, L.; Nations, S.P.; Mitsumoto, H.; Fernandes, J.A.; Saperstein, D.; et al. Rasagiline Investigators of the Muscle Study Group and Western ALS Consortium. Rasagiline for amyotrophic lateral sclerosis: A randomized, controlled trial. Muscle Nerve 2019, 59, 201–207. [Google Scholar] [CrossRef]



{kind=link}
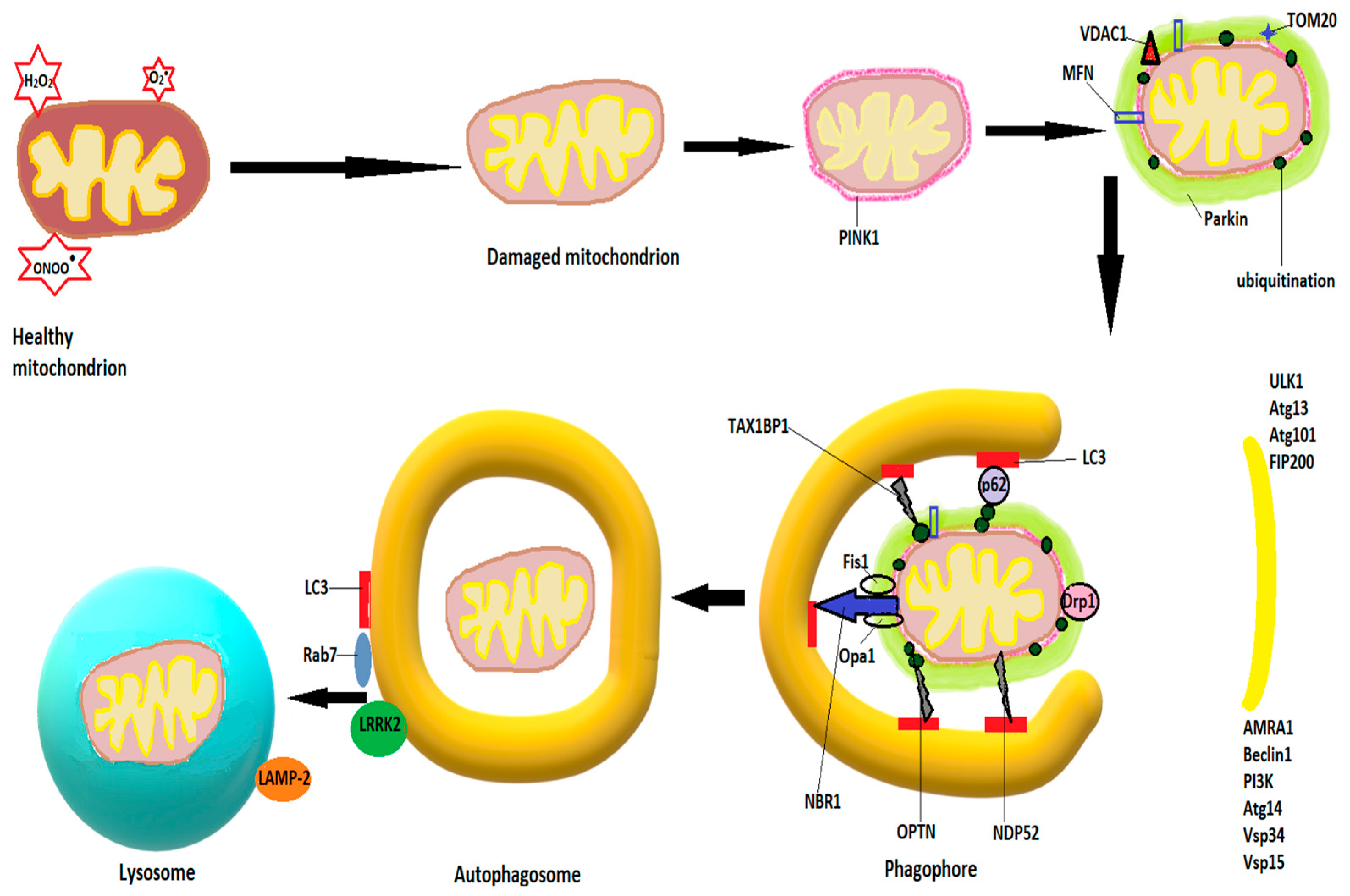{kind=link}
| Protein | Biological Process | Reference |
|---|---|---|
| Glutamate dehydrogenase 1 | TCA cycle (energy metabolism) | [294,295] |
| Malate dehydrogenase | TCA cycle (energy metabolism) | [295,296] |
| Subunit 5a of cytochrome c oxidase | ETC (energy metabolism) | [297] |
| NADH dehydrogenase (ubiquinone) | ETC (energy metabolism) | [297] |
| Subunits alpha, beta, d, and o of ATP synthase | OXPHOS (energy metabolism) | [291,297,298] |
| Core protein 1 of ubiquinol-cytochrome c reductase complex | ETC (energy metabolism) | [298] |
| Glutamine synthetase | Neurotransmission | [297,298] |
| Manganese superoxide dismutase | Antioxidant | [290,295] |
| Protein 1 of voltage-dependent anion-selective channels | Ion channel | [297] |
| Protein | Associated Disease | Result of Malfunction | References |
|---|---|---|---|
| Mitochondrial Fission | |||
| Aβ | AD | ↑ Drp1 levels | [371] |
| LRRK2 | PD | ↑ mitochondrial fission | [372] |
| SOD-1, TDP-43 | ALS | ↑ Drp1 and FIS1 protein levels (increases mitochondrial fission) | [373] |
| VPS35 | PD | Increased turnover of Drp1 and increased mitochondrial fission | [374] |
| Mitochondrial Fusion | |||
| Aβ | AD | ↑ Mfn1, Mfn2, OPA1 protein levels | [371] |
| α-synuclein | PD | ↓ mitochondrial fusion | [338] |
| LRRK2 | PD | ↓ OPA1 protein levels | [335] |
| SOD-1, TDP-43 | ALS | ↓ Mfn1 and OPA1 protein levels (decreased mitochondrial fusion) | [373,375] |
| VPS35 | PD | ↓ Mfn2 protein levels (decreased mitochondrial fusion) | [376] |
| Mitochondrial Transport | |||
| Aβ | AD | ↓ mitochondrial transport | [303] |
| LRRK2 | PD | Accumulation of MIRO1 (decreased mitochondrial transport) | [377] |
| Tau | AD | ↓ axonal transport | [378] |
| Mitochondrial Degradation | |||
| Aβ | AD | Delayed removal of damaged mitochondria | [379] |
| LRRK2 | PD | Delayed removal of damaged mitochondria | [377] |
| OPTN | ALS | Delayed removal and accumulation of damaged mitochondria | [380,381] |
| p62 | ALS | Impaired LC3 recognition Decreased autophagy | [382] |
| Parkin | PD | Impaired mitophagy in neurons and axons Degeneration of nigral dopaminergic neurons with impaired mtDNA replication | [383,384] |
| PINK1 | PD | Decrease of mitochondrial membrane potential | [385] |
| VPS35 | PD | Delayed removal of damaged mitochondria | [386] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurcau, A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 11847. https://doi.org/10.3390/ijms222111847
Jurcau A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. International Journal of Molecular Sciences. 2021; 22(21):11847. https://doi.org/10.3390/ijms222111847
Chicago/Turabian StyleJurcau, Anamaria. 2021. "Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress" International Journal of Molecular Sciences 22, no. 21: 11847. https://doi.org/10.3390/ijms222111847
APA StyleJurcau, A. (2021). Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. International Journal of Molecular Sciences, 22(21), 11847. https://doi.org/10.3390/ijms222111847