
Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Amiodarone Toxicity

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
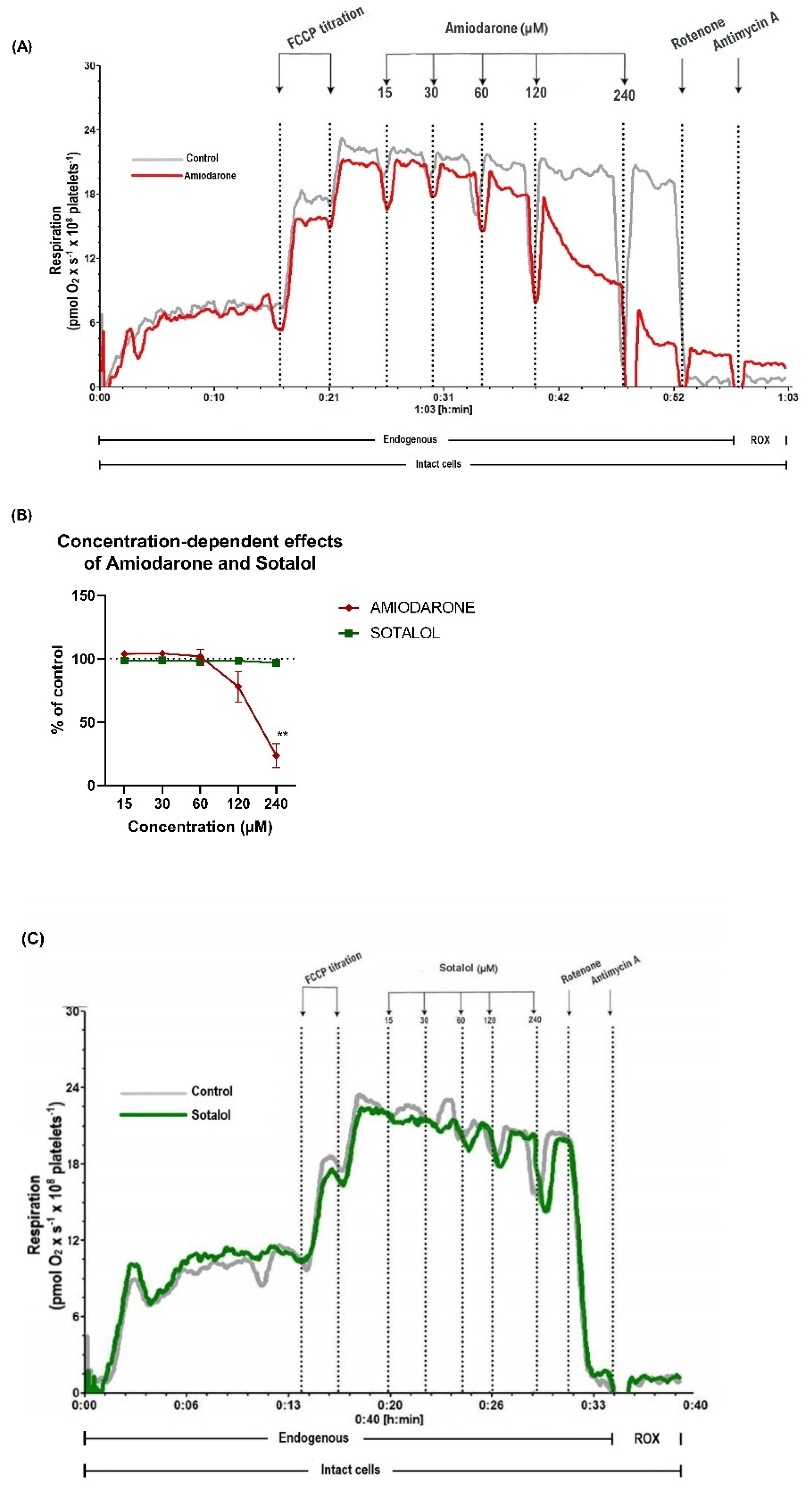
2.1. Concentration-Dependent Decrease of Mitochondrial Respiration by Amiodarone, but Not Sotalol in Intact Human Platelets
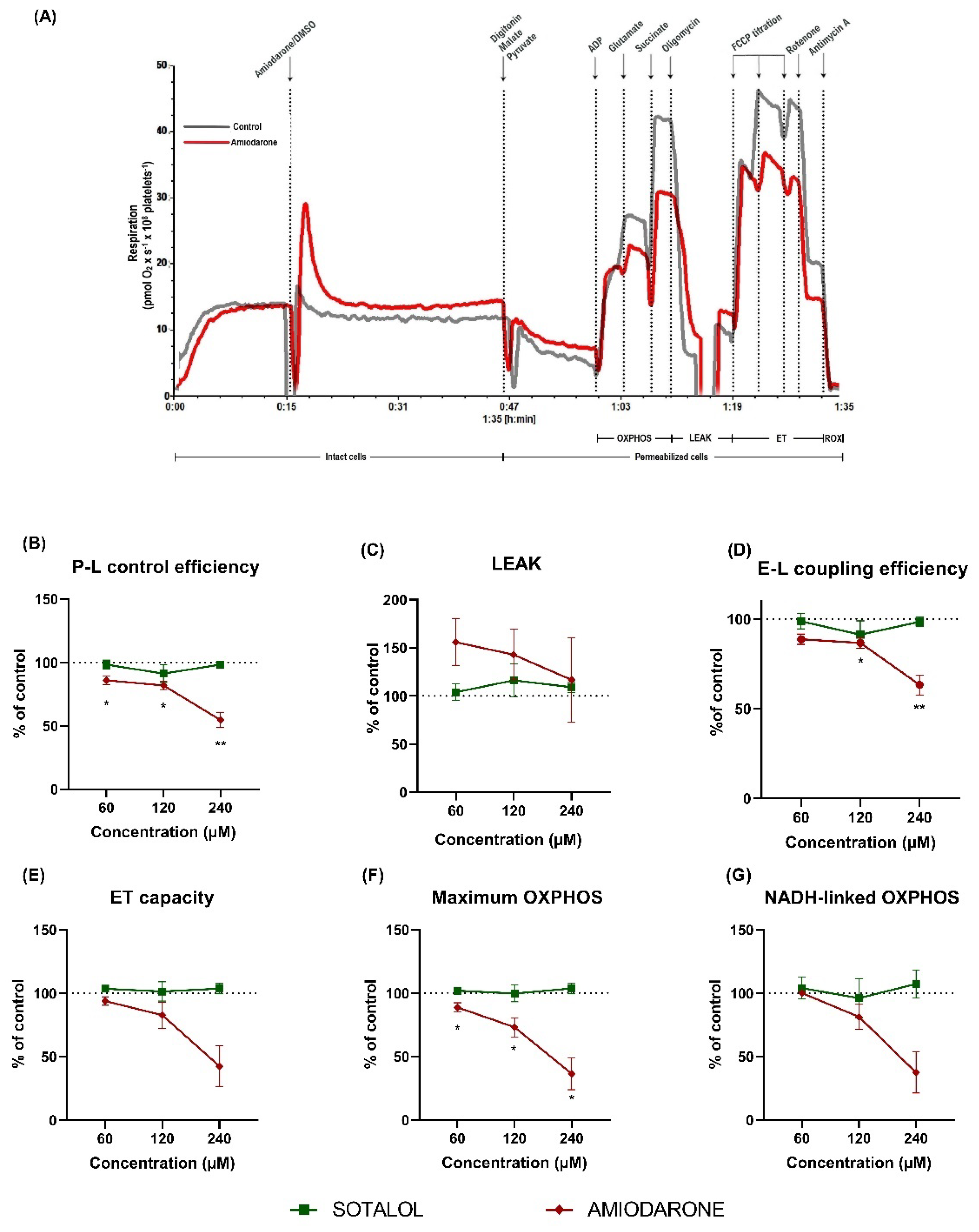
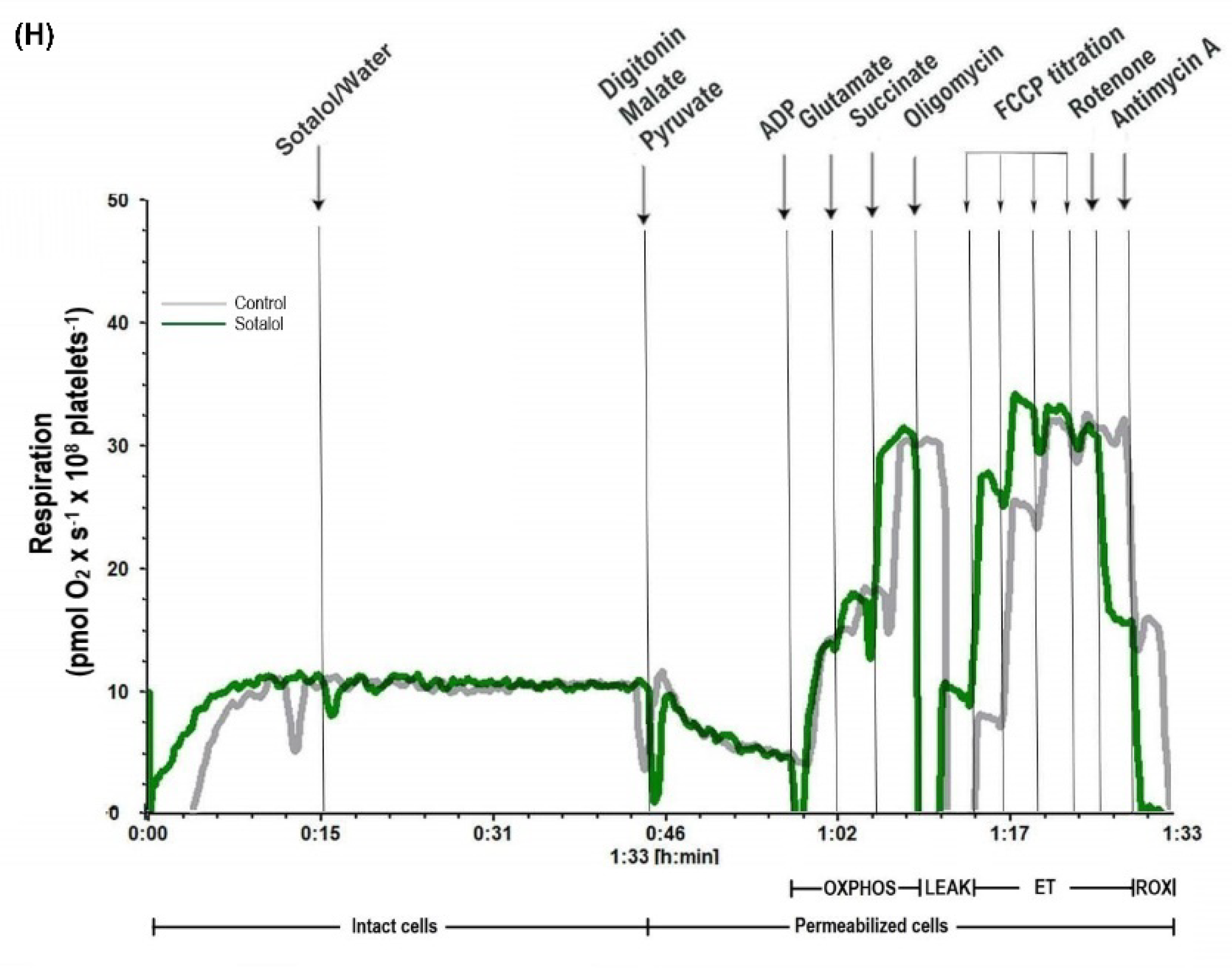
2.2. Amiodarone, but Not Sotalol, Elicited a Concentration-Dependent Decrease of Mitochondrial Respiration in Permeabilized Human Platelets
2.3. Characterization of DEA-Induced Mitochondrial Dysfunction in Permeabilized Human Platelets
2.4. Amiodarone Elicited a Time- and Concentration-Dependent Decrease in Mitochondrial Respiration Combined with ATP Depletion in Intact PBMCs
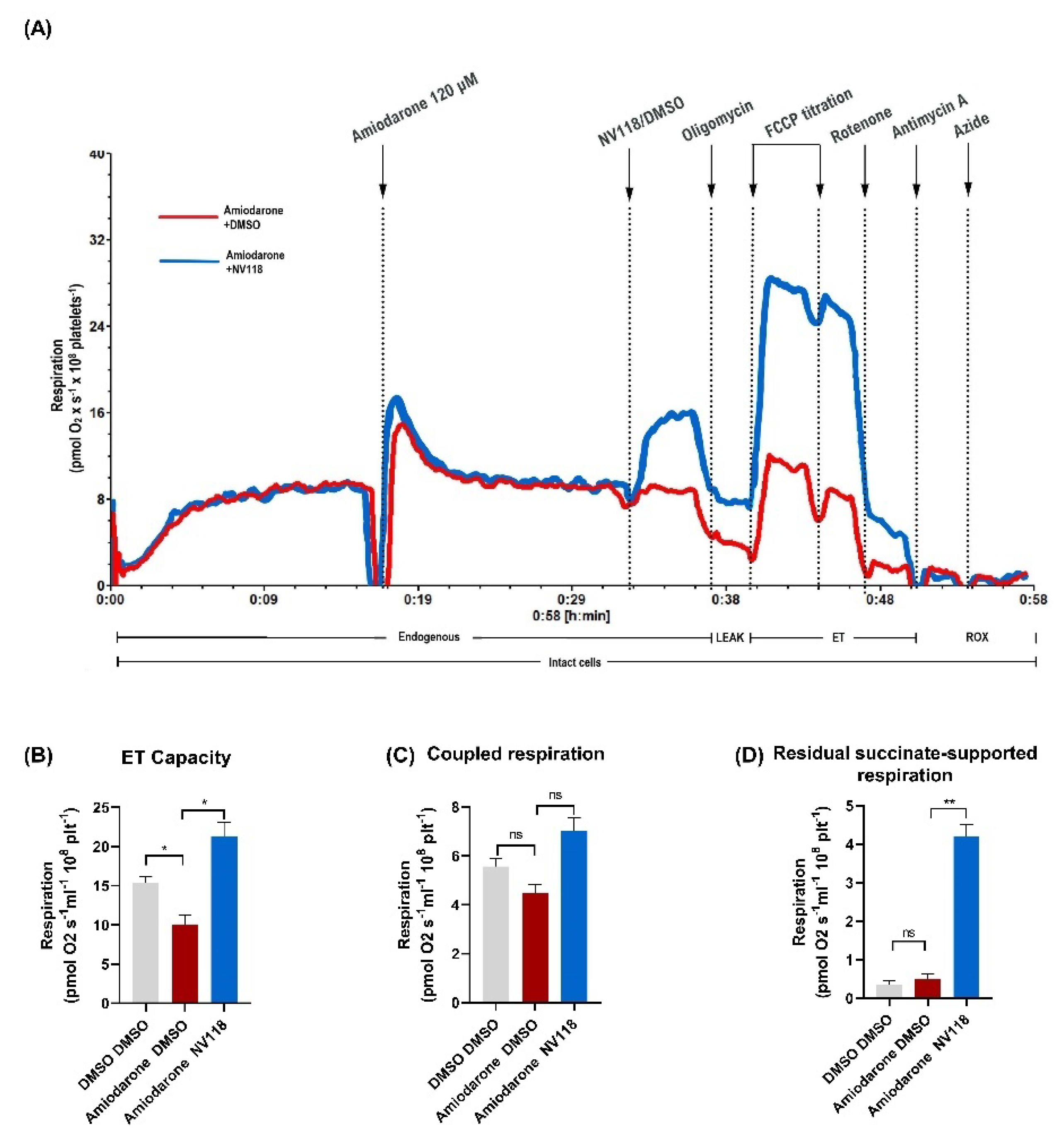
2.4.1. Treatment Effect of a Cell-Permeable Succinate Prodrug on Amiodarone-Induced Mitochondrial Dysfunction in Human Platelets
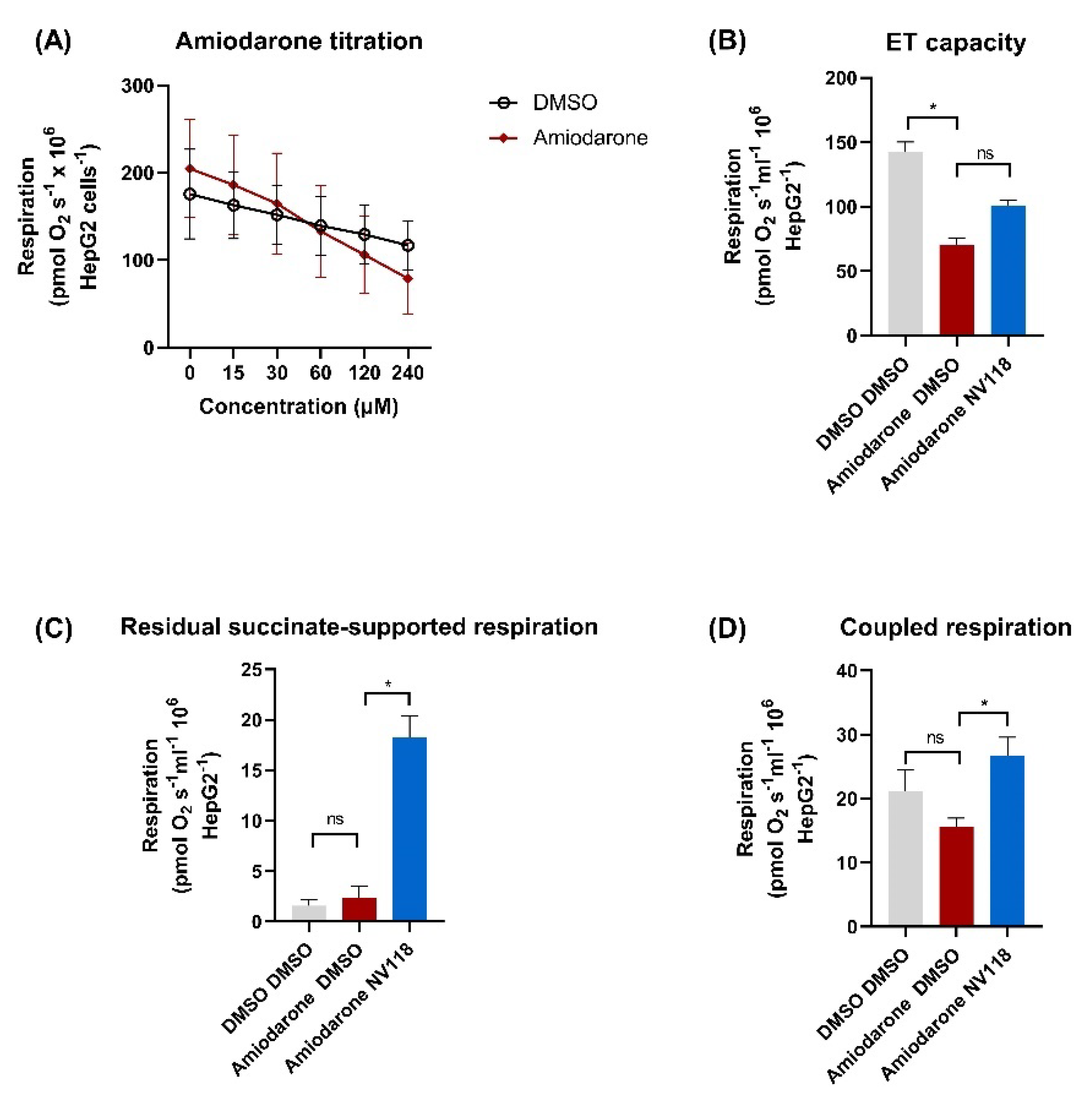
2.4.2. Cell-Permeable Succinate-Alleviated Mitochondrial Dysfunction Induced by Amiodarone in HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Human Samples
4.2. Cell Lines and Cell Culture
4.3. High-Resolution Respirometry (HRR)
- -
- ROUTINE respiration: mitochondrial oxygen consumption in the physiological coupling state;
- -
- LEAK respiration (non-phosphorylating respiration): mitochondrial oxygen consumption after inhibition of ATP-synthase;
- -
- ET capacity: mitochondrial oxygen consumption in a fully uncoupled state achieved by the titration of optimum concentration of FCCP (protonophore);
- -
- NADH-linked OXPHOS capacity: mitochondrial oxygen consumption at saturating concentrations of ADP and complex I substrates;
- -
- OXPHOS capacity (phosphorylating respiration): mitochondrial oxygen consumption at saturating concentrations of ADP with both complex I and complex II substrates;
- -
- Residual succinate-supported respiration: mitochondrial oxygen consumption after inhibition of complex I using rotenone;
- -
- P-L control efficiency: calculated by subtracting LEAK respiration from OXPHOS capacity and then dividing the result by the OXPHOS capacity, it is a measure of the state of coupling (ATP generation) of the ETS;
- -
- E-L coupling efficiency: calculated by subtracting LEAK respiration from ET capacity and then dividing the result by the ET capacity, as a measure of the degree of coupling (ATP generation).
4.4. ATP Quantification
4.5. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| Cyt. c | Cytochrome c |
| CI | Complex I |
| CII | Complex II |
| DEA | Desethylamiodarone |
| DMSO | Dimethyl sulfoxide |
| ET | Electron transport |
| ETS | Electron transport system |
| FCCP | Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone |
| LEAK | Non-phosphorylating respiration |
| NADH | Nicotinamide adenine dinucleotide reduced form |
| OXPHOS | Oxidative phosphorylation |
| PBMCs | Peripheral blood mononuclear cells |
| ROS | Reactive oxygen species |
| ROX | Residual oxygen consumption |
References
- Fu, D.G. Cardiac Arrhythmias: Diagnosis, Symptoms, and Treatments. Cell Biochem. Biophys. 2015, 73, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Soar, J.; Perkins, G.D.; Maconochie, I.; Böttiger, B.W.; Deakin, C.D.; Sandroni, C.; Olasveengen, T.M.; Wyllie, J.; Greif, R.; Lockey, A.; et al. European Resuscitation Council Guidelines for Resuscitation: 2018 Update—Antiarrhythmic drugs for cardiac arrest. Resuscitation 2019, 134, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2020, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Pallandi, R.T.; Campbell, T.J. Resting, and rate-dependent depression of Vmax of guinea-pig ventricular action potentials by amiodarone and desethylamiodarone. Br. J. Pharmacol. 1987, 92, 97–103. [Google Scholar] [CrossRef]
- Wu, Q.; Ning, B.; Xuan, J.; Ren, Z.; Guo, L.; Bryant, M.S. The role of CYP 3A4 and 1A1 in amiodarone-induced hepatocellular toxicity. Toxicol. Lett. 2016, 253, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.C.; Holt, D.W.; Storey, G.C.; Morley, A.R.; Callaghan, J.; Campbell, R.W. Amiodarone and its desethyl metabolite: Tissue distribution and morphologic changes during long-term therapy. Circulation 1985, 72, 1064–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brien, J.F.; Jimmo, S.; Brennan, F.J.; Ford, S.E.; Armstrong, P.W. Distribution of amiodarone and its metabolite, desethylamiodarone, in human tissues. Can. J. Physiol. Pharmacol. 1987, 65, 360–364. [Google Scholar] [CrossRef]
- Kannan, R.; Sarma, J.S.; Guha, M.; Venkataraman, K. Tissue drug accumulation and ultrastructural changes during amiodarone administration in rats. Fundam. Appl. Toxicol. Off. J. Soc. Toxicol. 1989, 13, 793–803. [Google Scholar] [CrossRef]
- Basaria, S.; Cooper, D.S. Amiodarone and the thyroid. Am. J. Med. 2005, 118, 706–714. [Google Scholar] [CrossRef]
- Colby, R.; Geyer, H. Amiodarone-induced pulmonary toxicity. JAAPA 2017, 30, 23–26. [Google Scholar] [CrossRef]
- Epstein, A.E.; Olshansky, B.; Naccarelli, G.V.; Kennedy, J.I., Jr.; Murphy, E.J.; Goldschlager, N. Practical Management Guide for Clinicians Who Treat Patients with Amiodarone. Am. J. Med. 2016, 129, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Visschers, R.G.J.; Duan, L.; Akakpo, J.Y.; Jaeschke, H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: Current understanding and future perspectives. J. Clin. Transl. Res. 2018, 4, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Silva Santos, L.F.; Stolfo, A.; Calloni, C.; Salvador, M. Catechin and epicatechin reduce mitochondrial dysfunction and oxidative stress induced by amiodarone in human lung fibroblasts. J. Arrhythmia 2017, 33, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Bolt, M.W.; Card, J.W.; Racz, W.J.; Brien, J.F.; Massey, T.E. Disruption of mitochondrial function and cellular ATP levels by amiodarone and N-desethylamiodarone in initiation of amiodarone-induced pulmonary cytotoxicity. J. Pharmacol. Exp. Ther. 2001, 298, 1280–1289. [Google Scholar] [PubMed]
- Felser, A.; Blum, K.; Lindinger, P.W.; Bouitbir, J.; Krähenbühl, S. Mechanisms of hepatocellular toxicity associated with dronedarone—A comparison to amiodarone. Toxicol. Sci. 2013, 131, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Fromenty, B.; Fisch, C.; Berson, A.; Letteron, P.; Larrey, D.; Pessayre, D. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J. Pharmacol. Exp. Ther. 1990, 255, 1377–1384. [Google Scholar]
- Varbiro, G.; Toth, A.; Tapodi, A.; Veres, B.; Sumegi, B.; Gallyas, F., Jr. Concentration dependent mitochondrial effect of amiodarone. Biochem. Pharm. 2003, 65, 1115–1128. [Google Scholar] [CrossRef]
- Ehinger, J.K.; Piel, S.; Ford, R.; Karlsson, M.; Sjövall, F.; Frostner, E.; Morota, S.; Taylor, R.W.; Turnbull, D.M.; Cornell, C.; et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun. 2016, 7, 12317. [Google Scholar] [CrossRef] [Green Version]
- Piel, S.; Ehinger, J.K.; Chamkha, I.; Frostner, E.; Sjövall, F.; Elmér, E.; Hansson, M.J. Bioenergetic bypass using cell-permeable succinate, but not methylene blue, attenuates metformin-induced lactate production. Intensive Care Med. Exp. 2018, 6, 22. [Google Scholar] [CrossRef]
- Avram, V.F.; Chamkha, I.; Åsander-Frostner, E.; Ehinger, J.K.; Timar, R.Z.; Hansson, M.J.; Muntean, D.M.; Elmér, E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Statin Toxicity. Int. J. Mol. Sci. 2021, 22, 424. [Google Scholar] [CrossRef]
- Owiredu, S.; Ranganathan, A.; Eckmann, D.M.; Shofer, F.S.; Hardy, K.; Lambert, D.S.; Kelly, M.; Jang, D.H. Ex vivo use of cell-permeable succinate prodrug attenuates mitochondrial dysfunction in blood cells obtained from carbon monoxide-poisoned individuals. Am. J. Physiol. Cell Physiol. 2020, 319, C129–C135. [Google Scholar] [CrossRef]
- Kerin, N.Z. Intravenous Sotalol: An Under Used Treatment Strategy. Cardiology 2018, 140, 143–145. [Google Scholar] [CrossRef]
- Petrus, A.T.; Lighezan, D.L.; Danila, M.D.; Duicu, O.M.; Sturza, A.; Muntean, D.M.; Ionita, I. Assessment of platelet respiration as emerging biomarker of disease. Physiol. Res. 2019, 68, 347–363. [Google Scholar] [CrossRef]
- Sjövall, F.; Ehinger, J.K.; Marelsson, S.E.; Morota, S.; Frostner, E.A.; Uchino, H.; Lundgren, J.; Arnbjörnsson, E.; Hansson, M.J.; Fellman, V.; et al. Mitochondrial respiration in human viable platelets--methodology and influence of gender, age and storage. Mitochondrion 2013, 13, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control an Introduction to OXPHOS Analysis. Bioenerg. Commun. 2020, 2020, 2. [Google Scholar] [CrossRef]
- Wallace, K.B. Multiple Targets for Drug-Induced Mitochondrial Toxicity. Curr. Med. Chem. 2015, 22, 2488–2492. [Google Scholar] [CrossRef]
- Fromenty, B.; Fisch, C.; Labbe, G.; Degott, C.; Deschamps, D.; Berson, A.; Letteron, P.; Pessayre, D. Amiodarone inhibits the mitochondrial beta-oxidation of fatty acids and produces microvesicular steatosis of the liver in mice. J. Pharmacol. Exp. Ther. 1990, 255, 1371–1376. [Google Scholar]
- Kaufmann, P.; Török, M.; Hänni, A.; Roberts, P.; Gasser, R.; Krähenbühl, S. Mechanisms of benzarone and benzbromarone-induced hepatic toxicity. Hepatology 2005, 41, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Spaniol, M.; Bracher, R.; Ha, H.R.; Follath, F.; Krähenbühl, S. Toxicity of amiodarone and amiodarone analogues on isolated rat liver mitochondria. J. Hepatol. 2001, 35, 628–636. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Giudetti, A.M.; Gnoni, G.V.; Capitanio, N.; Tamborra, R.; Romano, A.D.; Quinto, M.; Blonda, M.; Vendemiale, G.; et al. Mitochondrial oxidative stress and respiratory chain dysfunction account for liver toxicity during amiodarone but not dronedarone administration. Free Radic. Biol. Med. 2011, 51, 2234–2242. [Google Scholar] [CrossRef]
- Kushnareva, Y.; Newmeyer, D.D. Bioenergetics and cell death. Ann. N. Y. Acad. Sci. 2010, 1201, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Waldhauser, K.M.; Török, M.; Ha, H.R.; Thomet, U.; Konrad, D.; Brecht, K.; Follath, F.; Krähenbühl, S. Hepatocellular toxicity and pharmacological effect of amiodarone and amiodarone derivatives. J. Pharmacol. Exp. Ther. 2006, 319, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Stepanova, A.; Shurubor, Y.; Valsecchi, F.; Manfredi, G.; Galkin, A. Differential susceptibility of mitochondrial complex II to inhibition by oxaloacetate in brain and heart. Biochim. Biophys. Acta 2016, 1857, 1561–1568. [Google Scholar] [CrossRef] [Green Version]
- Risiglione, P.; Leggio, L.; Cubisino, S.A.M.; Reina, S.; Paternò, G.; Marchetti, B.; Magrì, A.; Iraci, N.; Messina, A. High-Resolution Respirometry Reveals MPP(+) Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7809. [Google Scholar] [CrossRef]
- Karkhanis, A.; Leow, J.W.H.; Hagen, T.; Chan, E.C.Y. Dronedarone-Induced Cardiac Mitochondrial Dysfunction and Its Mitigation by Epoxyeicosatrienoic Acids. Toxicol. Sci. 2018, 163, 79–91. [Google Scholar] [CrossRef]
- Takai, S.; Oda, S.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Establishment of a mouse model for amiodarone-induced liver injury and analyses of its hepatotoxic mechanism. J. Appl. Toxicol. JAT 2016, 36, 35–47. [Google Scholar] [CrossRef]
- Guo, L.; Rondina, M.T. The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front. Immunol. 2019, 10, 2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjövall, F.; Morota, S.; Persson, J.; Hansson, M.J.; Elmér, E. Patients with sepsis exhibit increased mitochondrial respiratory capacity in peripheral blood immune cells. Crit Care 2013, 17, R152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromenty, B.; Letteron, P.; Fisch, C.; Berson, A.; Deschamps, D.; Pessayre, D. Evaluation of human blood lymphocytes as a model to study the effects of drugs on human mitochondria. Effects of low concentrations of amiodarone on fatty acid oxidation, ATP levels and cell survival. Biochem. Pharm. 1993, 46, 421–432. [Google Scholar] [CrossRef]
- Lafuente-Lafuente, C.; Alvarez, J.C.; Leenhardt, A.; Mouly, S.; Extramiana, F.; Caulin, C.; Funck-Brentano, C.; Bergmann, J.F. Amiodarone concentrations in plasma and fat tissue during chronic treatment and related toxicity. Br. J. Clin. Pharmacol. 2009, 67, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahno, A.; Brecht, K.; Morand, R.; Maseneni, S.; Török, M.; Lindinger, P.W.; Krähenbühl, S. The role of CYP3A4 in amiodarone-associated toxicity on HepG2 cells. Biochem. Pharm. 2011, 81, 432–441. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Ogu, C.C.; Maxa, J.L. Drug interactions due to cytochrome P450. In Baylor University Medical Center Proceedings; Taylor & Francis: Dallas, TX, USA, 2000; Volume 13, pp. 421–423. [Google Scholar] [CrossRef]
- Novotna, A.; Dvorak, Z. Omeprazole and lansoprazole enantiomers induce CYP3A4 in human hepatocytes and cell lines via glucocorticoid receptor and pregnane X receptor axis. PLoS ONE 2014, 9, e105580. [Google Scholar] [CrossRef]
- Pascussi, J.-M.; Drocourt, L.; Gerbal-Chaloin, S.; Fabre, J.-M.; Maurel, P.; Vilarem, M.-J. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Eur. J. Biochem. 2001, 268, 6346–6358. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.-F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid Redox Signal 2012, 16, 1492–1526. [Google Scholar] [CrossRef] [Green Version]
- Muscari, C.; Caldarera, C.M.; Guarnieri, C. Age-dependent production of mitochondrial hydrogen peroxide, lipid peroxides and fluorescent pigments in the rat heart. Basic Res. Cardiol. 1990, 85, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, K.; Fazmin, I.T. Mitochondrial Dysfunction Increases Arrhythmic Triggers and Substrates; Potential Anti-arrhythmic Pharmacological Targets. Front. Cardiovasc. Med. 2021, 8, 646932. [Google Scholar] [CrossRef]
- Kashani, A.; Phillips, C.O.; Foody, J.M.; Wang, Y.; Mangalmurti, S.; Ko, D.T.; Krumholz, H.M. Risks Associated With Statin Therapy. Circulation 2006, 114, 2788–2797. [Google Scholar] [CrossRef] [Green Version]
- Hirota, T.; Ieiri, I. Drug-drug interactions that interfere with statin metabolism. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1435–1447. [Google Scholar] [CrossRef]
- Bouitbir, J.; Sanvee, G.M.; Panajatovic, M.V.; Singh, F.; Krahenbuhl, S. Mechanisms of statin-associated skeletal muscle-associated symptoms. Pharmacol. Res. 2019, 104201. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durhuus, J.A.; Hansson, S.; Morville, T.; Kuhlman, A.B.; Dohlmann, T.L.; Larsen, S.; Helge, J.W.; Angleys, M.; Muniesa-Vargas, A.; Bundgaard, J.R.; et al. Simvastatin improves mitochondrial respiration in peripheral blood cells. Sci. Rep. 2020, 10, 17012. [Google Scholar] [CrossRef]
- Piel, S.; Chamkha, I.; Dehlin, A.K.; Ehinger, J.K.; Sjövall, F.; Elmér, E.; Hansson, M.J. Cell-permeable succinate prodrugs rescue mitochondrial respiration in cellular models of acute acetaminophen overdose. PLoS ONE 2020, 15, e0231173. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E.; Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Steurer, W.; Margreiter, R. Mitochondria in the Cold. Proceedings of Life in the Cold, Berlin, Heidelberg, 13–18 August 2000; pp. 431–442. [Google Scholar]
- Avram, V.F.; Bîna, A.M.; Sima, A.; Aburel, O.M.; Sturza, A.; Burlacu, O.; Timar, R.Z.; Muntean, D.M.; Elmér, E.; Crețu, O.M. Improvement of Platelet Respiration by Cell-Permeable Succinate in Diabetic Patients Treated with Statins. Life 2021, 11, 288. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bețiu, A.M.; Chamkha, I.; Gustafsson, E.; Meijer, E.; Avram, V.F.; Åsander Frostner, E.; Ehinger, J.K.; Petrescu, L.; Muntean, D.M.; Elmér, E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Amiodarone Toxicity. Int. J. Mol. Sci. 2021, 22, 11786. https://doi.org/10.3390/ijms222111786
Bețiu AM, Chamkha I, Gustafsson E, Meijer E, Avram VF, Åsander Frostner E, Ehinger JK, Petrescu L, Muntean DM, Elmér E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Amiodarone Toxicity. International Journal of Molecular Sciences. 2021; 22(21):11786. https://doi.org/10.3390/ijms222111786
Chicago/Turabian StyleBețiu, Alina M., Imen Chamkha, Ellen Gustafsson, Elna Meijer, Vlad F. Avram, Eleonor Åsander Frostner, Johannes K. Ehinger, Lucian Petrescu, Danina M. Muntean, and Eskil Elmér. 2021. "Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Amiodarone Toxicity" International Journal of Molecular Sciences 22, no. 21: 11786. https://doi.org/10.3390/ijms222111786
APA StyleBețiu, A. M., Chamkha, I., Gustafsson, E., Meijer, E., Avram, V. F., Åsander Frostner, E., Ehinger, J. K., Petrescu, L., Muntean, D. M., & Elmér, E. (2021). Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Amiodarone Toxicity. International Journal of Molecular Sciences, 22(21), 11786. https://doi.org/10.3390/ijms222111786