
Autophagy-Related Chemoprotection against Sorafenib in Human Hepatocarcinoma: Role of FOXO3 Upregulation and Modulation by Regorafenib

 ,
,  ,
,  ,
,  , , , and
, , , and 
Abstract
:
1. Introduction
2. Results
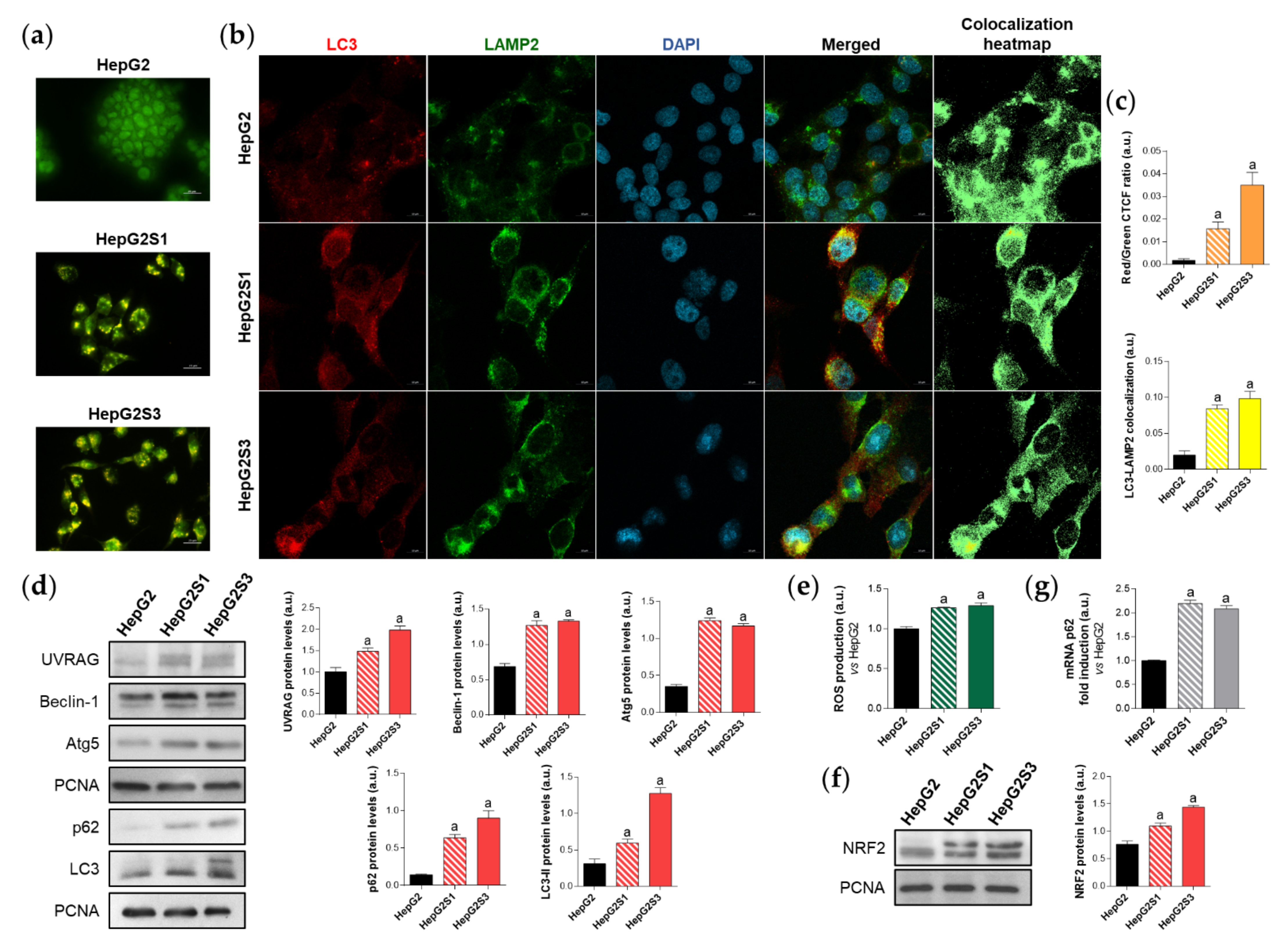
2.1. Sorafenib-Resistant HCC In Vitro Models Display an Enhanced Basal Autophagic Flux
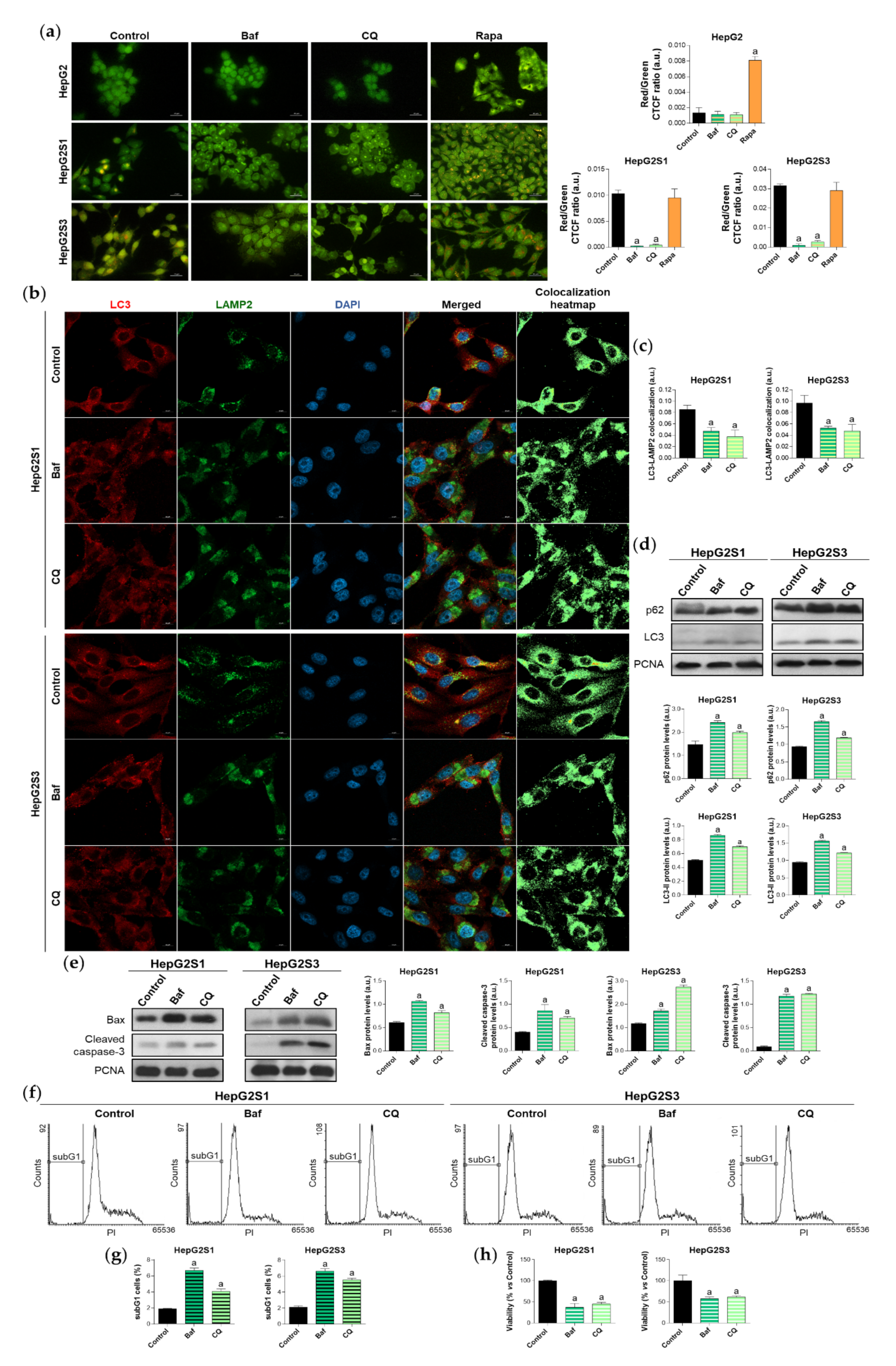
2.2. Inhibition of Autophagy Leads to Sorafenib-Resistant Cell Death
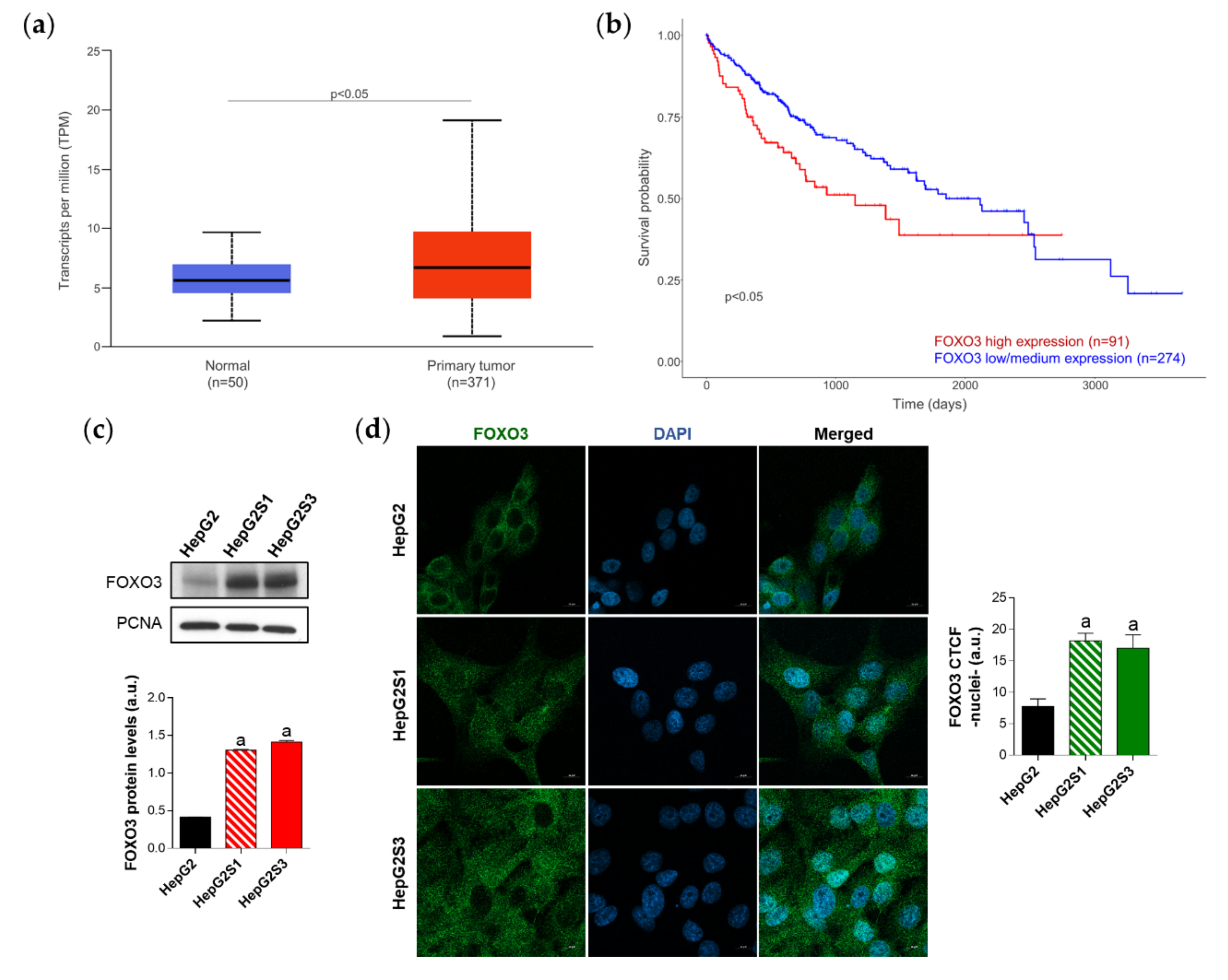
2.3. FOXO3 Is Upregulated in HCC Samples and Sorafenib-Resistant Lines, Being also Correlated with Autophagy-Related Genes Expression and Linked to Poor Patient Survival
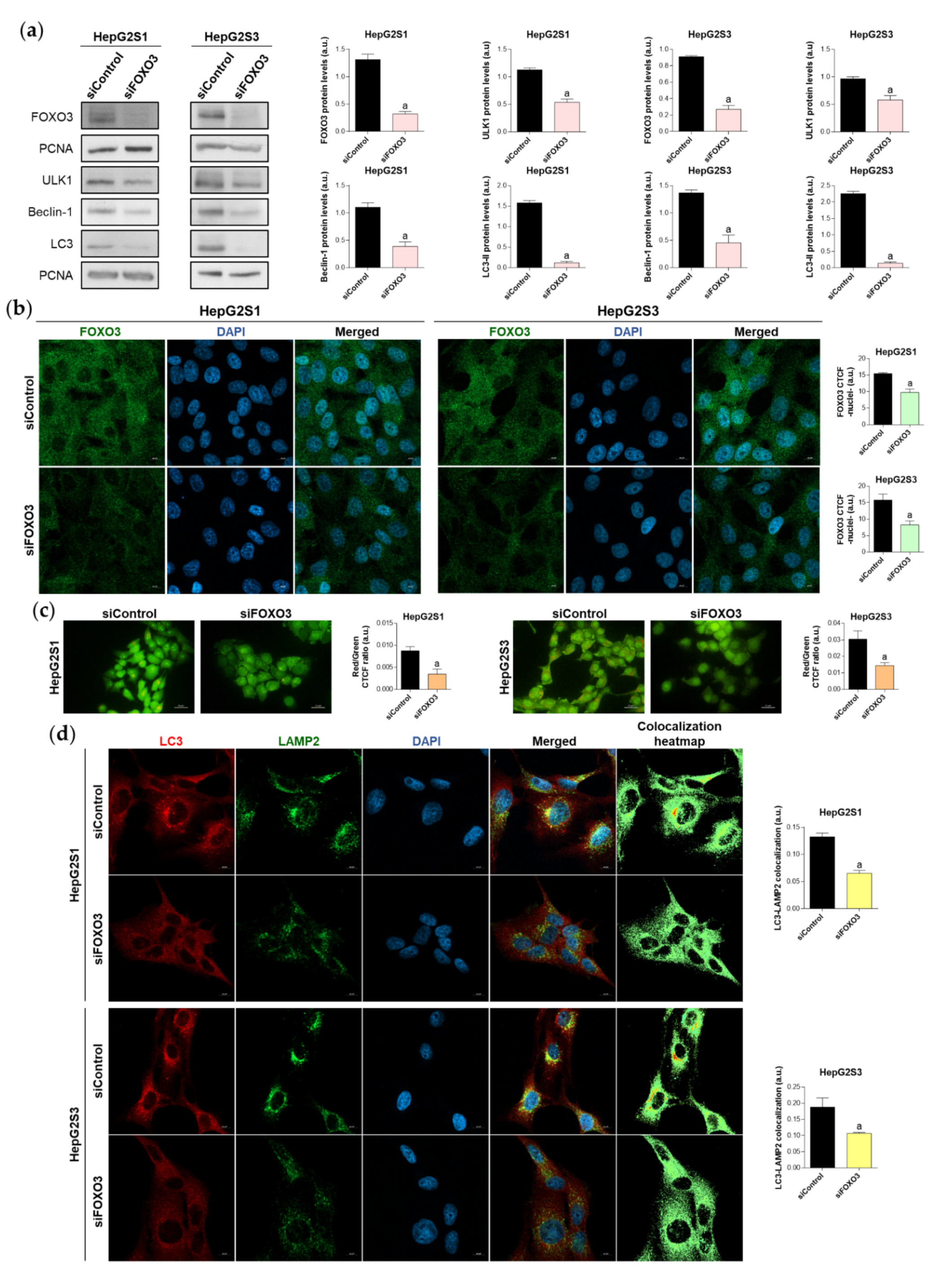
2.4. FOXO3 Mediates the Overactivation of Autophagy in Sorafenib-Resistant HCC Cells
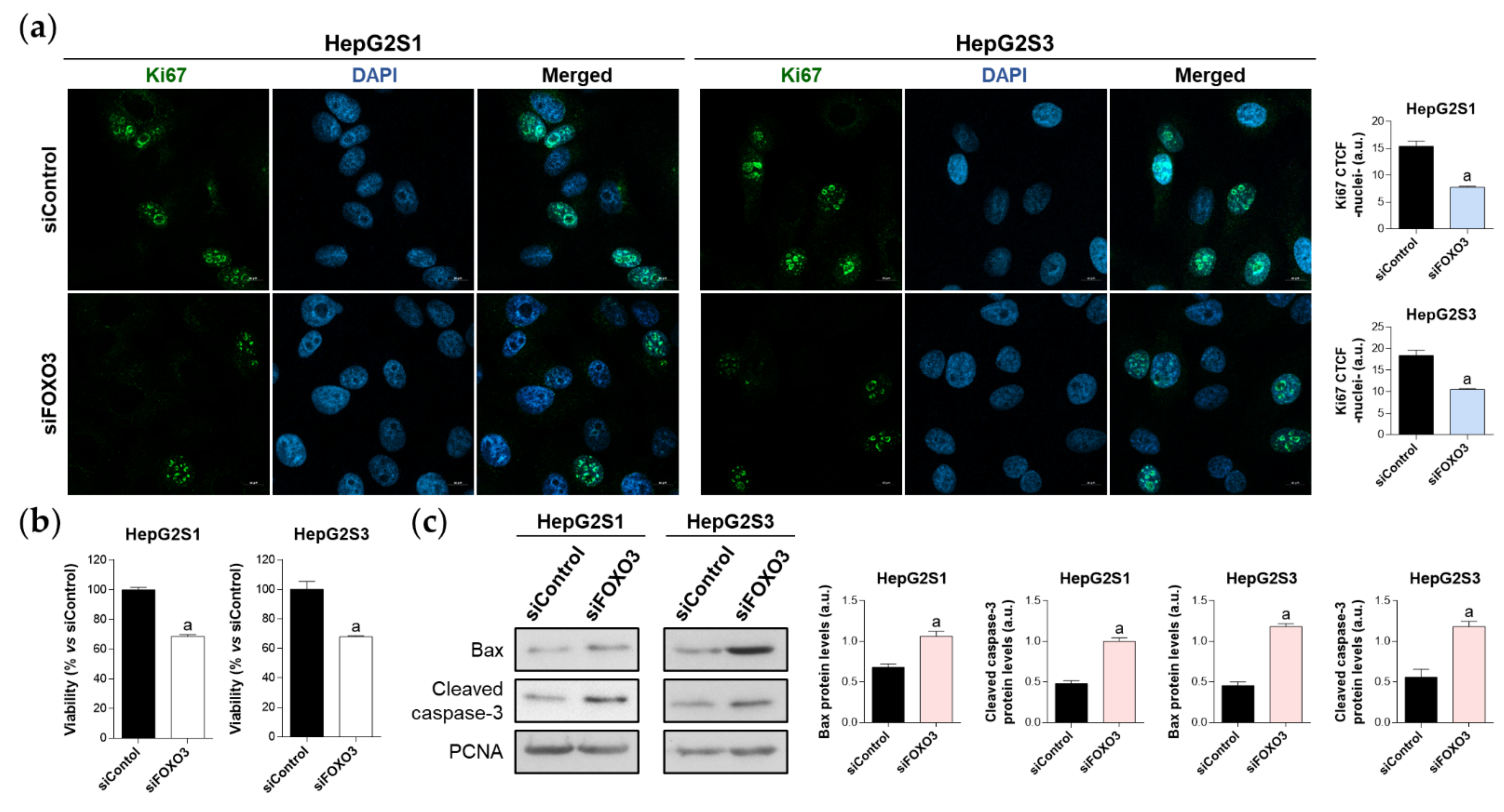
2.5. Survival of Sorafenib-Resistant Cell Lines Is FOXO3-Dependent
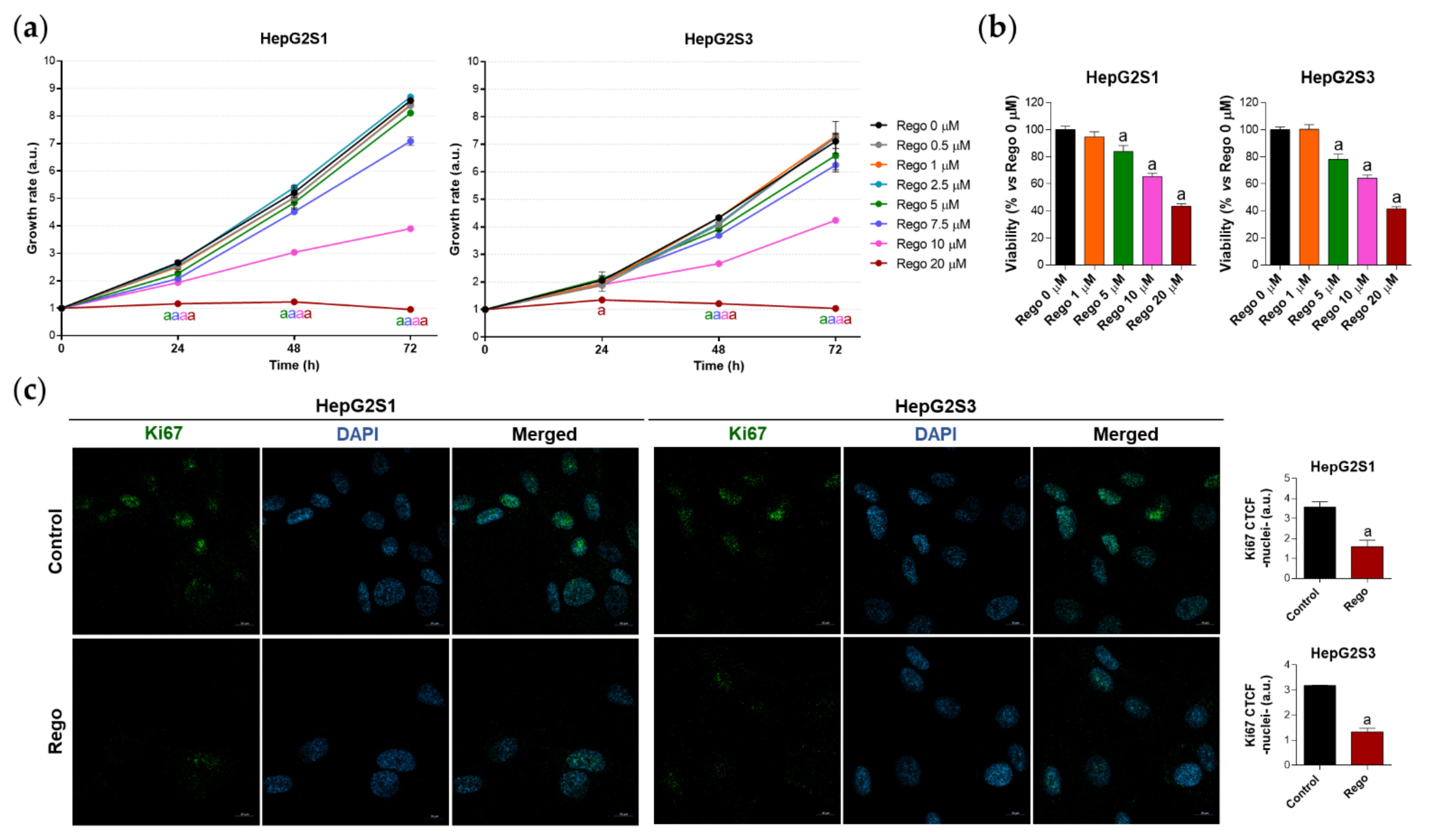
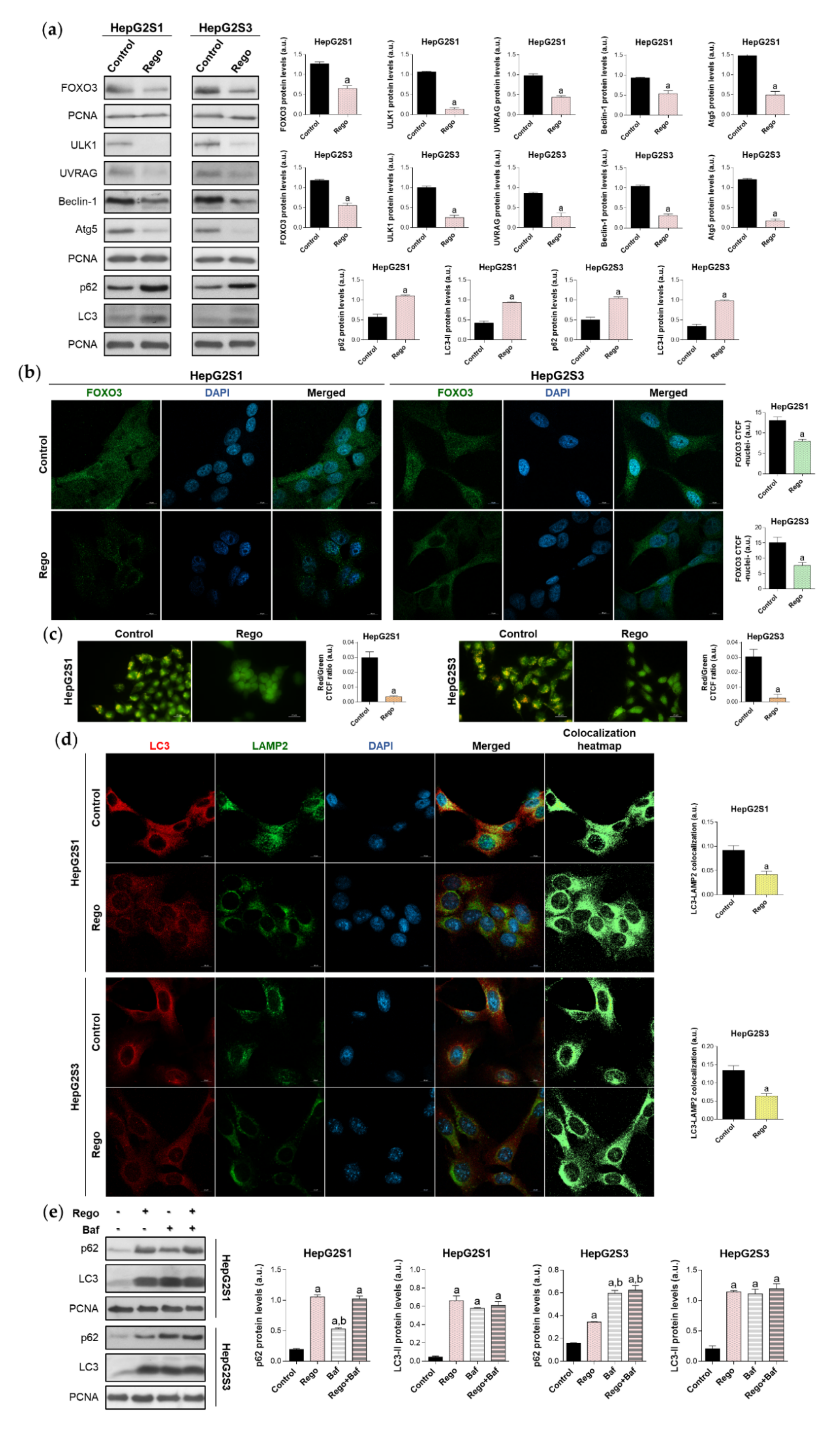
2.6. Regorafenib Impairs FOXO3-Mediated Autophagy in Sorafenib-Resistant HCC Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Acridine Orange Staining
4.3. Immunofluorescence and Laser Confocal Imaging
4.4. Western Blot
4.5. ROS Measurement
4.6. Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.7. Assessment of SubG1 Cell Population by Flow Cytometry
4.8. Cell Viability Assays
4.9. Expression, Survival and Gene Correlation Analysis of FOXO3 in HCC Patient Samples
4.10. Gene Silencing
4.11. Crystal Violet Staining
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Fan, G.; Wei, X.; Xu, X. Is the era of sorafenib over? A review of the literature. Ther. Adv. Med. Oncol. 2020, 12, 1758835920927602. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, L.K.D.; Tiribelli, C.; Sukowati, C.H.C. Sorafenib resistance in hepatocellular carcinoma: The relevance of genetic heterogeneity. Cancers 2020, 12, 1576. [Google Scholar] [CrossRef]
- Méndez-Blanco, C.; Fondevila, F.; Fernández-Palanca, P.; García-Palomo, A.; van Pelt, J.; Verslype, C.; González-Gallego, J.; Mauriz, J.L. Stabilization of hypoxia-inducible factors and BNIP3 promoter methylation contribute to acquired sorafenib resistance in human hepatocarcinoma cells. Cancers 2019, 11, 1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Blanco, C.; Fernández-Palanca, P.; Fondevila, F.; González-Gallego, J.; Mauriz, J.L. Prognostic and clinicopathological significance of hypoxia-inducible factors 1α and 2α in hepatocellular carcinoma: A systematic review with meta-analysis. Ther. Adv. Med. Oncol. 2021, 13, 1758835920987071. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, J.; Li, Q. Autophagy, an accomplice or antagonist of drug resistance in HCC? Cell Death Dis. 2021, 12, 266. [Google Scholar] [CrossRef]
- Wong, M.M.-T.; Chan, H.-Y.; Aziz, N.A.; Ramasamy, T.S.; Bong, J.-J.; Ch’ng, E.S.; Armon, S.; Peh, S.-C.; Teow, S.-Y. Interplay of autophagy and cancer stem cells in hepatocellular carcinoma. Mol. Biol. Rep. 2021, 48, 3695–3717. [Google Scholar] [CrossRef]
- Yang, S.; Yang, L.; Li, X.; Li, B.; Li, Y.; Zhang, X.; Ma, Y.; Peng, X.; Jin, H.; Li, H. New insights into autophagy in hepatocellular carcinoma: Mechanisms and therapeutic strategies. Am. J. Cancer Res. 2019, 9, 1329–1353. [Google Scholar]
- Kouroumalis, E.; Voumvouraki, A.; Augoustaki, A.; Samonakis, D.N. Autophagy in liver diseases. World J. Hepatol. 2021, 13, 6–65. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional regulation of autophagy: Mechanisms and diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef] [Green Version]
- Coomans De Brachène, A.; Demoulin, J.-B. FOXO transcription factors in cancer development and therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Steinmetz, C.; Kashyap, A.; Lorenz, S.; Mauriz, J.L.; Heise, M.; Galle, P.R.; Gonzalez-Gallego, J.; Strand, S. Melatonin induces transcriptional regulation of Bim by FoxO3a in HepG2 cells. Br. J. Cancer 2013, 108, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Hernández, M.A.; González, R.; de la Rosa, Á.J.; Gallego, P.; Ordóñez, R.; Navarro-Villarán, E.; Contreras, L.; Rodríguez-Arribas, M.; González-Gallego, J.; Álamo-Martínez, J.M.; et al. Molecular characterization of autophagic and apoptotic signaling induced by sorafenib in liver cancer cells. J. Cell. Physiol. 2018, 234, 692–708. [Google Scholar] [CrossRef] [Green Version]
- Salcher, S.; Hermann, M.; Kiechl-Kohlendorfer, U.; Ausserlechner, M.J.; Obexer, P. C10ORF10/DEPP-mediated ROS accumulation is a critical modulator of FOXO3-induced autophagy. Mol. Cancer 2017, 16, 95. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chen, E.; Tang, Y.; Mao, J.; Shen, J.; Zheng, X.; Xie, S.; Zhang, S.; Wu, Y.; Liu, H.; et al. miR-223 overexpression inhibits doxorubicin-induced autophagy by targeting FOXO3a and reverses chemoresistance in hepatocellular carcinoma cells. Cell Death Dis. 2019, 10, 843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Fan, X.; Tang, M.; Chen, R.; Wang, H.; Jia, R.; Zhou, X.; Jing, W.; Wang, H.; Yang, Y.; et al. Osteopontin induces autophagy to promote chemo-resistance in human hepatocellular carcinoma cells. Cancer Lett. 2016, 383, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Lee, S.Y.; Kim, C.Y.; Kim, Y.H.; Ju, W.; Kim, S.C. Subcellular localization of FOXO3a as a potential biomarker of response to combined treatment with inhibitors of PI3K and autophagy in PIK3CA-mutant cancer cells. Oncotarget 2017, 8, 6608–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Niu, Y.; Wan, A.; Chen, D.; Liang, H.; Chen, X.; Sun, L.; Zhan, S.; Chen, L.; Cheng, C.; et al. RNA m6A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 2020, 39, e103181. [Google Scholar] [CrossRef]
- Liang, C.; Dong, Z.; Cai, X.; Shen, J.; Xu, Y.; Zhang, M.; Li, H.; Yu, W.; Chen, W. Hypoxia induces sorafenib resistance mediated by autophagy via activating FOXO3a in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 1017. [Google Scholar] [CrossRef]
- Prieto-Domínguez, N.; Ordóñez, R.; Fernández, A.; García-Palomo, A.; Muntané, J.; González-Gallego, J.; Mauriz, J.L. Modulation of autophagy by sorafenib: Effects on treatment response. Front. Pharmacol. 2016, 7, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Ho, C.J.; Gorski, S.M. Molecular mechanisms underlying autophagy-mediated treatment resistance in cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and autophagy: Interactions and molecular regulatory mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Pang, L.; Dai, W.; Wu, S.; Ren, T.; Duan, Y.; Zheng, Y.; Bi, S.; Zhang, X.; Kong, J. Role of forkhead box O proteins in hepatocellular carcinoma biology and progression (review). Front. Oncol. 2021, 11, 667730. [Google Scholar] [CrossRef] [PubMed]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev. Cell 2018, 44, 555–565.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granito, A.; Forgione, A.; Marinelli, S.; Renzulli, M.; Ielasi, L.; Sansone, V.; Benevento, F.; Piscaglia, F.; Tovoli, F. Experience with regorafenib in the treatment of hepatocellular carcinoma. Therap. Adv. Gastroenterol. 2021, 14, 17562848211016960. [Google Scholar] [CrossRef]
- Fondevila, F.; Méndez-Blanco, C.; Fernández-Palanca, P.; González-Gallego, J.; Mauriz, J.L. Anti-tumoral activity of single and combined regorafenib treatments in preclinical models of liver and gastrointestinal cancers. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The dual role of autophagy in cancer development and a therapeutic strategy for cancer by targeting autophagy. Int. J. Mol. Sci. 2020, 22, 179. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, K.; Luan, X.; Zhang, J.; Liu, R.; Zheng, X.; Xie, S.; Ke, H.; Zhang, X.; Chen, W. Knockdown of eukaryotic translation initiation factor 5A2 enhances the therapeutic efficiency of doxorubicin in hepatocellular carcinoma cells by triggering lethal autophagy. Int. J. Oncol. 2020, 57, 1368–1380. [Google Scholar] [CrossRef]
- Zhang, C.; Lei, J.-L.; Zhang, H.; Xia, Y.-Z.; Yu, P.; Yang, L.; Kong, L.-Y. Calyxin Y sensitizes cisplatin-sensitive and resistant hepatocellular carcinoma cells to cisplatin through apoptotic and autophagic cell death via SCF βTrCP-mediated eEF2K degradation. Oncotarget 2017, 8, 70595–70616. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Y.; Zhou, W.; Chen, T.; Wu, Q.; Chutturghoon, V.K.; Lin, B.; Geng, L.; Yang, Z.; Zhou, L.; et al. YAP promotes multi-drug resistance and inhibits autophagy-related cell death in hepatocellular carcinoma via the RAC1-ROS-mTOR pathway. Cancer Cell Int. 2019, 19, 179. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhang, Q.; Wu, Z.; Xue, N. miR-101-3p sensitizes hepatocellular carcinoma cells to oxaliplatin by inhibiting Beclin-1-mediated autophagy. Int. J. Clin. Exp. Pathol. 2019, 12, 2056–2065. [Google Scholar] [PubMed]
- Song, B.; Bian, Q.; Shao, C.H.; Li, G.; Liu, A.A.; Jing, W.; Liu, R.; Zhang, Y.-J.; Zhou, Y.-Q.; Hu, X.-G.; et al. Ulinastatin reduces the resistance of liver cancer cells to epirubicin by inhibiting autophagy. PLoS ONE 2015, 10, e0120694. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Ye, X.; Ma, X.; Hu, X.; Yang, W.; Shi, J.; Chen, G.; Gong, L. SNGH16 regulates cell autophagy to promote sorafenib resistance through suppressing miR-23b-3p via sponging EGR1 in hepatocellular carcinoma. Cancer Med. 2020, 9, 4324–4338. [Google Scholar] [CrossRef]
- Song, Y.; He, S.; Ma, X.; Zhang, M.; Zhuang, J.; Wang, G.; Ye, Y.; Xia, W. RBMX contributes to hepatocellular carcinoma progression and sorafenib resistance by specifically binding and stabilizing BLACAT1. Am. J. Cancer Res. 2020, 10, 3644–3665. [Google Scholar]
- Shi, Y.; Yang, X.; Xue, X.; Sun, D.; Cai, P.; Song, Q.; Zhang, B.; Qin, L. HANR enhances autophagy-associated sorafenib resistance through miR-29b/ATG9A axis in hepatocellular carcinoma. OncoTargets Ther. 2020, 13, 2127–2137. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Liu, X.; Zhang, Y.; Li, A.; Xie, Y.; Zhou, S.; Song, L.; Xu, R.; Ma, Y.; Cai, S.; et al. BEZ235 increases the sensitivity of hepatocellular carcinoma to sorafenib by inhibiting PI3K/AKT/mTOR and inducing autophagy. Biomed Res. Int. 2021, 2021, 5556306. [Google Scholar] [CrossRef]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Lu, C.; Jun, W.; Wu, Y.; Shi, X.; Ding, Y. The up-regulation of P62 levels is associated with resistance of sorafenib in hepatocarcinoma cells. Int. J. Clin. Exp. Pathol. 2019, 12, 2622–2630. [Google Scholar]
- Xiang, X.; Qin, H.-G.; You, X.-M.; Wang, Y.-Y.; Qi, L.-N.; Ma, L.; Xiang, B.-D.; Zhong, J.-H.; Li, L.-Q. Expression of P62 in hepatocellular carcinoma involving hepatitis B virus infection and aflatoxin B1 exposure. Cancer Med. 2017, 6, 2357–2369. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, C.; Qin, L.; Xu, J.; Li, X.; Wang, W.; Kong, L.; Zhou, T.; Li, X. The prognostic value of NRF2 in solid tumor patients: A meta-analysis. Oncotarget 2017, 9, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Dong, X.; Zhai, B.; Jiang, X.; Dong, D.; Li, B.; Jiang, H.; Xu, S.; Sun, X. MiR-21 mediates sorafenib resistance of hepatocellular carcinoma cells by inhibiting autophagy via the PTEN/Akt pathway. Oncotarget 2015, 6, 28867–28881. [Google Scholar] [CrossRef] [Green Version]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.; Wang, Y.; Li, M.; Zhu, Y.; Liang, H.; Wang, C.; Wang, F.; Zhang, C.-Y.; Zen, K.; Li, L. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017, 8, e2540. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P.; Cho, H.J.; Kim, J.-T.; Baek, K.E.; Lee, H.G.; Kang, S.C. Morin hydrate reverses cisplatin resistance by impairing PARP1/HMGB1-dependent autophagy in hepatocellular carcinoma. Cancers 2019, 11, 986. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-Y.; Yadav, V.K.; Chu, Y.C.; Ong, J.R.; Huang, T.-Y.; Lee, K.-F.; Lee, K.-H.; Yeh, C.-T.; Lee, W.-H. Hydroxychloroquine (HCQ) modulates autophagy and oxidative DNA damage stress in hepatocellular carcinoma to overcome sorafenib resistance via TLR9/SOD1/hsa-miR-30a-5p/Beclin-1 axis. Cancers 2021, 13, 3227. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef]
- Zhao, F.; Feng, G.; Zhu, J.; Su, Z.; Guo, R.; Liu, J.; Zhang, H.; Zhai, Y. 3-Methyladenine-enhanced susceptibility to sorafenib in hepatocellular carcinoma cells by inhibiting autophagy. Anticancer Drugs 2021, 32, 386–393. [Google Scholar] [CrossRef]
- Ahn, H.; Kim, H.; Abdul, R.; Kim, Y.; Sim, J.; Choi, D.; Paik, S.S.; Shin, S.-J.; Kim, D.-H.; Jang, K. Overexpression of forkhead box O3a and its association with aggressive phenotypes and poor prognosis in human hepatocellular carcinoma. Am. J. Clin. Pathol. 2018, 149, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-S.; Ying, J.-F.; Zhang, Y.-N.; Pan, H.-Y.; He, X.-L.; Hu, Z.-M.; Wang, H.-J.; Dou, X.-B.; Mou, X.-Z. High expression of FOXO3 is associated with poor prognosis in patients with hepatocellular carcinoma. Oncol. Lett. 2020, 19, 3181–3188. [Google Scholar] [CrossRef]
- Li, J.; Qin, X.; Wu, R.; Wan, L.; Zhang, L.; Liu, R. Circular RNA circFBXO11 modulates hepatocellular carcinoma progress and oxaliplatin resistance through miR-605/FOXO3/ABCB1 axis. J. Cell. Mol. Med. 2020, 24, 5152–5161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Weng, M.-C.; Wang, M.-H.; Tsai, J.-J.; Kuo, Y.-C.; Liu, Y.-C.; Hsu, F.-T.; Wang, H.-E. Regorafenib inhibits tumor progression through suppression of ERK/NF-κB activation in hepatocellular carcinoma bearing mice. Biosci. Rep. 2018, 38, BSR20171264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yang, J.; Zhang, Y.; Cai, H.; Chen, X.; Sun, D. Regorafenib reverses HGF-induced sorafenib resistance by inhibiting epithelial-mesenchymal transition in hepatocellular carcinoma. FEBS Open Bio 2019, 9, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Van Malenstein, H.; Dekervel, J.; Verslype, C.; Van Cutsem, E.; Windmolders, P.; Nevens, F.; van Pelt, J. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013, 329, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell. Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Full Name | Pearson-CC |
|---|---|---|
| ATG5 | Autophagy-related 5 | +0.62 |
| RAB33B | RAB33B, member RAS oncogene family | +0.6 |
| STX17 | Syntaxin 17 | +0.56 |
| ATG2B | Autophagy-related 2B | +0.55 |
| SMCR8 | SMCR8-C9orf72 complex subunit | +0.55 |
| TRAF6 | TNF receptor associated factor 6 | +0.55 |
| UVRAG | UV radiation resistance associated | +0.55 |
| PIK3C3 | Phosphatidylinositol 3-kinase catalytic subunit type 3 | +0.51 |
| PIK3R4 | Phosphoinositide-3-kinase regulatory subunit 4 | +0.51 |
| TANK | TRAF family member associated NFKB activator | +0.51 |
| ATG16L1 | Autophagy-related 16 like 1 | +0.48 |
| HMGB1 | High mobility group box 1 | +0.48 |
| AMBRA1 | Autophagy and beclin-1 regulator 1 | +0.47 |
| TBK1 | TANK binding kinase 1 | +0.47 |
| RAB1A | RAB1A, member RAS oncogene family | +0.44 |
| RAB7A | RAB7A, member RAS oncogene family | +0.42 |
| ULK1 | Unc-51 like autophagy activating kinase 1 | +0.41 |
| SH3GLB1 | SH3 domain containing GRB2 like, endophilin B1 | +0.4 |
| ATG16L2 | Autophagy-related 16 like 2 | +0.38 |
| ATG4C | Autophagy-related 4C cysteine peptidase | +0.38 |
| ATG9A | Autophagy-related 9A | +0.38 |
| ATG12 | Autophagy-related 12 | +0.37 |
| NRBF2 | Nuclear receptor binding factor 2 | +0.37 |
| SNAP29 | Synaptosome associated protein 29 | +0.37 |
| C9orf72 | C9orf72-SMCR8 complex subunit | +0.36 |
| DAPK1 | Death associated protein kinase 1 | +0.36 |
| ATG3 | Autophagy-related 3 | +0.35 |
| WDR41 | WD repeat domain 41 | +0.35 |
| RB1CC1 | RB1 inducible coiled-coil 1 | +0.34 |
| BECN1 | Beclin-1 | +0.31 |
| ATG4B | Autophagy-related 4B cysteine peptidase | +0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fondevila, F.; Méndez-Blanco, C.; Fernández-Palanca, P.; Payo-Serafín, T.; van Pelt, J.; Verslype, C.; González-Gallego, J.; Mauriz, J.L. Autophagy-Related Chemoprotection against Sorafenib in Human Hepatocarcinoma: Role of FOXO3 Upregulation and Modulation by Regorafenib. Int. J. Mol. Sci. 2021, 22, 11770. https://doi.org/10.3390/ijms222111770
Fondevila F, Méndez-Blanco C, Fernández-Palanca P, Payo-Serafín T, van Pelt J, Verslype C, González-Gallego J, Mauriz JL. Autophagy-Related Chemoprotection against Sorafenib in Human Hepatocarcinoma: Role of FOXO3 Upregulation and Modulation by Regorafenib. International Journal of Molecular Sciences. 2021; 22(21):11770. https://doi.org/10.3390/ijms222111770
Chicago/Turabian StyleFondevila, Flavia, Carolina Méndez-Blanco, Paula Fernández-Palanca, Tania Payo-Serafín, Jos van Pelt, Chris Verslype, Javier González-Gallego, and José L. Mauriz. 2021. "Autophagy-Related Chemoprotection against Sorafenib in Human Hepatocarcinoma: Role of FOXO3 Upregulation and Modulation by Regorafenib" International Journal of Molecular Sciences 22, no. 21: 11770. https://doi.org/10.3390/ijms222111770
APA StyleFondevila, F., Méndez-Blanco, C., Fernández-Palanca, P., Payo-Serafín, T., van Pelt, J., Verslype, C., González-Gallego, J., & Mauriz, J. L. (2021). Autophagy-Related Chemoprotection against Sorafenib in Human Hepatocarcinoma: Role of FOXO3 Upregulation and Modulation by Regorafenib. International Journal of Molecular Sciences, 22(21), 11770. https://doi.org/10.3390/ijms222111770