
Deacetylation of Transcription Factors in Carcinogenesis

 , ,
, ,  and
and 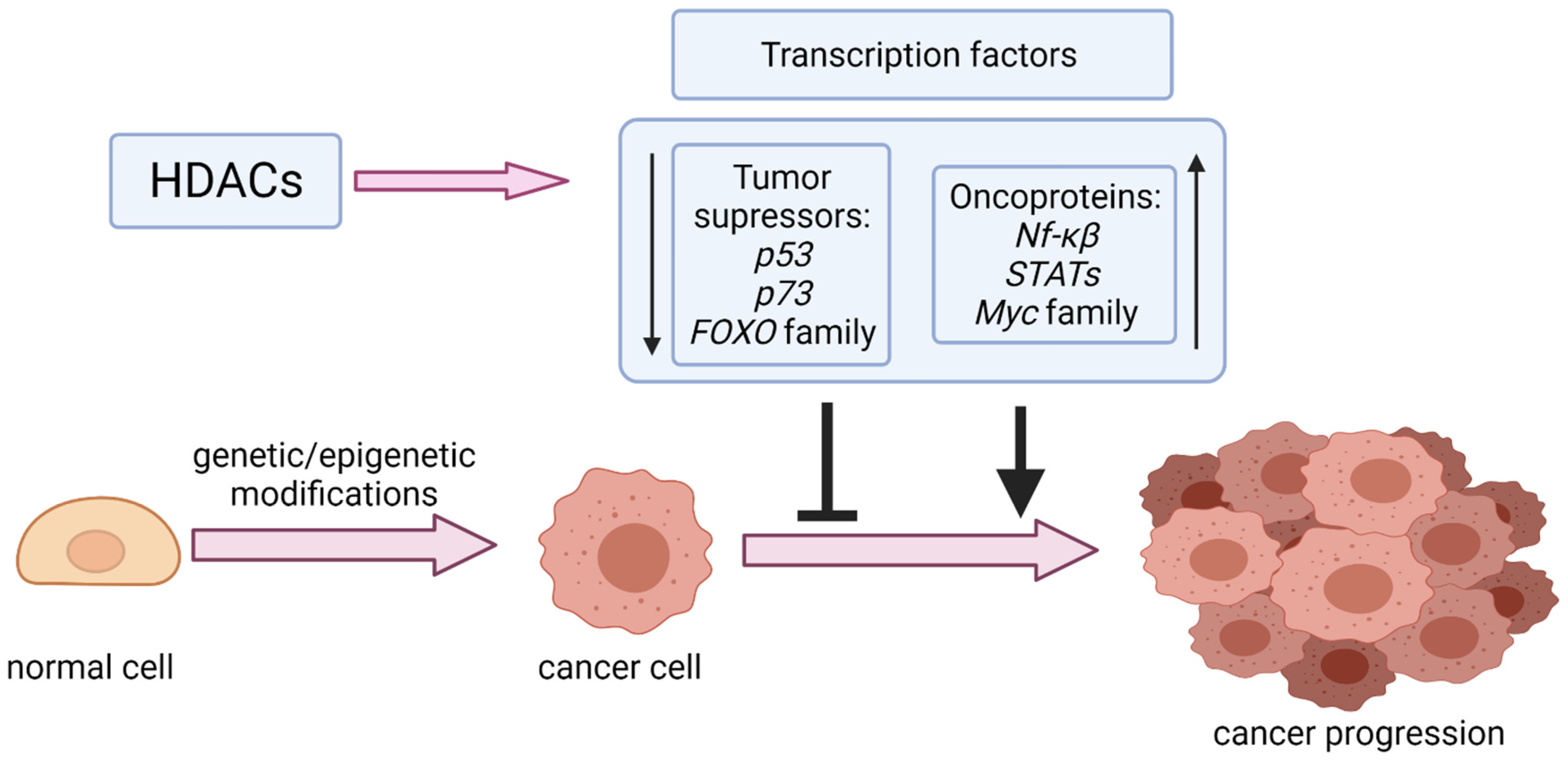{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tumor Suppressors
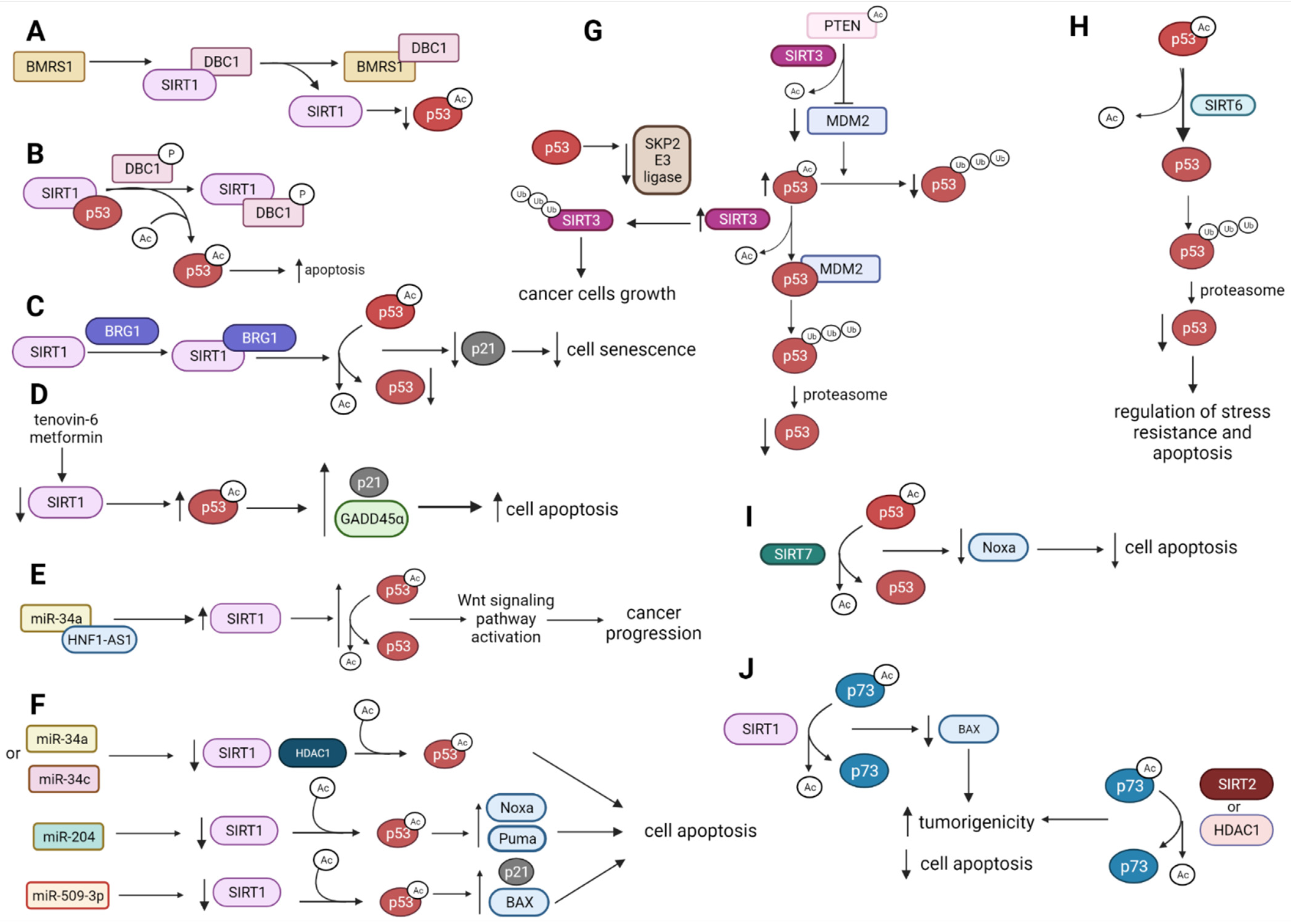
2.1. p53 Deacetylation by HDACs
2.1.1. p53 Is Deacetylated and Destabilized by SIRT1
2.1.2. p53, SIRT1 and HDAC1 Expression Is Affected by miRNAs Influencing Cell Apoptosis
2.1.3. SIRT3 Modulates p53 Tumor-Suppressive Functions
2.1.4. SIRT6 and SIRT7 Directly Deacetylate p53 to Regulate p53-Mediated Apoptosis
2.1.5. HDAC1 and HDAC8 Deacetylate and Suppress p53 Activity
2.1.6. HDAC1 and HDAC2 Control p53 Activity and Contribute to p53 Mutant Expression
2.2. SIRT1, SIRT2, and HDAC1 Deacetylate p73 and Suppress Its Activity
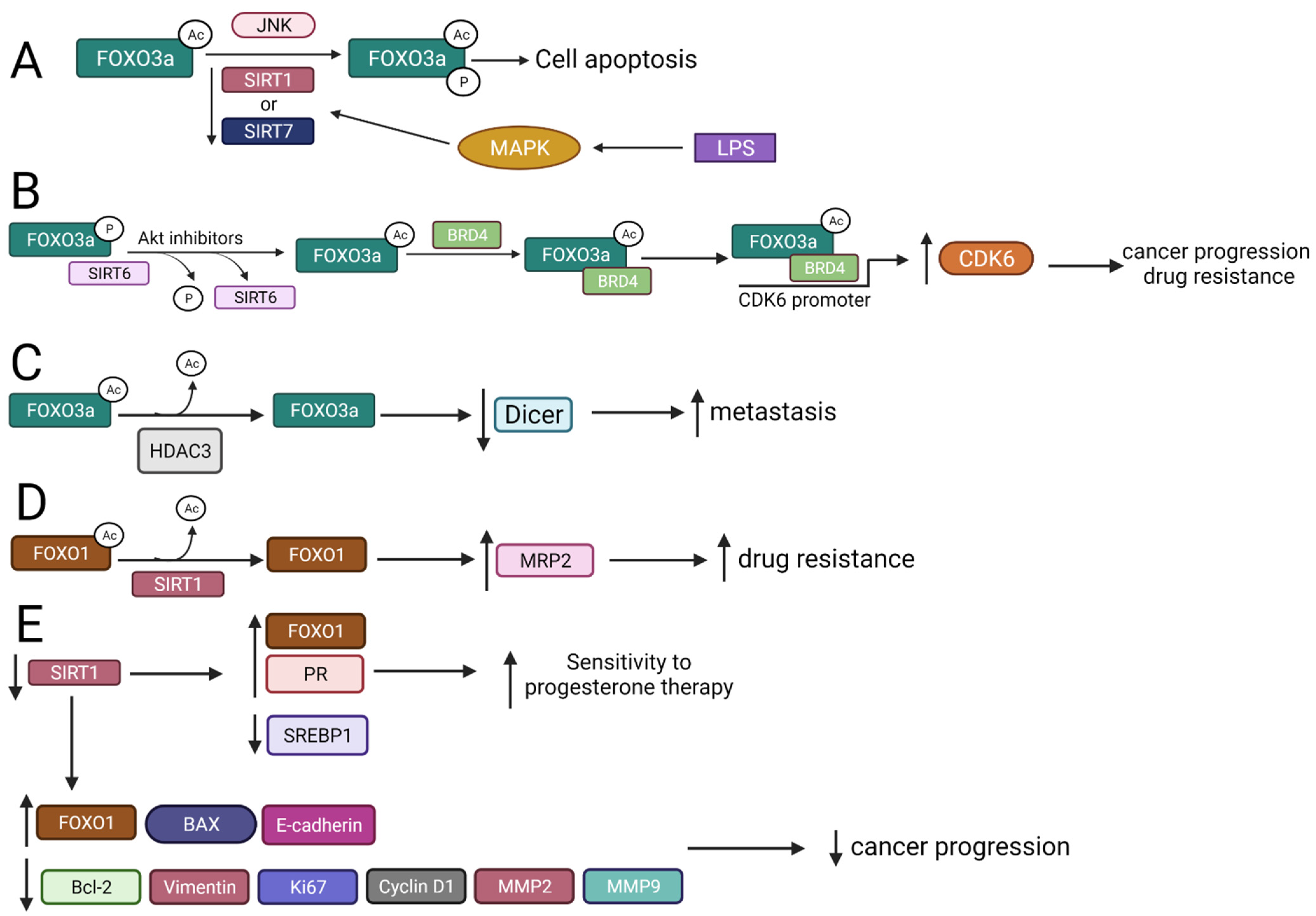
2.3. FOXO Deacetylation by HDACs Gives Different Biological Effects
2.3.1. FOXO3a Is Deacetylated by SIRT1 and SIRT7 That Regulates Apoptosis
2.3.2. SIRT6 Interacts with FOXO3a to Regulate Cancer Progression, Drug Resistance, and Apoptosis
2.3.3. HDAC3 Interplays with FOXO3 Increasing Metastasis
2.3.4. SIRT1 Mediates FOXO1 Deacetylation
2.3.5. FOXOM1 Is Acetylated by CBP/p300
3. Oncoproteins
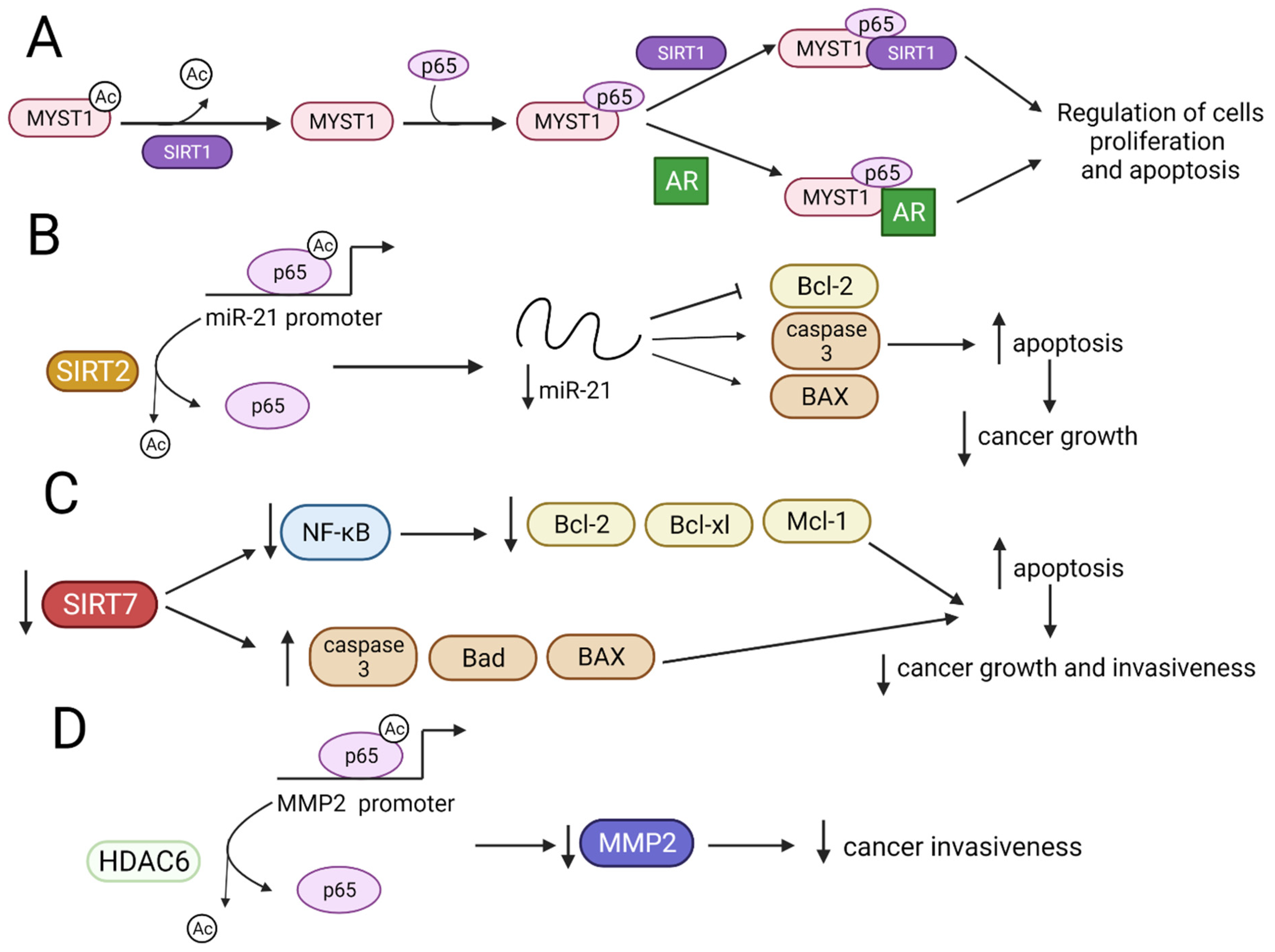
3.1. HDACs Deacetylate NF-ĸB Family Member p65 Modulating Its Tumor-Suppressive Functions
3.1.1. p65 Activity Is Modulated by SIRT1 and SIRT2 That Inhibits Cell Cancer Growth
3.1.2. Downregulation of SIRT7 Gene Decreases Expression of NF-κB and Inhibits the Growth and Invasiveness of Cancer Cells
3.1.3. HDIs Regulate Expression of NF-κB Partly through Inhibition of HDACs Activity
3.2. Signal Transducers and Activators of Transcription (STATs)
3.2.1. HDAC1 and HDAC4 Inhibit STAT3 Activity and Interfere with Its Stability
3.2.2. SIRT1 and Its Activators Affect STAT3 Transcriptional Function
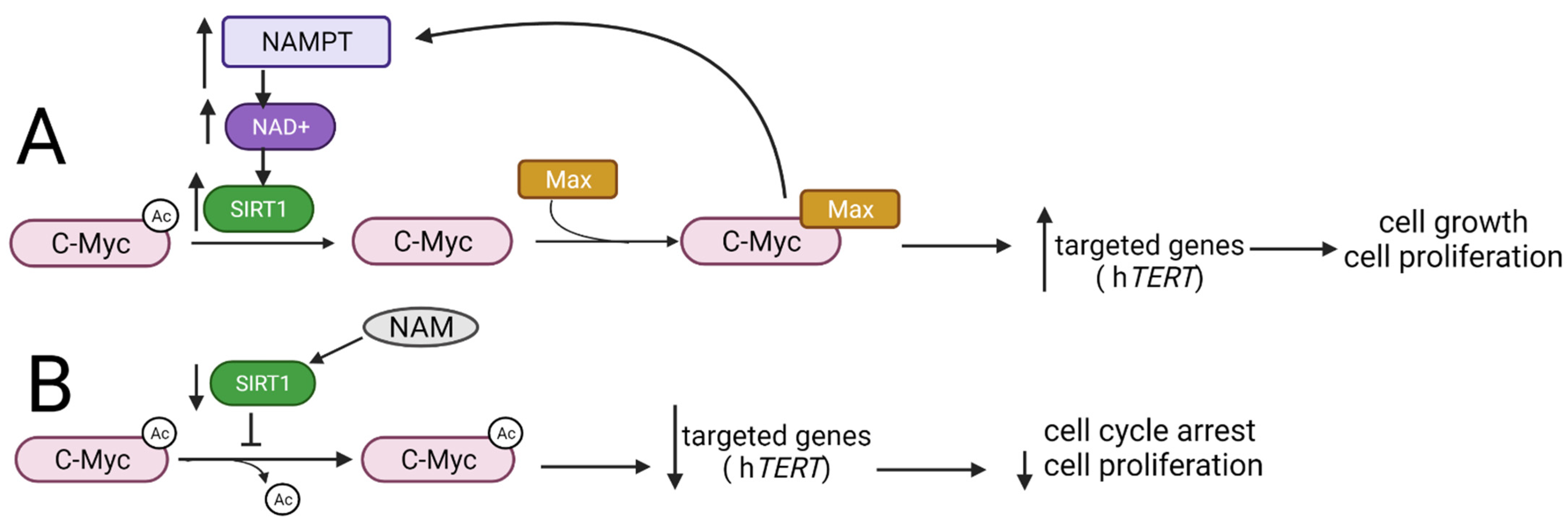
3.3. Myc Family
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.B.; Anderson, R.N. The Leading Causes of Death in the US for 2020. JAMA 2021, 325, 1829–1830. [Google Scholar] [CrossRef]
- (US) NI of H, Study BSC. Understanding Cancer. 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK20362/ (accessed on 3 September 2021).
- Jin, J.; Wu, X.; Yin, J.; Li, M.; Shen, J.; Li, J.; Zhao, Y.; Zhao, Q.; Wu, J.; Wen, Q. Identification of Genetic Mutations in Cancer: Challenge and Opportunity in the New Era of Targeted Therapy. Front. Oncol. 2019, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Tulsyan, S.; Dar, S.A.; Sisodiya, S.; Abiha, U.; Kumar, R.; Mishra, B.N.; Haque, S. Role of Epigenetics in carcinogenesis: Recent Advancements in Anticancer Therapy. Semin. Cancer Biol. 2021. in Press. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1044579X21001930 (accessed on 30 June 2021).
- Liu, Y.; Wang, B.; Shi, S.; Li, Z.; Wang, Y.; Yang, J. Construction of methylation-associated nomogram for predicting the recurrence-free survival risk of stage I-III lung adenocarcinoma. Future Oncol. 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/34476982/ (accessed on 6 September 2021). [CrossRef] [PubMed]
- Seok, H.J.; Choi, Y.E.; Choi, J.Y.; Yi, J.M.; Kim, E.J.; Choi, M.Y.; Lee, S.J.; Bae, I.H. Novel miR-5088-5p promotes malignancy of breast cancer by inhibiting DBC2. Mol. Ther. Nucleic Acids 2021, 25, 127–142. Available online: https://pubmed.ncbi.nlm.nih.gov/34457998/ (accessed on 6 September 2021). [CrossRef]
- Ghalkhani, E.; Akbari, M.T.; Izadi, P.; Mahmoodzadeh, H.; Kamali, F. Assessment of DAPK1 and CAVIN3 Gene Promoter Methylation in Breast Invasive Ductal Carcinoma and Metastasis. Cell J. 2021, 23, 397–405. Available online: https://pubmed.ncbi.nlm.nih.gov/34455714/ (accessed on 6 September 2021).
- Hałasa, M.; Wawruszak, A.; Przybyszewska, A.; Jaruga, A.; Guz, M.; Kałafut, J.; Stepulak, A.; Cybulski, M. H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy. Cells 2019, 8, 485. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31121824 (accessed on 16 March 2020). [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 1004. [Google Scholar]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M. A subcellular map of the human proteome. Science 2017, 356, 806. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Zhang, D.; Wang, X.; Lin, D.; Chen, Y.; Xu, X. Targeting SIRT1 to inhibit the proliferation of multiple myeloma cells. Oncol. Lett. 2021, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Kim, Y.; Kim, D.; Cho, E.Y.; Han, J.; Kim, H.K.; Shim, Y.M.; Kim, D.H. Metformin and tenovin-6 synergistically induces apoptosis through LKB1-independent SIRT1 down-regulation in non-small cell lung cancer cells. J. Cell Mol. Med. 2019, 23, 2872–2889. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Kashiwagi, E.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Seki, N.; Naito, S. Foxo3a expression and acetylation regulate cancer cell growth and sensitivity to cisplatin. Cancer Sci. 2010, 101, 1177–1185. Available online: https://pubmed.ncbi.nlm.nih.gov/20210796/ (accessed on 18 June 2021). [CrossRef] [PubMed]
- Jaganathan, A.; Chaurasia, P.; Xiao, G.Q.; Philizaire, M.; Lv, X.; Yao, S.; Burnstein, K.L.; Liu, D.P.; Levine, A.C. Coactivator MYST1 regulates nuclear factor-κB and androgen receptor functions during proliferation of prostate cancer cells. Mol. Endocrinol. 2014, 28, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-B.; Lee, S.-H.; Um, J.-H.; Kim, M.-J.; Hyun, S.-K.; Gong, E.-J.; Oh, W.K.; Kang, C.D.; Kim, S.H. Sensitization of Chemo-Resistant Human Chronic Myeloid Leukemia Stem-Like Cells to Hsp90 Inhibitor by SIRT1 Inhibition. Int. J. Biol. Sci. 2015, 11, 923. [Google Scholar] [CrossRef][Green Version]
- Zhang, S.; Yang, Y.; Huang, S.; Deng, C.; Zhou, S.; Yang, J. SIRT1 inhibits gastric cancer proliferation and metastasis via STAT3/MMP-13 signaling. J. Cell Physiol. 2019, 234, 15395–15406. Available online: https://pubmed.ncbi.nlm.nih.gov/30710340/ (accessed on 15 March 2021). [CrossRef] [PubMed]
- Ma, J.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal 2020, 181, 1–13. Available online: https://biosignaling.biomedcentral.com/articles/10.1186/s12964-020-0527-z (accessed on 6 September 2021). [CrossRef] [PubMed]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4, ra46. Available online: https://pubmed.ncbi.nlm.nih.gov/21775285/ (accessed on 6 September 2021). [CrossRef] [PubMed]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; Buchholz, T.A.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/β-catenin signaling. Stem Cells 2012, 30, 2366–2377. Available online: https://pubmed.ncbi.nlm.nih.gov/22961641/ (accessed on 6 September 2021). [CrossRef] [PubMed]
- Al Emam, A.; Arbon, D.; Jeeves, M.; Kysela, B. Ku70 N-terminal lysines acetylation/deacetylation is required for radiation-induced DNA-double strand breaks repair. Neoplasma 2018, 65, 708–719. Available online: https://pubmed.ncbi.nlm.nih.gov/30249103/ (accessed on 6 September 2021). [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, X.; Han, C.; Wang, L.; Zhang, X.; He, X.; Lu, X. Targeting tumor suppressor genes for cancer therapy. BioEssays 2015, 37, 1277–1286. Available online: https://pubmed.ncbi.nlm.nih.gov/26445307/ (accessed on 13 April 2021). [CrossRef] [PubMed]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. Available online: https://ar.iiarjournals.org/content/40/11/6009 (accessed on 4 October 2021). [CrossRef] [PubMed]
- Adcock, I.M.; Caramori, G. Transcription Factors; Asthma COPD: London, UK, 2009; pp. 373–380. [Google Scholar]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell. Biol. 2001, 2, 599–609. Available online: https://www.nature.com/articles/35085068 (accessed on 15 September 2021). [CrossRef] [PubMed]
- Xu, R.; Won, J.Y.; Kim, C.H.; Kim, D.E.; Yim, H. Roles of the Phosphorylation of Transcriptional Factors in Epithelial-Mesenchymal Transition. J. Oncol. 2019, 2019, 5810465. [Google Scholar] [CrossRef]
- Thiagarajan, D.; Vedantham, S.; Ananthakrishnan, R.; Schmidt, A.M.; Ramasamy, R. Mechanisms of transcription factor acetylation and consequences in hearts. Biochim. Biophys. Acta—Mol. Basis Dis. 2016, 1862, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.M.; Johnson, R.W. Targeting Histone Modifications in Bone and Lung Metastatic Cancers. Curr. Osteoporos Rep. 2021, 19, 230–246. Available online: https://link.springer.com/article/10.1007/s11914-021-00670-2 (accessed on 6 September 2021). [CrossRef]
- Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers 2021, 13, 3409. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30691229 (accessed on 9 October 2019). [CrossRef] [PubMed]
- Kanapathipillai, M. Treating p53 mutant aggregation-associated cancer. Cancers 2018, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, E.; Stuendl, N.; Kaltenegger, H.; Windpassinger, C.; Donohue, N.; Leithner, A.; Lohberger, B. Histone deacetylase inhibitors vorinostat and panobinostat induce G1 cell cycle arrest and apoptosis in multidrug resistant sarcoma cell lines. Oncotarget 2017, 8, 77254–77267. Available online: https://www.oncotarget.com/article/20460/text/ (accessed on 16 September 2021). [CrossRef]
- Sonnemann, J.; Marchx, C.; Becker, S.; Wittig, S.; Palani, C.D.; Krämer, O.H.; Beck, J.F. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br. J. Cancer 2014, 110, 656–667. Available online: https://www.nature.com/articles/bjc2013742 (accessed on 16 September 2021). [CrossRef] [PubMed]
- Peltonen, K.; Kiviharju, T.M.; Järvinen, P.M.; Ra, R.; Laiho, M. Melanoma cell lines are susceptible to histone deacetylase inhibitor TSA provoked cell cycle arrest and apoptosis. Pigment cell Res. 2005, 18, 196–202. Available online: https://pubmed.ncbi.nlm.nih.gov/15892716/ (accessed on 16 September 2021). [CrossRef]
- Fang, Q.; Bellanti, J.A.; Zheng, S.G. Advances on the role of the deleted in breast cancer (DBC1) in cancer and autoimmune diseases. J. Leukoc. Biol. John Wiley Sons Inc. 2021, 109, 449–454. Available online: https://pubmed.ncbi.nlm.nih.gov/32337788/ (accessed on 13 April 2021). [CrossRef] [PubMed]
- Liu, X.; Ehmed, E.; Li, B.; Dou, J.; Qiao, X.; Jiang, W.; Yang, X.; Qiao, S.; Wu, Y. Breast cancer metastasis suppressor 1 modulates SIRT1-dependent p53 deacetylation through interacting with DBC1. Am. J. Cancer Res. 2016, 6, 1441–1449. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27429856 (accessed on 9 October 2019).
- Zannini, L.; Buscemi, G.; Kim, J.E.; Fontanella, E.; Delia, D. DBC1 phosphorylation by ATM/ATR inhibits SIRT1 deacetylase in response to DNA damage. J. Mol. Cell Biol. 2012, 4, 294–303. [Google Scholar] [CrossRef]
- Pan, J.H.; Zhou, H.; Zhu, S.B.; Huang, J.L.; Zhao, X.X.; Ding, H.; Qin, L.; Pan, Y.L. Nicotinamide phosphoribosyl transferase regulates cell growth via the Sirt1/P53 signaling pathway and is a prognosis Marker in colorectal cancer. J. Cell Physiol. 2019, 234, 4385–4395. [Google Scholar] [CrossRef]
- Wang, G.; Fu, Y.; Hu, F.; Lan, J.; Xu, F.; Yang, X.; Luo, X.; Wang, J.; Hu, J. Loss of BRG1 induces CRC cell senescence by regulating p53/p21 pathway. Cell Death Dis. 2017, 8, e2607. [Google Scholar] [CrossRef]
- Fang, C.; Qiu, S.; Sun, F.; Li, W.; Wang, Z.; Yue, B.; Wu, X.; Yan, D. Long non-coding RNA HNF1A-AS1 mediated repression of miR-34a/SIRT1/p53 feedback loop promotes the metastatic progression of colon cancer by functioning as a competing endogenous RNA. Cancer Lett. 2017, 410, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef]
- Okada, N.; Lin, C.P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C.; et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Ren, L.; Xie, B.; Liang, Z.; Chen, J. MiR-204 enhances mitochondrial apoptosis in doxorubicin-treated prostate cancer cells by targeting SIRT1/p53 pathway. Oncotarget 2017, 8, 97313–97322. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29228612 (accessed on 9 October 2021). [CrossRef]
- Abdolvahabi, Z.; Nourbakhsh, M.; Hosseinkhani, S.; Hesari, Z.; Alipour, M.; Jafarzadeh, M.; Ghorbanhosseini, S.S.; Seiri, P.; Yousefi, Z.; Yarahmadi, S.; et al. MicroRNA-590-3P suppresses cell survival and triggers breast cancer cell apoptosis via targeting sirtuin-1 and deacetylation of p53. J. Cell Biochem. 2019, 120, 9356–9368. [Google Scholar] [CrossRef]
- Ilisso, C.P.; Delle Cave, D.; Mosca, L.; Pagano, M.; Coppola, A.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine regulates apoptosis and autophagy in MCF-7 breast cancer cells through the modulation of specific microRNAs. Cancer Cell Int. 2018, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhou, Y.; Qiao, C.; Ni, T.; Li, Z.; Wang, X.; Guo, Q.; Lu, N.; Wei, L. Oroxylin A promotes PTEN-mediated negative regulation of MDM2 transcription via SIRT3-mediated deacetylation to stabilize p53 and inhibit glycolysis in wt-p53 cancer cells. J. Hematol. Oncol. 2015, 8, 41. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25902914 (accessed on 9 October 2019). [CrossRef] [PubMed]
- Zhang, Y.Y.; Zhou, L.M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, L.; Wang, S.; Wang, M.; Zhao, J.; Zhang, Z.; Li, X.; Jia, L.; Han, Y. SIRT3 deacetylates and promotes degradation of P53 in PTEN-defective non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 189–198. [Google Scholar] [CrossRef] [PubMed]
- KuMarch, A.; Corey, C.; Scott, I.; Shiva, S.; D’Cunha, J. Minnelide/Triptolide impairs mitochondrial function by regulating SIRT3 in P53-dependent manner in non-small cell lung cancer. PLoS ONE 2016, 11, e0160783. [Google Scholar]
- Wood, M.; RyMarchchyk, S.; Zheng, S.; Cen, Y. Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6. Arch. Biochem. Biophys. 2018, 638, 8–17. [Google Scholar] [CrossRef]
- Zhao, J.; Wozniak, A.; Adams, A.; Cox, J.; Vittal, A.; Voss, J.; Bridges, B.; Weinman, S.A.; Li, Z. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, H.; Zhu, M.; Hu, P.; Liu, X.; Qing, Y.; Wang, X.; Wang, H.; Wang, Z.; Xu, J.; et al. Involvement of p53 acetylation in growth suppression of Cutaneous T-cell lymphomas induced by HDAC inhibition. J. Invest. Dermatol. 2020, 140, 2009–2022. Available online: http://www.ncbi.nlm.nih.gov/pubmed/32119867 (accessed on 16 March 2020). [CrossRef]
- Luo, S.-M.; Tsai, W.-C.; Tsai, C.-K.; Chen, Y.; Hueng, D.-Y. ARID4B Knockdown Suppresses PI3K/AKT Signaling and Induces Apoptosis in Human Glioma Cells. Onco Targets Ther. 2021, 14, 1843. [Google Scholar] [CrossRef]
- Zhu, X.; Leboeuf, M.; Liu, F.; Grachtchouk, M.; Seykora, J.T.; Morrisey, E.E.; Dlugosz, A.A.; Millar, S.E. HDAC1/2 Control Proliferation and Survival in Adult Epidermis and Pre‒Basal Cell Carcinoma through p16 and p53. J. Investig. Dermatol. 2021, in press. [Google Scholar] [CrossRef]
- Mirza, A.N.; Fry, M.A.; Urman, N.M.; Atwood, S.X.; Roffey, J.; Ott, G.R.; Chen, B.; Lee, A.; Brown, A.S.; Aasi, S.Z.; et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight 2017, 2, e97071. Available online: https://pubmed.ncbi.nlm.nih.gov/29093271/ (accessed on 17 September 2021). [CrossRef] [PubMed]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schäfer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. Available online: https://pubmed.ncbi.nlm.nih.gov/27721407/ (accessed on 16 September 2021). [CrossRef]
- Cascio, C.L.; McNamara, J.B.; Melendez, E.L.; Lewis, E.M.; Dufault, M.E.; Sanai, N.; Plaisier, C.L.; Mehta, S. Nonredundant, isoform-specific roles of HDAC1 in glioma stem cells. JCI Insight 2021, 6, e149232. Available online: https://insight.jci.org/articles/view/149232 (accessed on 30 September 2021). [CrossRef]
- Choi, M.; Choi, Y.M.; An, I.S.; Bae, S.; Jung, J.H.; An, S. E3 ligase RCHY1 negatively regulates HDAC2. Biochem. Biophys. Res. Commun. 2020, 521, 37–41. [Google Scholar] [CrossRef]
- Sun, D.; Yu, M.; Li, Y.; Xing, H.; Gao, Y.; Huang, Z.; Hao, W.; Lu, K.; Kong, C.; Shimozato, O.; et al. Histone deacetylase 2 is involved in DNA damage-mediated cell death of human osteosarcoma cells through stimulation of the ATM/p53 pathway. FEBS Open Bio. 2019, 9, 478. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, S.; Brody, L.; Rahman, S.; Mahul-Mellier, A.-L.; Mercado, N.; Ito, K.; El-Bahrawy, M.; Silver, A.; Boobis, A.; Bell, J.D.; et al. Synergy between histone deacetylase inhibitors and DNA-damaging agents is mediated by histone deacetylase 2 in colorectal cancer. Oncotarget 2016, 7, 44505. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F. Effect of Valproic Acid on the Class I Histone Deacetylase 1, 2 and 3, Tumor Suppressor Genes p21WAF1/CIP1 and p53, and Intrinsic Mitochondrial Apoptotic Pathway, Pro- (Bax, Bak, and Bim) and anti- (Bcl-2, Bcl-xL, and Mcl-1) Apoptotic Genes Expression, Cell Viability, and Apoptosis Induction in Hepatocellular Carcinoma HepG2 Cell Line. Asian Pac. J. Cancer Prev. 2021, 22, 89–95. Available online: https://pubmed.ncbi.nlm.nih.gov/33576217/ (accessed on 30 September 2021).
- Jost, C.A.; Marchin, M.C.; Kaelin, W.G. p73 is a human p53-related protein that can induce apoptosis. Nature 1997, 389, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. Available online: https://pubmed.ncbi.nlm.nih.gov/9288759/ (accessed on 9 October 2021). [CrossRef]
- Wu, H.H.; Abou Zeinab, R.; Leng, R.P. Regulation of p73 by Pirh2-AIP4 loop. Aging 2021, 13, 1–2. Available online: https://pubmed.ncbi.nlm.nih.gov/34520394/ (accessed on 9 October 2021). [CrossRef]
- Olmos, Y.; Brosens, J.J.; Lam, E.W. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updates 2011, 14, 35–44. [Google Scholar] [CrossRef]
- Dai, J.M.; Wang, Z.Y.; Sun, D.C.; Lin, R.X.; Wang, S.Q. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J. Cell Physiol. 2007, 210, 161–166. Available online: http://www.ncbi.nlm.nih.gov/pubmed/16998810 (accessed on 9 October 2021). [CrossRef]
- Funato, K.; Hayashi, T.; Echizen, K.; Negishi, L.; Shimizu, N.; Koyama-Nasu, R.; Nasu-Nishimura, Y.; Morishita, Y.; Tabar, V.; Todo, T.; et al. SIRT 2-mediated inactivation of p73 is required for glioblastoma tumorigenicity. EMBO Rep. 2018, 19, e45587. [Google Scholar] [CrossRef]
- Schipper, H.; Alla, V.; Meier, C.; Nettelbeck, D.M.; Pützer, B.M. Eradication of metastatic melanoma through cooperative expression of RNA-based HDAC1 inhibitor and p73 by oncolytic adenovirus. Oncotarget 2014, 5, 5893–5907. [Google Scholar] [CrossRef]
- Grossi, V.; Fasano, C.; Celestini, V.; Signorile, M.L.; Sanese, P.; Simone, C. Chasing the Foxo3: Insights into its new mitochondrial lair in colorectal cancer landscape. Cancers 2019, 11, 414. [Google Scholar] [CrossRef]
- Li, Z.; Bridges, B.; Olson, J.; Weinman, S.A. The interaction between acetylation and serine-574 phosphorylation regulates the apoptotic function of FOXO3. Oncogene 2017, 36, 1887–1898. [Google Scholar] [CrossRef]
- Jeung, Y.-J.; Kim, H.-G.; Ahn, J.; Lee, H.-J.; Lee, S.-B.; Won, M.; Jung, C.R.; Im, J.Y.; Kim, B.K.; Park, S.K.; et al. Shikonin induces apoptosis of lung cancer cells via activation of FOXO3a/EGR1/SIRT1 signaling antagonized by p300. Biochim. Biophys. Acta 2016, 1863, 2584–2593. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27452907 (accessed on 9 October 2021). [CrossRef]
- Huo, L.; Bai, X.; Wang, Y.; Wang, M. Betulinic acid derivative B10 inhibits glioma cell proliferation through suppression of SIRT1, acetylation of FOXO3a and upregulation of Bim/PUMA. Biomed. Pharmacother. 2017, 92, 347–355. Available online: http://www.ncbi.nlm.nih.gov/pubmed/28554130 (accessed on 9 October 2021). [CrossRef]
- Liu, J.; Duan, Z.; Guo, W.; Zeng, L.; Wu, Y.; Chen, Y.; Tai, F.; Wang, Y.; Lin, Y.; Zhang, Q.; et al. Targeting the BRD4/FOXO3a/CDK6 axis sensitizes AKT inhibition in luminal breast cancer. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nie, L.; Xu, K.; Fu, Y.; Zhong, J.; Gu, K.; Zhang, L. SIRT6, a novel direct transcriptional target of FoxO3a, mediates colon cancer therapy. Theranostics 2019, 9, 2380–2394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cai, M.; Gong, Z.; Zhang, B.; Li, Y.; Guan, L.; Hou, X.; Li, Q.; Liu, G.; Xue, Z.; et al. Geminin facilitates FoxO3 deacetylation to promote breast cancer cell metastasis. J. Clin. Investig. 2017, 127, 2159–2175. [Google Scholar] [CrossRef]
- Choi, H.K.; Cho, K.B.; Phuong, N.T.T.; Han, C.Y.; Han, H.K.; Hien, T.T.; Choi, H.S.; Kang, K.W. SIRT1-mediated foxo1 deacetylation is essential for multidrug resistance-associated protein 2 expression in tamoxifen-resistant breast cancer cells. Mol. Pharm. 2013, 10, 2517–2527. Available online: https://pubmed.ncbi.nlm.nih.gov/23763570/ (accessed on 22 July 2020). [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Che, X.; Li, W.; Liu, Z.; Jiang, J. Roles of SIRT1/FoxO1/SREBP-1 in the development of progestin resistance in endometrial cancer. Arch. Gynecol. Obstet. 2018, 298, 961–969. Available online: https://pubmed.ncbi.nlm.nih.gov/30206735/ (accessed on 22 July 2020). [CrossRef]
- Liu, H.; Liu, N.; Zhao, Y.; Zhu, X.; Wang, C.; Liu, Q.; Gao, C.; Zhao, X.; Li, J. Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging 2019, 11, 9643–9660. Available online: https://pubmed.ncbi.nlm.nih.gov/31689236/ (accessed on 22 July 2020). [CrossRef]
- Ceballos, M.P.; Decándido, G.; Quiroga, A.D.; Comanzo, C.G.; Livore, V.I.; Lorenzetti, F.; Lambertucci, F.; Chazarreta-Cifre, L.; Banchio, C.; Alvarez, M.L.; et al. Inhibition of sirtuins 1 and 2 impairs cell survival and migration and modulates the expression of P-glycoprotein and MRP3 in hepatocellular carcinoma cell lines. Toxicol. Lett. 2018, 289, 63–74. Available online: https://pubmed.ncbi.nlm.nih.gov/29545174/ (accessed on 22 July 2020). [CrossRef]
- Lv, C.; Zhao, G.; Sun, X.; Wang, P.; Xie, N.; Luo, J.; Tong, T. Acetylation of FOXM1 is essential for its transactivation and tumor growth stimulation. Oncotarget 2016, 7, 60366–66382. [Google Scholar] [CrossRef]
- Wang, W.; Nag, S.A.; Zhang, R. Targeting the NFκB Signaling Pathways for Breast Cancer Prevention and Therapy. Curr. Med. Chem. 2015, 22, 264. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, D.; Lu, Q.; Fei, M.; Li, M.; Wu, X. Sirt2 suppresses glioma cell growth through targeting NF-κB-miR-21 axis. Biochem. Biophys. Res. Commun. 2013, 441, 661–667. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24161395 (accessed on 9 October 2021). [CrossRef] [PubMed]
- Mao, S.; Ma, J.; Yu, H. Sirtuin-7 knockdown inhibits the growth of endometrial cancer cells by inducing apoptosis via the NF-κB signaling pathway. Oncol. Lett. 2019, 17, 937–943. [Google Scholar] [CrossRef]
- Yang, C.-J.; Liu, Y.-P.; Dai, H.-Y.; Shiue, Y.-L.; Tsai, C.-J.; Huang, M.-S.; Yeh, Y.T. Nuclear HDAC6 inhibits invasion by suppressing NF-κB/MMP2 and is inversely correlated with metastasis of non-small cell lung cancer. Oncotarget 2015, 6, 30263–30276. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26388610 (accessed on 9 October 2021). [CrossRef] [PubMed]
- Chen, C.-Y.; Chen, C.-C.; Chuang, W.-Y.; Leu, Y.-L.; Ueng, S.-H.; Hsueh, C.; Yeh, C.T.; Wang, T.H. Hydroxygenkwanin Inhibits Class I HDAC Expression and Synergistically Enhances the Antitumor Activity of Sorafenib in Liver Cancer Cells. Front. Oncol. 2020, 10, 216. [Google Scholar] [CrossRef]
- Okabe, S.; Tanaka, Y.; Gotoh, A. Targeting phosphoinositide 3-kinases and histone deacetylases in multiple myeloma. Exp. Hematol. Oncol. 2021, 10, 1–12. Available online: https://pubmed.ncbi.nlm.nih.gov/33663586/ (accessed on 30 September 2021). [CrossRef]
- Ishikawa, C.; Mori, N. The role of CUDC-907, a dual phosphoinositide-3 kinase and histone deacetylase inhibitor, in inhibiting proliferation of adult T-cell leukemia. Eur. J. Haematol. 2020, 105, 763–772. Available online: https://onlinelibrary.wiley.com/doi/full/10.1111/ejh.13508 (accessed on 2 October 2021). [CrossRef]
- Afaloniati, H.; Angelopoulou, K.; Giakoustidis, A.; Hardas, A.; Pseftogas, A.; Makedou, K.; Gargavanis, A.; Goulopoulos, T.; Iliadis, S.; Papadopoulos, V.; et al. HDAC1/2 Inhibitor Romidepsin Suppresses DEN-Induced Hepatocellular Carcinogenesis in Mice. Onco Targets Ther. 2020, 13, 5575. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Xu, J.; Wang, C.; Feng, Q.; Wang, Q.; Yang, Y.; Lu, H.; Wang, F.; Zhu, K.; Li, W.; et al. Suppression of the SAP18/HDAC1 complex by targeting TRIM56 and Nanog is essential for oncogenic viral FLICE-inhibitory protein-induced acetylation of p65/RelA, NF-κB activation, and promotion of cell invasion and angiogenesis. Cell Death Differ. 2019, 26, 1970. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Daniel, J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Guan, Y.; Chatterjee, D. Stat3 Dimerization Regulated by Reversible Acetylation of a Single Lysine Residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Boldogh, I.; Brasier, A.R. STAT3 NH 2 -Terminal Acetylation Is Activated by the Hepatic Acute-Phase Response and Required for IL-6 Induction of Angiotensinogen. Gastroenterology 2005, 129, 1616–1632. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, Y.; Sheng, Y.; Xiao, J.; Xiao, Y.; Cheng, N.; Chai, Y.; Wu, X.; Zhang, S.; Xiang, T. SIRT1 downregulated FGB expression to inhibit RCC tumorigenesis by destabilizing STAT3. Exp. Cell Res. 2019, 382, 111466. [Google Scholar] [CrossRef] [PubMed]
- Scuto, A.; Kirschbaum, M.; Buettner, R.; Kujawski, M.; Cermak, J.M.; Atadja, P.; Jove, R. SIRT1 activation enhances HDAC inhibition-mediated upregulation of GADD45G by repressing the binding of NF- j B/STAT3 complex to its promoter in malignant lymphoid cells. Cell Death Dis. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Hou, X.; Rooklin, D.; Fang, H.; Zhang, Y. Resveratrol serves as a protein- substrate interaction stabilizer in human SIRT1 activation. Nat. Publ. Gr. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, L.; Chen, X.; Lu, Q.; Yang, Y.; Liu, J.; Ma, X. SIRT1 counteracted the activation of STAT3 and NF-κB to repress the gastric cancer growth. Int. J. Clin. Exp. Med. 2014, 7, 5050–5058. [Google Scholar]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Apetoh, L.; Delmas, D.; Deacetylation, S. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef]
- Hee, M.; Nickerson, S.; Kim, E.T.; Liot, C.; Laurent, G.; Spang, R.; Philips, M.R.; Shan, Y.; Shaw, D.E.; Bar-Sagi, D.; et al. Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. USA 2012, 109, 10843–10848. [Google Scholar]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target 2018, 3, 1–7. Available online: https://www.nature.com/articles/s41392-018-0008-7 (accessed on 9 October 2021).
- Yuan, J.; Minter-dykhouse, K.; Lou, Z. A c-Myc–SIRT1 feedback loop regulates cell growth and transformation. J. Cell Biol. 2009, 185, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Zhao, G.; Lv, X.; Chen, H.; Xue, Z.; Yang, B.; Liu, D.P.; Liang, C.C. Sirt1 deacetylates c-Myc and promotes c-Myc/Max association. Int. J. Biochem. Cell Biol. 2011, 43, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Guccione, E.; Tergaonkar, V.; Koh, C.M.; Khattar, E.; Leow, S.C.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig 2015, 125, 2109–2122. [Google Scholar] [CrossRef]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Lüscher, B. The c-MYC oncoprotein, the NAMPT enzyme, the form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, 187–196. [Google Scholar] [CrossRef]
- Jing, H.; Hu, J.; He, B.; Negrón Abril, Y.L.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. Available online: https://pubmed.ncbi.nlm.nih.gov/30031797/ (accessed on 18 March 2021). [CrossRef] [PubMed]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors in cancer therapy. Cancer Biol. Ther. Curr Top Med. Chem. 2003, 2, 30–37. Available online: https://pubmed.ncbi.nlm.nih.gov/30526462/ (accessed on 18 March 2021). [CrossRef]
- Lahusen, T.J.; Kim, S.J.; Miao, K.; Huang, Z.; Xu, X.; Deng, C.X. BRCA1 function in the intra-S checkpoint is activated by acetylation via a pCAF/SIRT1 axis. Oncogene 2018, 37, 2343–2350. [Google Scholar] [CrossRef]
- Choi, Y.H.; Kim, H.; Lee, S.H.; Jin, Y.H.; Lee, K.Y. Src regulates the activity of SIRT2. Biochem. Biophys Res. Commun. 2014, 450, 1120–1125. Available online: https://pubmed.ncbi.nlm.nih.gov/24996174/ (accessed on 15 December 2020). [CrossRef] [PubMed]
- Seo, J.; Guk, G.; Park, S.H.; Jeong, M.H.; Jeong, J.H.; Yoon, H.G.; Choi, K.C. Tyrosine phosphorylation of HDAC3 by Src kinase mediates proliferation of HER2-positive breast cancer cells. J. Cell Physiol. 2019, 234, 6428–6436. Available online: https://pubmed.ncbi.nlm.nih.gov/30317579/ (accessed on 15 December 2020). [CrossRef]
- Yousafzai, N.A.; Zhou, Q.; Xu, W.; Shi, Q.; Xu, J.; Feng, L.; Chen, H.; Shin, V.Y.; Jin, H.; Wang, X. SIRT1 deacetylated and stabilized XRCC1 to promote chemoresistance in lung cancer. Cell Death Dis. 2019, 10, 1–12. Available online: https://pubmed.ncbi.nlm.nih.gov/31043584/ (accessed on 8 August 2020). [CrossRef]
- Dell’Omo, G.; Crescenti, D.; Vantaggiato, C.; Parravicini, C.; Borroni, A.P.; Rizzi, N.; Garofalo, M.; Pinto, A.; Recordati, C.; Scanziani, E.; et al. Inhibition of SIRT1 deacetylase and p53 activation uncouples the anti-inflammatory and chemopreventive actions of NSAIDs. Br. J. Cancer 2019, 120, 537–546. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021. Available online: https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bcp.14889 (accessed on 6 July 2021). [CrossRef]
- Amengual, J.E.; Clark-Garvey, S.; Kalac, M.; Scotto, L.; Marchchi, E.; Neylon, E.; Johannet, P.; Wei, Y.; Zain, J.; O’Connor, O.A. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood 2013, 122, 2104–2113. Available online: https://ashpublications.org/blood/article-pdf/122/12/2104/1369349/2104.pdf (accessed on 8 August 2020). [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. Available online: www.frontiersin.org (accessed on 15 March 2021). [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. P53 at the crossroads between different types of hdac inhibitor-mediated cancer cell death. Int. J. Mol. Sci. 2019, 20, 2415. Available online: https://pubmed.ncbi.nlm.nih.gov/31096697/ (accessed on 15 March 2021). [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halasa, M.; Adamczuk, K.; Adamczuk, G.; Afshan, S.; Stepulak, A.; Cybulski, M.; Wawruszak, A. Deacetylation of Transcription Factors in Carcinogenesis. Int. J. Mol. Sci. 2021, 22, 11810. https://doi.org/10.3390/ijms222111810
Halasa M, Adamczuk K, Adamczuk G, Afshan S, Stepulak A, Cybulski M, Wawruszak A. Deacetylation of Transcription Factors in Carcinogenesis. International Journal of Molecular Sciences. 2021; 22(21):11810. https://doi.org/10.3390/ijms222111810
Chicago/Turabian StyleHalasa, Marta, Kamila Adamczuk, Grzegorz Adamczuk, Syeda Afshan, Andrzej Stepulak, Marek Cybulski, and Anna Wawruszak. 2021. "Deacetylation of Transcription Factors in Carcinogenesis" International Journal of Molecular Sciences 22, no. 21: 11810. https://doi.org/10.3390/ijms222111810
APA StyleHalasa, M., Adamczuk, K., Adamczuk, G., Afshan, S., Stepulak, A., Cybulski, M., & Wawruszak, A. (2021). Deacetylation of Transcription Factors in Carcinogenesis. International Journal of Molecular Sciences, 22(21), 11810. https://doi.org/10.3390/ijms222111810