
Combination of the Glutaminyl Cyclase Inhibitor PQ912 (Varoglutamstat) and the Murine Monoclonal Antibody PBD-C06 (m6) Shows Additive Effects on Brain Aβ Pathology in Transgenic Mice

 , ,
, , 

Abstract
:1. Introduction
2. Results
2.1. Dose Finding Study with m6 Antibody
2.2. Combination Treatment of PQ912 and m6 Antibody
2.2.1. Effect on Animal Health and Drug Levels
2.2.2. Development of Aβ Pathology in 9- to 12.5-Month-Old APPslxhQC Mice
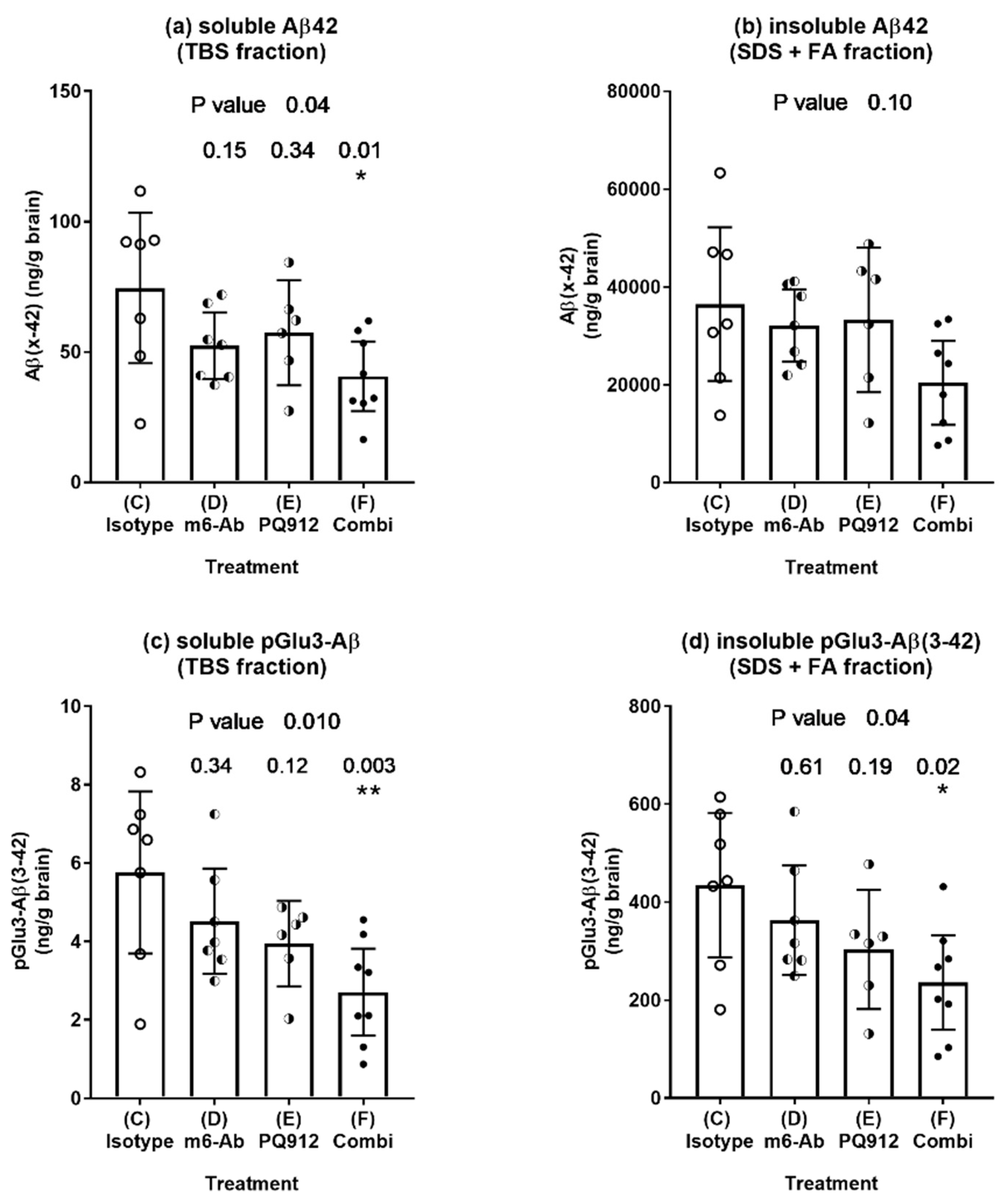
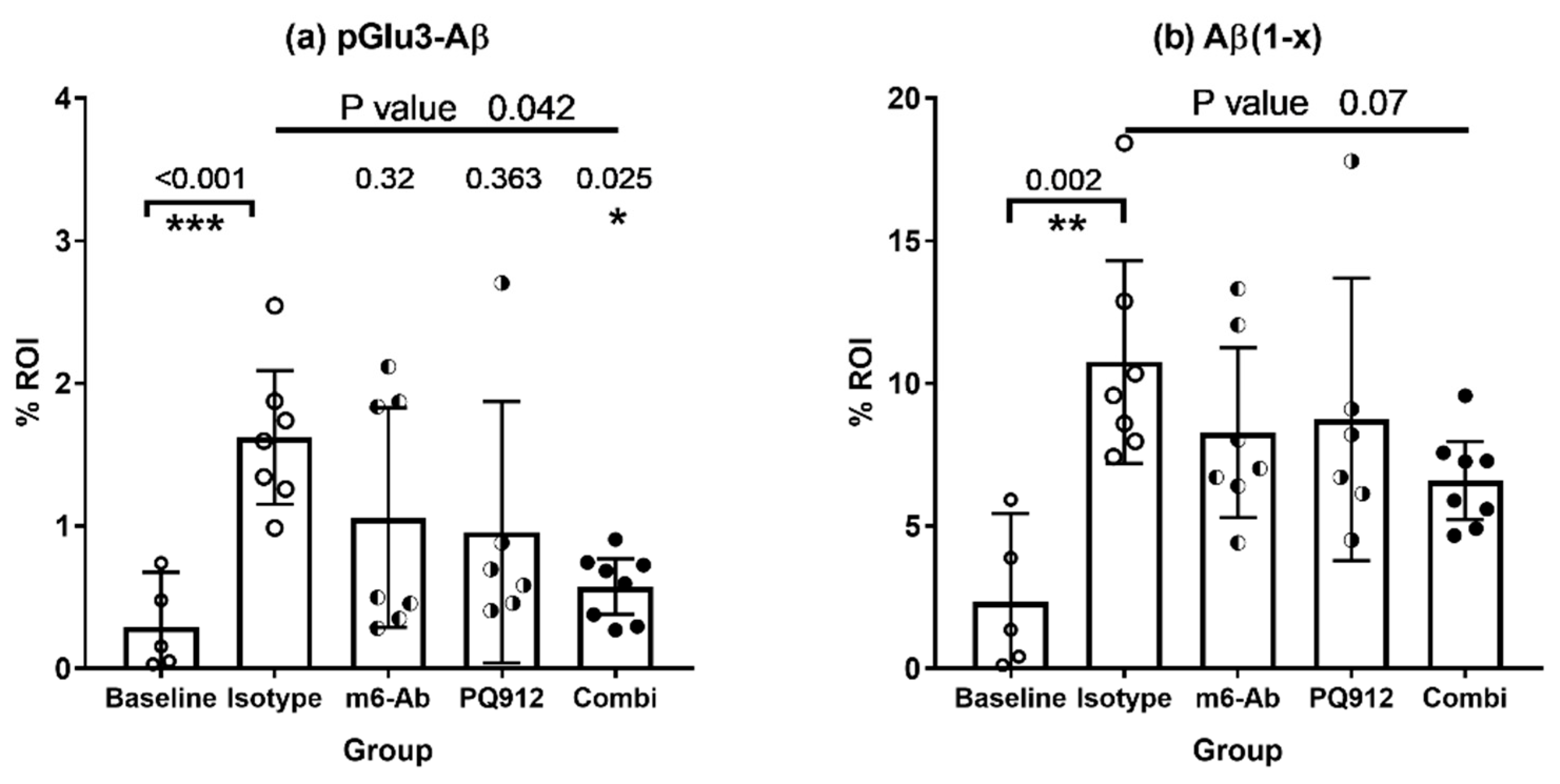
2.2.3. Effect of Single and Combination Treatment on Aβ Deposition
2.3. Evaluation of Combination Effect According to Bliss
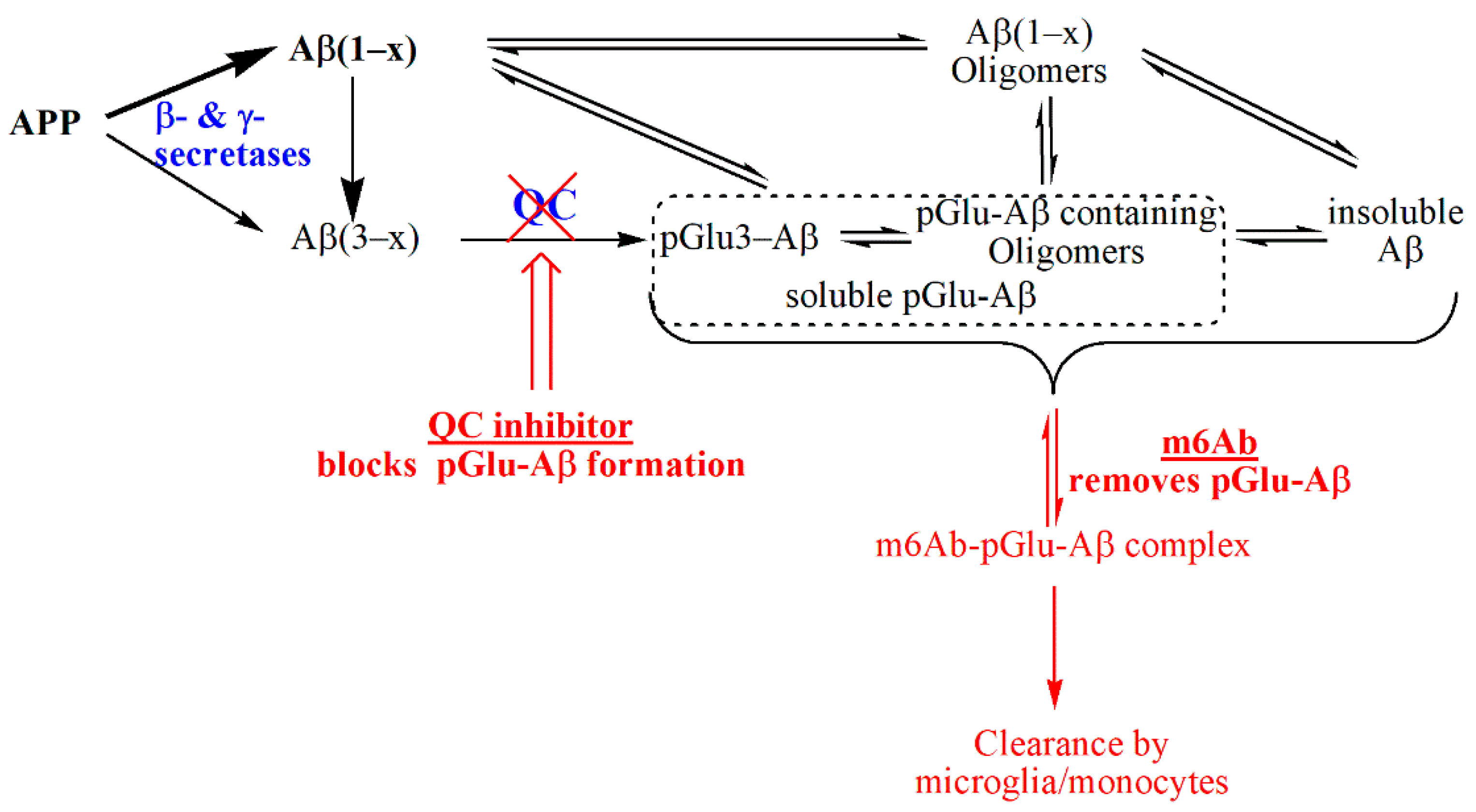
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Experiments and Tissue Collection
4.3. Aβ Extraction and ELISAs
4.4. Immunohistochemistry
4.5. Data Handling, Statistics, and Calculations
Evaluation of Additivity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet. Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Budd, H.S.; O’Gorman, J.; Chiao, P.; Bussiere, T.; von Rosenstiel, P.; Tian, Y.; Zhu, Y.; von Hehn, C.; Gheuens, S.; Skordos, L.; et al. Clinical Development of Aducanumab, an Anti-Abeta Human Monoclonal Antibody Being Investigated for the Treatment of Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2017, 4, 255–263. [Google Scholar]
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimer’s Res. Ther. 2021, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.L.; Willis, B.A.; Hawdon, A.; Natanegara, F.; Chua, L.; Foster, J.; Shcherbinin, S.; Ardayfio, P.; Sims, J.R. Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12112. [Google Scholar] [CrossRef]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Ronicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittnam, J.L.; Portelius, E.; Zetterberg, H.; Gustavsson, M.K.; Schilling, S.; Koch, B.; Demuth, H.U.; Blennow, K.; Wirths, O.; Bayer, T.A. Pyroglutamate amyloid beta (Abeta) aggravates behavioral deficits in transgenic amyloid mouse model for Alzheimer disease. J. Biol. Chem. 2012, 287, 8154–8162. [Google Scholar] [CrossRef] [Green Version]
- Becker, A.; Kohlmann, S.; Alexandru, A.; Jagla, W.; Canneva, F.; Bauscher, C.; Cynis, H.; Sedlmeier, R.; Graubner, S.; Schilling, S.; et al. Glutaminyl cyclase-mediated toxicity of pyroglutamate-beta amyloid induces striatal neurodegeneration. BMC Neurosci. 2013, 14, 108. [Google Scholar] [CrossRef] [Green Version]
- Schlenzig, D.; Manhart, S.; Cinar, Y.; Kleinschmidt, M.; Hause, G.; Willbold, D.; Funke, S.A.; Schilling, S.; Demuth, H.U. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry 2009, 48, 7072–7078. [Google Scholar] [CrossRef] [PubMed]
- Morawski, M.; Schilling, S.; Kreuzberger, M.; Waniek, A.; Jager, C.; Koch, B.; Cynis, H.; Kehlen, A.; Arendt, T.; Hartlage-Rubsamen, M.; et al. Glutaminyl cyclase in human cortex: Correlation with (pGlu)-amyloid-beta load and cognitive decline in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Morawski, M.; Hartlage-Rubsamen, M.; Jager, C.; Waniek, A.; Schilling, S.; Schwab, C.; McGeer, P.L.; Arendt, T.; Demuth, H.U.; Rossner, S. Distinct glutaminyl cyclase expression in Edinger-Westphal nucleus, locus coeruleus and nucleus basalis Meynert contributes to pGlu-Abeta pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 120, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirths, O.; Breyhan, H.; Cynis, H.; Schilling, S.; Demuth, H.U.; Bayer, T.A. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009, 118, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Jawhar, S.; Wirths, O.; Schilling, S.; Graubner, S.; Demuth, H.U.; Bayer, T.A. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J. Biol. Chem. 2011, 286, 4454–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valverde, A.; Dunys, J.; Lorivel, T.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Roques, B.P.; Chami, M.; Checler, F. Aminopeptidase A contributes to biochemical, anatomical and cognitive defects in Alzheimer’s disease (AD) mouse model and is increased at early stage in sporadic AD brain. Acta Neuropathol. 2021, 141, 823–839. [Google Scholar] [CrossRef]
- Antonyan, A.; Schlenzig, D.; Schilling, S.; Naumann, M.; Sharoyan, S.; Mardanyan, S.; Demuth, H.U. Concerted action of dipeptidyl peptidase IV and glutaminyl cyclase results in formation of pyroglutamate-modified amyloid peptides in vitro. Neurochem. Int. 2018, 113, 112–119. [Google Scholar] [CrossRef]
- Schlenzig, D.; Cynis, H.; Hartlage-Rübsamen, M.; Zeitschel, U.; Menge, K.; Fothe, A.; Ramsbeck, D.; Spahn, C.; Wermann, M.; Roßner, S.; et al. Dipeptidyl-Peptidase Activity of Meprin β Links N-truncation of Aβ with Glutaminyl Cyclase-Catalyzed pGlu-Aβ Formation. J. Alzheimers’s Dis. 2018, 66, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Schlenzig, D.; Ronicke, R.; Cynis, H.; Ludwig, H.H.; Scheel, E.; Reymann, K.; Saido, T.; Hause, G.; Schilling, S.; Demuth, H.U. N-Terminal pyroglutamate formation of Abeta38 and Abeta40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J. Neurochem. 2012, 121, 774–784. [Google Scholar] [CrossRef]
- Grochowska, K.M.; Yuanxiang, P.; Bär, J.; Raman, R.; Brugal, G.; Sahu, G.; Schweizer, M.; Bikbaev, A.; Schilling, S.; Demuth, H.; et al. Posttranslational modification impact on the mechanism by which amyloid-β induces synaptic dysfunction. EMBO Rep. 2017, 18, 962–981. [Google Scholar] [CrossRef]
- Russo, C.; Violani, E.; Salis, S.; Venezia, V.; Dolcini, V.; Damonte, G.; Benatti, U.; Arrigo, C.; Patrone, E.; Carlo, P.; et al. Pyroglutamate-modified amyloid beta-peptides--AbetaN3(pE)--strongly affect cultured neuron and astrocyte survival. J. Neurochem. 2002, 82, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- Rijal, U.A.; Kosterin, I.; Kumar, S.; von Arnim, C.A.; Yamaguchi, H.; Fandrich, M.; Walter, J.; Thal, D.R. Biochemical stages of amyloid-beta peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 2014, 137, 887–903. [Google Scholar] [CrossRef]
- Mandler, M.; Walker, L.; Santic, R.; Hanson, P.; Upadhaya, A.R.; Colloby, S.J.; Morris, C.M.; Thal, D.R.; Thomas, A.J.; Schneeberger, A.; et al. Pyroglutamylated amyloid-beta is associated with hyperphosphorylated tau and severity of Alzheimer’s disease. Acta Neuropathol. 2014, 128, 67–79. [Google Scholar] [CrossRef]
- Schilling, S.; Zeitschel, U.; Hoffmann, T.; Heiser, U.; Francke, M.; Kehlen, A.; Holzer, M.; Hutter-Paier, B.; Prokesch, M.; Windisch, M.; et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat. Med. 2008, 14, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.L.; Liu, B.; Kleinschmidt, M.; Schilling, S.; Demuth, H.U.; Lemere, C.A. Passive immunization against pyroglutamate-3 amyloid-beta reduces plaque burden in Alzheimer-like transgenic mice: A pilot study. Neurodegener. Dis. 2012, 10, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, J.L.; Liu, B.; Rahfeld, J.U.; Kleinschmidt, M.; O’Nuallain, B.; Le, K.X.; Lues, I.; Caldarone, B.J.; Schilling, S.; Demuth, H.U.; et al. An anti-pyroglutamate-3 Abeta vaccine reduces plaques and improves cognition in APPswe/PS1DeltaE9 mice. Neurobiol. Aging 2015, 36, 3187–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, T.; Meyer, A.; Heiser, U.; Kurat, S.; Bohme, L.; Kleinschmidt, M.; Buhring, K.U.; Hutter-Paier, B.; Farcher, M.; Demuth, H.U.; et al. Glutaminyl Cyclase Inhibitor PQ912 Improves Cognition in Mouse Models of Alzheimer’s Disease-Studies on Relation to Effective Target Occupancy. J. Pharmacol. Exp. Ther. 2017, 362, 119–130. [Google Scholar] [CrossRef]
- Demattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; Delong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A Plaque-Specific Antibody Clears Existing beta-amyloid Plaques in Alzheimer’s Disease Mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, T.; Zimmer, M.; Mugele, K.; Spiess, J. Primary structure and functional expression of a glutaminyl cyclase. Proc. Natl. Acad. Sci. USA 1991, 88, 10059–10063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockers, T.M.; Kreutz, M.R.; Pohl, T. Glutaminyl-cyclase expression in the bovine/porcine hypothalamus and pituitary. J. Neuroendocr. 1995, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- De Kimpe, L.; Bennis, A.; Zwart, R.; van Haastert, E.S.; Hoozemans, J.J.; Scheper, W. Disturbed Ca2+ homeostasis increases glutaminyl cyclase expression; connecting two early pathogenic events in Alzheimer’s disease in vitro. PLoS ONE 2012, 7, e44674. [Google Scholar] [CrossRef] [Green Version]
- Lues, I.; Weber, F.; Meyer, A.; Bühring, U.; Hoffmann, T.; Kühn-Wache, K.; Manhart, S.; Heiser, U.; Pokorny, R.; Chiesa, J.; et al. A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2015, 1, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Hallikainen, M.; Grimmer, T.; Duning, T.; Gouw, A.A.; Teunissen, C.E.; Wink, A.M.; Maruff, P.; Harrison, J.; Van Baal, C.M.; et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: Results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimer’s Res. Ther. 2018, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Hettmann, T.; Gillies, S.D.; Kleinschmidt, M.; Piechotta, A.; Makioka, K.; Lemere, C.A.; Schilling, S.; Rahfeld, J.U.; Lues, I. Development of the clinical candidate PBD-C06, a humanized pGlu3-Aβ-specific antibody against Alzheimer’s disease with reduced complement activation. Sci. Rep. 2020, 10, 3294. [Google Scholar] [CrossRef] [PubMed]
- Crehan, H.; Liu, B.; Kleinschmidt, M.; Rahfeld, J.U.; Le, K.X.; Caldarone, B.J.; Frost, J.L.; Hettmann, T.; Hutter-Paier, B.; O’Nuallain, B.; et al. Effector function of anti-pyroglutamate-3 Aβ antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimer’s Res. Ther. 2020, 12, 12. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Staufenbiel, M. Pathogenic mechanisms of Alzheimer’s disease analyzed in the APP23 transgenic mouse model. Ann. N. Y. Acad. Sci. 2000, 920, 134–139. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Rahfeld, J.U.; Lues, I.; Lemere, C.A. Passive Aβ immunotherapy: Current achievements and future perspectives. Molecules 2018, 23, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2017. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Piccini, A.; Russo, C.; Gliozzi, A.; Relini, A.; Vitali, A.; Borghi, R.; Giliberto, L.; Armirotti, A.; D’Arrigo, C.; Bachi, A.; et al. {beta}-Amyloid Is Different in Normal Aging and in Alzheimer Disease. J. Biol. Chem. 2005, 280, 34186–34192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Guntert, A.; Dobeli, H.; Bohrmann, B. High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience 2006, 143, 461–475. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, J.A.; Bateman, R.J.; Brashear, H.R.; Duggan, C.; Carrillo, M.C.; Bain, L.J.; DeMattos, R.; Katz, R.G.; Ostrowitzki, S.; Siemers, E.; et al. Challenges, solutions, and recommendations for Alzheimer’s disease combination therapy. Alzheimer’s Dement. 2016, 12, 623–630. [Google Scholar] [CrossRef]
- Chen, R.; Chan, P.T.; Chu, H.; Lin, Y.C.; Chang, P.C.; Chen, C.Y.; Chou, K.R. Treatment effects between monotherapy of donepezil versus combination with memantine for Alzheimer disease: A meta-analysis. PLoS ONE 2017, 12, e0183586. [Google Scholar] [CrossRef]
- Atri, A.; Hendrix, S.B.; Pejović, V.; Hofbauer, R.K.; Edwards, J.; Molinuevo, J.L.; Graham, S.M. Cumulative, additive benefits of memantine-donepezil combination over component monotherapies in moderate to severe Alzheimer’s dementia: A pooled area under the curve analysis. Alzheimer’s Res. Ther. 2015, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.-Q. Comparison of the efficacy of four cholinesterase inhibitors in combination with memantine for the treatment of Alzheimer’s disease. Int. J. Clin. Exp. Med. 2015, 8, 2944–2948. [Google Scholar]
- Riverol, M.; Slachevsky, A.; López, O.L. Efficacy and Tolerability of a Combination Treatment of Memantine and Donepezil for Alzheimer’s Disease: A Literature Review Evidence. Eur. Neurol. J. 2014, 23, 15–19. [Google Scholar]
- Strömberg, K.; Eketjäll, S.; Georgievska, B.; Tunblad, K.; Eliason, K.; Olsson, F.; Radesäter, A.C.; Klintenberg, R.; Arvidsson, P.I.; Von Berg, S.; et al. Combining an amyloid-beta (Aβ) cleaving enzyme inhibitor with a γ-secretase modulator results in an additive reduction of Aβ production. FEBS J. 2015, 282, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]
- Loewe, S.; Muischnek, H. Über Kombinationswirkungen. Naunyn. Schmiedebergs. Arch. Exp. Pathol. Pharmakol. 1926, 114, 313–326. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Tallarida, R.J. An overview of drug combination analysis with isobolograms. J. Pharmacol. Exp. Ther. 2006, 319, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BLISS, C.I. THE TOXICITY OF POISONS APPLIED JOINTLY. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Cynis, H.; Hoffmann, T.; Friedrich, D.; Kehlen, A.; Gans, K.; Kleinschmidt, M.; Rahfeld, J.U.; Wolf, R.; Wermann, M.; Stephan, A.; et al. The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. EMBO Mol. Med. 2011, 3, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Cynis, H.; Kehlen, A.; Haegele, M.; Hoffmann, T.; Heiser, U.; Fujii, M.; Shibazaki, Y.; Yoneyama, H.; Schilling, S.; Demuth, H.U. Inhibition of Glutaminyl Cyclases alleviates CCL2-mediated inflammation of non-alcoholic fatty liver disease in mice. Int. J. Exp. Pathol. 2013, 94, 217–225. [Google Scholar] [CrossRef]
- Piechotta, A.; Parthier, C.; Kleinschmidt, M.; Gnoth, K.; Pillot, T.; Lues, I.; Demuth, H.U.; Schilling, S.; Rahfeld, J.U.; Stubbs, M.T. Structural and functional analyses of pyroglutamate-amyloid-β-specific antibodies as a basis for Alzheimer immunotherapy. J. Biol. Chem. 2017, 292, 12713–12724. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| m6-Ab (ED) | PQ912 (EE) | Combi (EF) | Bliss Additivity (ED + EE − ED ∗ EE) | CIBliss (ED + EE − ED ∗ EE)/EF | Rating of CI According to Chou [37] | |
|---|---|---|---|---|---|---|
| Aβ biochemistry | ||||||
| soluble pE-Aβ42 | 21.6 | 31.5 | 53.0 | 46.3 | 0.87 | slight synergism |
| insoluble pE-Aβ42 | 16.4 | 30.2 | 45.7 | 41.6 | 0.91 | nearly additive |
| soluble Aβ42 | 29.8 | 23.1 | 45.4 | 46.0 | 0.99 | (nearly) additive |
| Aβ Histochemistry | ||||||
| pE3Aβ (K17) | 34.6 | 41.0 | 64.5 | 61.4 | 0.95 | nearly additive |
| Group | N | Treatment | Time of Tissue Sampling | |
|---|---|---|---|---|
| Chow | Injection (i.p.) | |||
| A | 10 | - | - | baseline |
| B | 10 | vehicle | PBS | at the end of treatment |
| C | 15 | vehicle | IgG2a isotype control (150 µg/week) | at the end of treatment |
| D | 15 | vehicle | m6 (150 µg/week) | at the end of treatment |
| E | 15 | PQ912 (0.8 g/kg pellets) | IgG2a isotype control (150 µg/week) | at the end of treatment |
| F | 15 + 1 | PQ912 (0.8 g/kg pellets) | m6 (150 µg/week) | at the end of treatment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, T.; Rahfeld, J.-U.; Schenk, M.; Ponath, F.; Makioka, K.; Hutter-Paier, B.; Lues, I.; Lemere, C.A.; Schilling, S. Combination of the Glutaminyl Cyclase Inhibitor PQ912 (Varoglutamstat) and the Murine Monoclonal Antibody PBD-C06 (m6) Shows Additive Effects on Brain Aβ Pathology in Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 11791. https://doi.org/10.3390/ijms222111791
Hoffmann T, Rahfeld J-U, Schenk M, Ponath F, Makioka K, Hutter-Paier B, Lues I, Lemere CA, Schilling S. Combination of the Glutaminyl Cyclase Inhibitor PQ912 (Varoglutamstat) and the Murine Monoclonal Antibody PBD-C06 (m6) Shows Additive Effects on Brain Aβ Pathology in Transgenic Mice. International Journal of Molecular Sciences. 2021; 22(21):11791. https://doi.org/10.3390/ijms222111791
Chicago/Turabian StyleHoffmann, Torsten, Jens-Ulrich Rahfeld, Mathias Schenk, Falk Ponath, Koki Makioka, Birgit Hutter-Paier, Inge Lues, Cynthia A. Lemere, and Stephan Schilling. 2021. "Combination of the Glutaminyl Cyclase Inhibitor PQ912 (Varoglutamstat) and the Murine Monoclonal Antibody PBD-C06 (m6) Shows Additive Effects on Brain Aβ Pathology in Transgenic Mice" International Journal of Molecular Sciences 22, no. 21: 11791. https://doi.org/10.3390/ijms222111791
APA StyleHoffmann, T., Rahfeld, J.-U., Schenk, M., Ponath, F., Makioka, K., Hutter-Paier, B., Lues, I., Lemere, C. A., & Schilling, S. (2021). Combination of the Glutaminyl Cyclase Inhibitor PQ912 (Varoglutamstat) and the Murine Monoclonal Antibody PBD-C06 (m6) Shows Additive Effects on Brain Aβ Pathology in Transgenic Mice. International Journal of Molecular Sciences, 22(21), 11791. https://doi.org/10.3390/ijms222111791