
Signal Transduction of Mineralocorticoid and Angiotensin II Receptors in the Central Control of Sodium Appetite: A Narrative Review


 ,
,  ,
, 
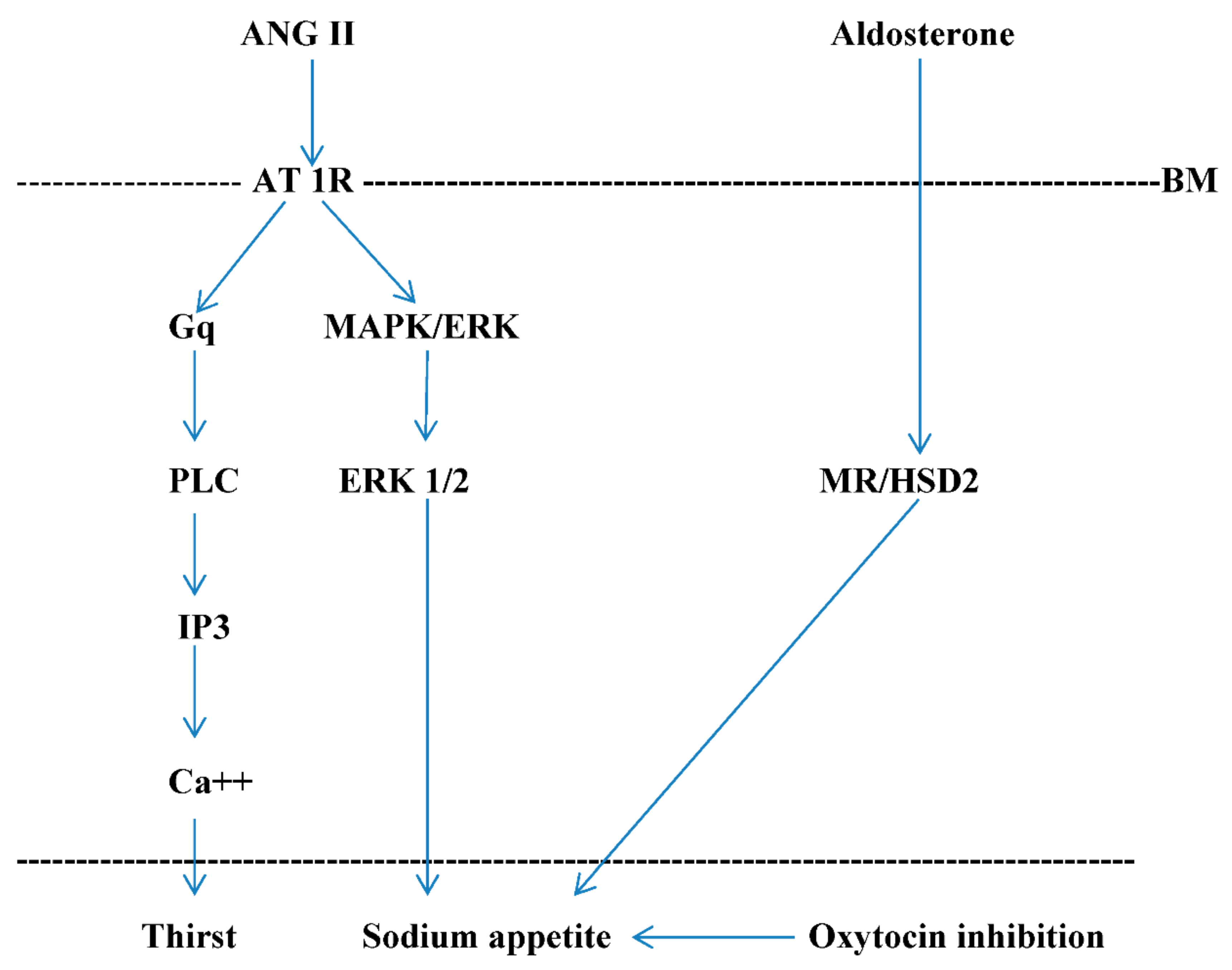{kind=link}
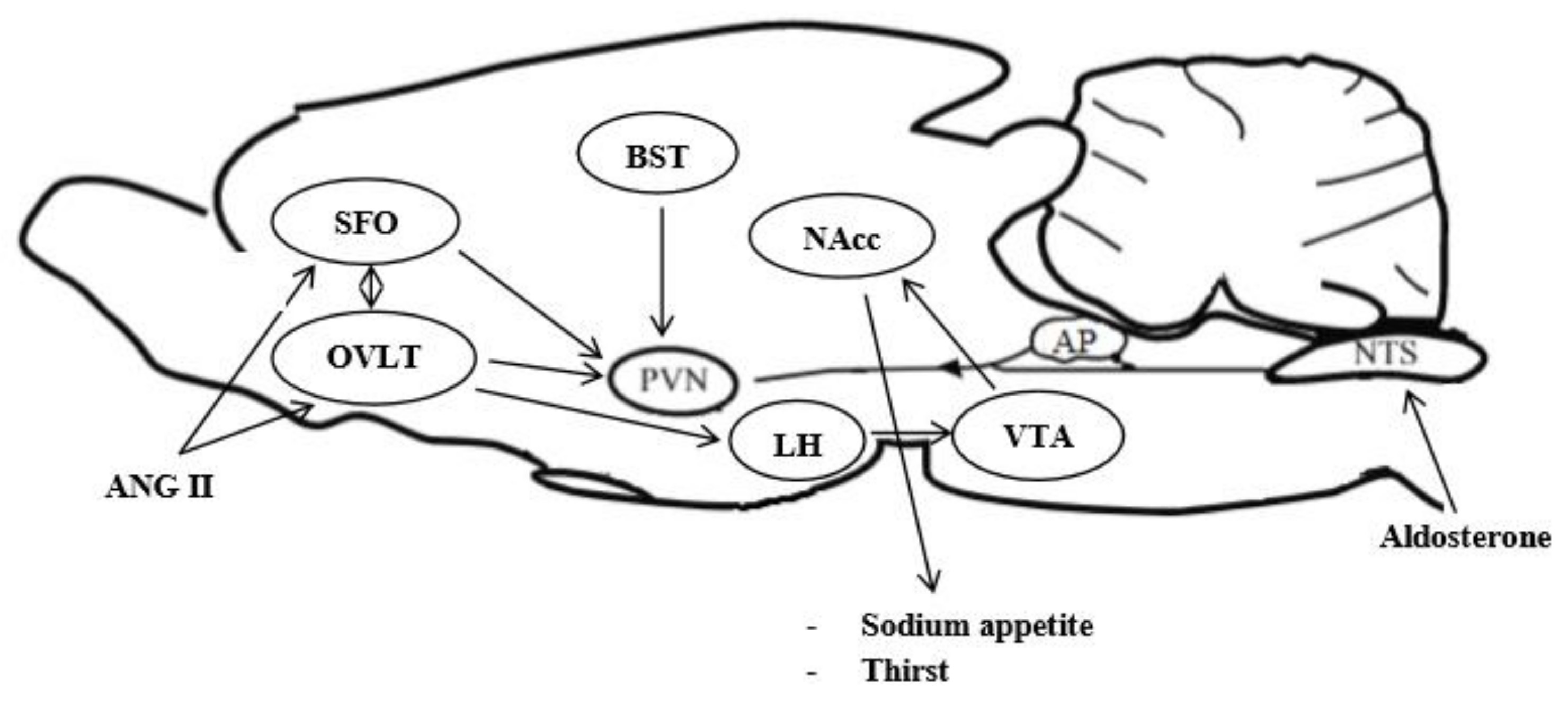{kind=link}
Abstract
1. Background
2. Fluid Balance
3. Forebrain Sites Are Involved in the Integration of Signals Regulating Sodium Appetite
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yosten, G.L.C.; Samson, K.W. Chapter 5—Sex Differences in the Central Control of Sodium Appetite and Blood Pressure. In Sex Differences in Cardiovascular Physiology and Pathophysiology; La Marca, B., Alexander, B.T., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 63–71. [Google Scholar] [CrossRef]
- Wolf, G. Effect of deoxycorticosterone on sodium appetite of intact and adrenalectomized rats. Am. J. Physiol. 1965, 205, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Gizowski, P.; Bourque, C.W. The neural basis of homeostatic an anticipatory thirst. Nat. Rev. Nephrol. 2018, 14, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, J.T. Angiotensin, thirst and sodium appetite. Physiol. Rev. 1998, 78, 583–686. [Google Scholar] [CrossRef] [PubMed]
- Iovino, M.; Guastamacchia, E.; Giagulli, V.A.; Licchelli, B.; Triggiani, V. Vasopressin secretion control: Central neural pathways, neurotransmitters and effects of drugs. Curr. Pharm. Des. 2012, 18, 4714–4724. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.L.; Athar, S.; Shelton, R.L. Osmotic control of vasopressin function. In Disturbance in Body Fluid Osmolality; Andreoli, T.E., Grantham, J.J., Floyd, C.R., Eds.; The Williams and Wilkins Co.: Baltimore, MD, USA, 1997; pp. 125–149. [Google Scholar]
- Iovino, M.; Guastamacchia, E.; Giagulli, V.A.; Licchelli, B.; Iovino, E.; Triggiani, V. Molecular mechanisms involved in the control of neurohypophyseal hormones secretion. Curr. Pharm. Des. 2014, 20, 6702–6713. [Google Scholar] [CrossRef]
- Caillens, H.; Pruszcynski, W.; Meyrier, A. Relationship between change in volemia at costant osmolality and plasma antidiuretic hormone. Mineral. Electrolyte Metab. 1980, 4, 161–171. [Google Scholar]
- Kalia, M.P. Localization of aortic and carotid baroreceptor and chemoreceptor primary afferents in the brain stem. In Central Nervous System Mechanisms in Hypertension; Buckeley, J.P., Ferrario, C.M., Eds.; Raven Press: New York, NY, USA, 1981; pp. 9–24. [Google Scholar]
- Dahlstroem, A.; Fuxe, K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol. Scand. 1964, 62 (Suppl. 232), 1–80. [Google Scholar]
- Azizi, S.A. Monoamines: Dopamine, Norepinephrine, and Serotonin, Beyond Modulation, “Switches” That Alter the State of Target Networks. Neuroscientist 2020, 1073858420974336. [Google Scholar] [CrossRef]
- Hokfelt, T.; Fuxe, K.; Goldstein, M.; Johansson, O. Immunohistochemical evidence for the existence of adrenaline neurons in the rat brain. Brain Res. 1974, 66, 235–251. [Google Scholar] [CrossRef]
- Di Francesco, D. The cardiac hyperpolarizing-activated current. Origin and developments. Prog. Biophys. Mol. Biol. 1985, 46, 163–183. [Google Scholar] [CrossRef]
- Sved, A.F.; Imaizumi, T.; Talman, W.T.; Reis, D.J. Vasopressin contributes to hypertension caused by nucleus tractus solitarius lesions. Hypertension 1985, 7, 262–267. [Google Scholar] [CrossRef]
- Iovino, M.; Vanacore, A.; Steardo, L. Alpha 2-adrenergic stimulation within the nucleus tractus solitarius attenuates vasopressin release induced by depletion of the cardiovascular volume. Pharmacol. Biochem. Behav. 1990, 37, 821–824. [Google Scholar] [CrossRef]
- Atlas, A.S. The renin-angiotensin-aldosterone system: Pathophysiological role and pharmacological inhibition. J. Manag. Care Pharm. 2007, 13, 9–20. [Google Scholar] [CrossRef]
- Iovino, M.; Messana, T.; De Pergola, G.; Iovino, E.; Guastamacchia, E.; Licchelli, B.; Vanacore, A.; Giagulli, V.A.; Triggiani, V. Brain Angiotensinergic Regulation of the Immune System: Implication for Cardiovascular and Neuroendocrine Responses. Endocr. Metab. Immune Disord. Drug Target 2020, 20, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Sayeski, P.P.; Bernstein, K.E. Signal transduction mechanisms of the angiotensin II type AT(1)-receptor: Looking beyond the heterometric G protein paradigm. J. Renin Angiotensin Aldosterone Syst. 2001, 2, 4–10. [Google Scholar] [CrossRef]
- Fillon, D.; Cabana, J.; Guillemette, G.; Leduc, R.; Lavigne, P.; Escher, E. Structure of the human angiotensin II type 1 (AT 1) receptor bound to angiotensin from multiple chemoselective photoprobe contacts reveals a unique peptide binding mode. J. Biol. Chem. 2013, 288, 8187–8197. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Corval, P.; Gasc, J.M. Ontogeny of the two angiotensin II type 1 receptor subtypes in rats. Am. J. Physiol. 1994, 267, E828–E836. [Google Scholar] [CrossRef]
- Iwai, N.; Inagami, T. Identification of two subtypes in the rat type 1 angiotensin II receptor. FEBS Lett. 1992, 298, 257–260. [Google Scholar] [CrossRef]
- Premer, C.; Lamondin, C.; Mitzey, A.; Speth, R.C.; Brownfield, M.S. Immunohistochemical Localization of AT1a, AT1b, and AT2 Angiotensin II Receptor Subtypes in the Rat Adrenal, Pituitary, and Brain with a Perspective Commentary. Int. J. Hypertens. 2013, 2013, 175428. [Google Scholar] [CrossRef] [PubMed]
- Pechlivanova, D.M.; Stoynev, A.G. Effect of chronic treatment with angiotensin receptor ligands on water-salt balance in Wistar and spontaneously hypertensive rats. Folia Med. 2013, 55, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, S.; Melo, M.R.; Nascimento, P.A.; Andrade-Franzé, G.M.F.; Antunes-Rodrigues, J.; Yosten, G.L.C.; Menani, J.V.; Samson, W.K.; Colombari, E. Interaction of central angiotensin II and aldosterone on sodium intake and blood pressure. Brain Res. 2019, 1720, 146299. [Google Scholar] [CrossRef]
- Bresford, M.J.; Fitzsimons, J.T. Intracerebroventricular angiotensin II-induced thirst and sodium appetite in rat are blocked by the AT 1 receptor antagonist losartan (DuP 753), but not by the AT 2 antagonist, CGP 42112B. Exp. Physiol. 1992, 77, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.; Mietlicki, E.G.; Nowak, E.L.; Fluharty, S.J. Angiotensin II stimulates water and salt intake through separate cell signaling pathway in rats. Exp. Physiol. 2009, 94, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Schraeder, L.A.; Bimbaum, S.G.; Nadin, B.M.; Bui, D.; Andersen, A.E. ERK/MAPK regulates the kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am. J. Physiol. Cell Physiol. 2006, 290, E852–E861. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.G.; Zhang, Z.H.; Felder, R.B. Angiotensin II up-regulates hypothalamic AT1 receptor expression in rats via mitogen-activated protein kinase pathway. Am. J. Physiol. Heart Circul Physiol. 2009, 296, H1425–H1433. [Google Scholar] [CrossRef] [PubMed]
- Alonso, G.; Gabrion, J.; Travers, E.; Asseumacher, I. Ultrastructural organization of actin filaments in neurosecretory axons of the rat. Cell Tissue Res. 1981, 214, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, P.C. Actin in axon: Stable scaffolds and dynamic filaments. Results Probl. Cell Differ. 2009, 48, 65–90. [Google Scholar] [PubMed]
- Saarikangas, J.; Zhao, H.; Lappalainen, P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinosytides. Physiol. Rev. 2010, 90, 259–289. [Google Scholar] [CrossRef]
- Geerling, I.C.; Loewy, A.D. Aldosterone-sensitive NTS neurons are inhibited by saline ingestion during chronic mineralcorticoid treatment. Brain Res. 2006, 1115, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Okubo, S.; Niimura, F.; Nishimura, H.; Takemato, F.; Fogo, A.; Matsusaka, T.; Ichikawa, I. Angiotensin-independent mechanism for aldosterone synthesis during chronic extracellular fluid volume depletion. J. Clin. Investig. 1997, 99, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Demura, M.; Yoneda, T.; Takeda, Y. DNA Methylation of the Angiotensinogen Gene, AGT, and the Aldosterone Synthase Gene, CYP11B2 in Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 27, 4587. [Google Scholar] [CrossRef] [PubMed]
- Makanova, N.; Sequeira-Lopez, M.L.; Gomez, R.A.; Kim, H.S.; Smithies, D. Disturbed homeostasis in sodium-restricted mice heterozygous for aldosterone synthase gene disruption. Hypertension 2006, 48, 1151–1159. [Google Scholar] [CrossRef]
- Francis, J.; Weiss, R.M.; Wei, S.G.; Johnson, A.K.; Beltz, T.G.; Zimmerman, K.; Felder, R.B. Central mineral corticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2241–H2251. [Google Scholar] [CrossRef]
- Viengchareun, S.; Le Menuet, D.; Mastinerie, L.; Munier, M.; Pascual-Le Tallec, L.; Lombes, M. The mineralcorticoid receptor: Insights into its molecular and (patho)physiological biology. Nucl. Recept. Signal 2007, 5, e012. [Google Scholar] [CrossRef]
- Fluharty, S.J.; Epstein, A.N. Sodium appetite elicited by intracerebroventricular infusion of angiotensin II in the rat. II. Synergistic interaction with systemic mineralcorticoids. Behav. Neurosci. 1983, 97, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Reis, W.L.; Saad, W.A.; Camargo, L.A.; Elias, L.L.; Antunes-Rodrigues, J. Central nitrergic system regulation of neuroendocrine secretion, fluid intake and blood pressure induced by angiotensin-II. Behav. Brain Funct. 2010, 25, 64. [Google Scholar] [CrossRef]
- Miselis, R.R. The efferent projections of the subfornical organ of the rat: A circumventricular organ within a neural network subserving water balance. Brain Res. 1981, 230, 1–23. [Google Scholar] [CrossRef]
- McKinley, M.J.; Badoer, E.; Olfield, B.J. Intravenous angiotensin II induces Fos-immunoreactivity in circumventricular organs of the lamina terminalis. Brain Res. 1992, 594, 295–300. [Google Scholar] [CrossRef]
- Fry, W.M.; Ferguson, A.V. The subfornical organ and organum vasculosum of the lamina terminalis: Critical roles in cardiovascular regulation and the control of fluid balance. Handb. Clin. Neurol. 2021, 180, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.B.; Smith, L.; Brown, E.R.; Barues, D.; Sabir, M.A.; Davis, J.S. Angiotensin rapidly increases phosphotidate-phosphoinositide synthesis, an phosphoinositide hydrolysis and metabolizes intracellular calcium in cultured arterial muscle cells. Proc. Natl. Acad. Sci. USA 1984, 81, 7812–7816. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Franzé, G.M.F.; Pereira, E.D., Jr.; Yosten, G.L.C.; Samson, W.K.; Menani, J.V.; De Luca, L.A., Jr.; Andrade, C.A.F. Blockade of ERK1/2 activation with U0126 or PEP7 reduces sodium appetite and angiotensin II-induced pressor responses in spontaneously hypertensive rats. Peptides 2021, 136, 170439. [Google Scholar] [CrossRef] [PubMed]
- Felgendreger, L.A.; Fluarthy, S.J.; Yen, D.K.; Flanagan-Cato, L.M. Endogenous angiotensin II-induced p44/42 mitogen-activated proteinkinase activation mediates sodium-appetite but not thirst or neurohypophyseal secretion in male rats. J. Neuroendocrinol. 2013, 25, 97–106. [Google Scholar] [CrossRef]
- Geerling, J.C.; Kawata, M.; Loewy, A.D. Aldosterone-sensitive neurons in the rat central nervous system. J. Comp. Neurol. 2006, 494, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Grafe, L.A.; Takacs, A.E.; Yen, D.K.; Flanagan-Cato, L.M. The role of hypothalamic paraventricular nucleus and the organum vasculosum of the lateral terminalis in the control of sodium appetite in male rats. J. Neurosci. 2014, 34, 9249–9260. [Google Scholar] [CrossRef] [PubMed]
- Roesch, D.M.; Balckburn-Monro, R.E.; Verbalis, J.G. Mineralcorticoid treatment attenuates activation of oxytocinergic and vasopressinergic neurons by icv ANG II. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R1853–R1864. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G. Disorders of body water homeostasis. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 471–503. [Google Scholar] [CrossRef]
- Guyton, A.C. Blood pressure control—Special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef]
- Kinsman, B.J.; Nation, H.N.; Stocker, S.D. Hypothalamic Signaling in Body Fluid Homeostasis and Hypertension. Curr. Hypertens. Rep. 2017, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Na, E.S.; Grippo, A.J.; Johnson, A.K. The effects of deoxycorticosterone-induced sodium appetite on hedonic behaviors in rats. Behav. Neurosci. 2006, 120, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, P.N.; Pereira, D.T.; Monra, F.V.; Menani, J.V.; DeLuca, L.A. Ingestion of hypertonic NaCl vs palatable drinks by sodium-depleted rats. Physiol. Behav. 2002, 75, 443–448. [Google Scholar] [CrossRef]
- Crystal, S.R.; Bernstein, I.L. Morning sickness: Impact on offspring salt preference. Appetite 1995, 25, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Crystal, S.R.; Bernstein, I.L. Infant salt preference and mother’s morning sickness. Appetite 1998, 30, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.J.; Cowart, B.J.; Beauchamp, G.K. The development of salty taste acceptance is related to dietary experience in human infants: A prospective study. Am. J. Clin. Nutr. 2012, 95, 123–129. [Google Scholar] [CrossRef]
- Frigley, M.J.; Watters, I.W. Effects of mineralcorticoids on spontaneous sodium chloride appetite of adrenalectomized rats. Physiol. Behav. 1966, 1, 65–74. [Google Scholar] [CrossRef]
- Tordoff, M.G.; Hughes, R.L.; Pilchak, D.M. Different effects of three aldosterone treatment on plasma aldosterone and salt appetite. Physiol. Behav. 1993, 54, 129–134. [Google Scholar] [CrossRef]
- Stricker, E.M.; Thiels, E.; Verbalis, J.G. Sodium appetite in rats after prolonged dietary sodium deprivation: A sexually dimorphic phenomenon. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991, 260, R1082–R1088. [Google Scholar] [CrossRef] [PubMed]
- Toth, E.; Stelfox, J.; Kaufman, S. Cardiac control of salt appetite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1987, 252, R925–R929. [Google Scholar] [CrossRef] [PubMed]
- Stricker, E.M. Thirst and sodium appetite after colloid treatment in rats. J. Comp. Physiol. Psychol. 1981, 95, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Jalowiec, J.E. Sodium appetite elicited by furosemide: Effects of differential dietary mainteinance. Behav. Biol. 1974, 10, 313–327. [Google Scholar] [CrossRef]
- Kochli, A.; Tenenbaum-Rakover, Y.; Leshem, M. Increased salt appetite in patients with congenital adrenal hyperplasia 21-hydroxylase deficiency. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1673–R1681. [Google Scholar] [CrossRef]
- Cruz, D.N.; Simon, D.B.; Nelson-Williams, C.; Farhi, A.; Finberg, K.; Burleson, L.; Gill, J.R.; Lifton, R.P. Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension 2001, 37, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zaunad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palunesky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Stricker, E.M. Some physiological and motivational properties of the hypovolemic stimulus for thirst. Physiol. Behav. 1968, 3, 379–385. [Google Scholar] [CrossRef]
- Wolf, G.; Stricker, E.M. Sodium appetite elicited by hypovolemia in adrenalectomized rats: Reevaluation of the “reservoir” hypothesis. J. Comp. Physiol. Psychol. 1967, 63, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Nose, H.; Morimoto, T. Changes in Na concentration in cerebrospinal fluid during acute hypernatremia and their effect on drinking in juvenile rats. Physiol. Behav. 1992, 52, 499–504. [Google Scholar] [CrossRef]
- Weisinger, R.S.; Considine, P.; Denton, D.A.; Leksell, L.; Mc Kinley, M.J.; Mouw, D.R.; Muller, A.F.; Tarjan, E. Role of sodium concentration of the cerebrospinal fluid in the salt appetite of sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1982, 242, R51–R63. [Google Scholar] [CrossRef] [PubMed]
- Chiaraviglio, E.; Perez-Guaita, M.F. The effect of intracerebroventricular hypertonic infusion on sodium appetite in rats after peritoneal dialysis. Physiol. Behav. 1986, 37, 695–699. [Google Scholar] [CrossRef]
- Denton, D.A.; Mc Kinley, M.J.; Weisinger, R.S. Hypothalamic integration of body fluid regulation. Proc. Natl. Acad. Sci. USA 1996, 93, 7397–7404. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, E.; Fujikawa, A.; Matsunaga, H.; Yasoshima, Y.; Sako, K.; Yamamoto, T.; Saegusa, C.; Noda, M. Nav2/NaG channel is involved in the control of salt-intake behavior in the CNS. J. Neurosci. 2000, 20, 7743–7751. [Google Scholar] [CrossRef]
- Sticker, E.M.; Verbalis, J.G. Central inhibitory control of sodium appetite in rats correlation with pituitary oxytocin secretion. Behav. Neurosci. 1987, 101, 560–567. [Google Scholar] [CrossRef]
- Sticker, E.M.; Verbalis, J.G. Central inhibition of salt appetite by oxytocin in rats. Regul. Pept. 1996, 66, 83–85. [Google Scholar] [CrossRef]
- Rigatto, K.; Puryear, R.; Bernastova, I.; Morris, M. Salt appetite and renin-angiotensin system. Effect of oxytocin deficiency. Hypertension 2003, 42, 793–797. [Google Scholar] [CrossRef]
- Yoshura, R.; Kiyama, H.; Kimura, T.; Araki, T.; Maeno, H.; Tanizawa, D. Localization of oxytocin receptor messanger ribonucleic acid in the brain. Endocrinology 1993, 133, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Buijs, R.M. Intra- and extra-hypothalamic vasopressin and oxytocin pathways in the rat. Cell Tissue Res. 1978, 192, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.K.; Thunhorst, R.L. The neuroendocrinology of thirst and salt appetite: Visceral sensory signals and mechanisms of central integration. Front. Neuroendocrinol. 1997, 18, 283–292. [Google Scholar] [CrossRef]
- Samson, W.K.; Murphy, T.C. Adrenomedullin inhibits salt appetite. Endocrinology 1997, 138, 613–616. [Google Scholar] [CrossRef][Green Version]
- Antunes-Rodrigues, J.; Mc Cann, S.M.; Samson, W.K. Central administration of atrial natriuretic factor inhibits saline preference in the rat. Endocrinology 1986, 118, 1726–1728. [Google Scholar] [CrossRef] [PubMed]
- Menani, J.V.; Johnson, A.K. Cholecystokinin actions in the parabrachial nucleus effects in thirst and salt appetite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1998, 275, R1431–R1437. [Google Scholar] [CrossRef]
- Menani, J.V.; Thunhorst, R.L.; Johnson, A.K. Lateral parabrachial nucleus and serotonergic mechanisms in the control of salt appetite in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1996, 270, R162–R168. [Google Scholar] [CrossRef]
- Weisinger, R.S.; Blair-Weat, J.R.; Denton, D.A.; Tarjan, E. Central administration of somatostatin suppress the stimulated sodium intake of sheep. Brain Res. 1991, 543, 213–218. [Google Scholar] [CrossRef]
- Simpson, J.B.; Routtemberg, A. Subfornical organ: Site of drinking elicitation by angiotensin II. Science 1973, 181, 1172–1175. [Google Scholar] [CrossRef]
- Iovino, M.; Steardo, L. Vasopressin release to central and peripheral angiotensin II in rats with lesions of the subfornical organ. Brain Res. 1984, 322, 365–368. [Google Scholar] [CrossRef]
- Iovino, M.; Steardo, L. Thirst and vasopressin secretion following central administration of angiotensin II in rats with lesions of the septal area and subfornical organ. Neuroscience 1985, 15, 61–67. [Google Scholar] [CrossRef]
- De Luca, L.A., Jr.; Galaverna, O.; Schulkin, J.; Yan, S.Z.; Epstein, A.N. The antero-ventral wall of the third ventricle and the angiotensinergic component of need-induced sodium intake in the rat. Brain Res. Bull. 1992, 28, 73–87. [Google Scholar] [CrossRef]
- Thunhorst, R.L.; Erlich, K.J.; Simpson, J.E. Subfornical organ participate in salt appetite. Behav. Neurosci. 1990, 104, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Fitts, A.; Freece, J.A.; Van Bebber, J.E.; Zierath, D.K.; Bassett, J.E. Effect of forebrain circumventricular organ ablation on drinking or salt appetite after sodium depletion or hypernatremia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1325–R1334. [Google Scholar] [CrossRef]
- Noda, M. The subfornical organ, a specialized sodium channel and the sensing of sodium levels in the brain. Neuroscientist 2006, 12, 80–91. [Google Scholar] [CrossRef]
- Hiyama, T.Y.; Watanabe, E.; Okada, H.; Noda, M. The subfornical organ is the primary locus of sodium-level sensing by Na, sodium channels for the control of salt-intake behavior. J. Neurosci. 2004, 24, 9276–9281. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Watanabe, E.; Hiyama, T.Y.; Nagakura, A.; Fujikama, A.; Okado, H.; Yanagawa, Y.; Obata, K.; Noda, M. Glial Nax channels control lactate signaling to neuron for brain [Na+] sensing. Neuron 2007, 54, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Iovino, M.; De Caro, G.; Massi, M.; Steardo, L.; Poenaru, S. Muscimol inhibits ADH release induced by hypertonic sodium chloride in rats. Pharmacol. Biochem. Behav. 1983, 19, 335–338. [Google Scholar] [CrossRef]
- Matsuda, T.; Hiyama, T.Y.; Niimura, T.; Fukamian, A.; Kobayashi, K.; Nada, M. Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nat. Neurosci. 2017, 20, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.E. The effect of remaining area postrema on the sodium and potassium balances and consumptions in the rat. Brain Res. 1985, 359, 224–232. [Google Scholar] [CrossRef]
- Iovino, M.; Papa, M.; Monteleone, P.; Steardo, L. Neuroanatomical and biochemical evidence for the involvement of the area postrema in the regulation of vasopressin release in rats. Brain Res. 1988, 447, 178–182. [Google Scholar] [CrossRef]
- Curtis, K.S.; Huang, W.; Sved, A.F.; Verbalis, J.G.; Stricker, E.M. Impaired osmoregulatory responses in rats with area postrema lesions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 277, R209–R219. [Google Scholar] [CrossRef] [PubMed]
- Geerling, J.C.; Loewy, A.D. Sodium depletion activates the aldosterone-sensitive neurons in the NTS independently of thirst. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1338–R1348. [Google Scholar] [CrossRef][Green Version]
- Fazan, F.S.; Colombari, E.; Loewy, A.D.; Geerling, J.C. Despite increasing aldosterone, elevated potassium is not necessary for activating aldosterone-sensitive HSD2 neurons or sodium appetite. Physiol. Rep. 2021, 9, e14714. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, S.; Resch, J.M.; Narayan, S.V.; Peltekian, L.; Iverson, G.N.; Karthik, S.; Geerling, J.C. Aldosterone-sensitive HSD2 neurons in mice. Brain Struct. Funct. 2019, 224, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Jarvie, B.C.; Palmiter, R. HSD2 neurons in the hindbrain drive sodium appetite. Nat. Neurosci. 2017, 20, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Resch, J.M.; Feuselam, H.; Madara, J.C.; Wu, C.; Campbell, J.N.; Lyubetskaia, A.; Dawes, B.A.; Tsai, L.T.; Li, M.M.; Livneh, Y. Aldosterone-sensing neurons in the nucleus tractus solitarius exhibit state-dependent pacemaker activity and drive sodium appetite via synergy with angiotensin II signaling. Neuron 2017, 96, 190–206. [Google Scholar] [CrossRef]
- Contreras, R.J.; Stetson, P.W. Changes in salt intake after lesions of the area postrema and the nucleus of the solitary tract in rats. Brain Res. 1981, 211, 355–366. [Google Scholar] [CrossRef]
- Sequeira, S.M.; Geerling, J.C.; Loewy, A.D. Local inputs to aldosterone-sensitive neurons of the nucleus tractus solitarius. Neuroscience 2006, 141, 1995–2005. [Google Scholar] [CrossRef]
- Geerling, J.C.; Loewy, A.D. Central regulation of sodium appetite. Exp. Physiol. 2008, 93, 177–209. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Augustine, V.; Zhao, Y.; Ebisen, H.; Ho, B.; Okay, Y. Chemosensory modulation of neural circuits for sodium appetite. Nature 2019, 568, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Augustine, V.; Lee, S.; Oka, Y. Neural control and modulation of thirst, sodium appetite and hunger. Cell 2020, 180, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sunn, N.; Mc Kinley, M.J.; Oldfield, B.J. Circulating angiotensin II activates neurons in circumventricular organs of the lamina terminalis that project to the bed nucleus of the stria terminalis. J. Neuroendocrinol. 2003, 15, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Alden, M.; Besson, J.M.; Bernard, J.F. Organization of the efferent projections from the pontine parabrachial area to the bed nucleus of the stria terminalis and neighboring regions: A PHA study in the rat. J. Comp. Neurol. 1994, 341, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Karimnamazi, H.; Travers, J.B. Differential projections from gustatory responsive regions of the parabrachial nucleus to the medulla and forebrain. Brain Res. 1998, 813, 283–302. [Google Scholar] [CrossRef]
- Geerling, J.C.; Loewy, A.D. Aldosterone-sensitive neurons in the nucleus of the solitary tract: Efferent projections. J. Comp. Neurol. 2006, 497, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.W.; Petrovich, G.D.; Watts, A.G.; Swanson, L.W. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J. Comp. Neurol. 2001, 436, 430–455. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Quartermain, D. Sodium chloride intake of adrenalectomized rats with lateral hypothalamic lesions. Am. J. Physiol. 1967, 212, 113–118. [Google Scholar] [CrossRef]
- Sticker, E.M.; Verbalis, J.G. Inhibition of salt appetite in rats by central oxytocin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R487–R488. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.J.; Maki, R.; Nardozzi, J.; Schulkin, J. The effects of lesions of the bed nucleus of the stria terminalis on sodium appetite. Acta Neurobiol. Exp. 1994, 54, 253–257. [Google Scholar]
- Zardetto-Smith, A.M.; Beltz, T.G.; Johnson, A.K. Role of the central nucleus of the amygdala and bed nucleus of the stria terminalis in experimentally-induced salt-appetite. Brain Res. 1994, 645, 123–134. [Google Scholar] [CrossRef]
- Gentil, C.G.; Antunes-Rorigues, J.; Negro-Vilar, A.; Covian, M.R. Role of amygdaloid complex in sodium chloride and motor intake in the rat. Ohysiol. Behav. 1968, 3, 981–985. [Google Scholar] [CrossRef]
- Gentil, C.G.; Mogenson, G.J.; Stevenson, J.A. Electrical stimulation of septum, hypothalamus and amygdala and sodium preferences. Am. J. Physiol. 1971, 220, 1172–1177. [Google Scholar] [CrossRef]
- Zhang, D.M.; Epstein, A.N.; Schulkin, J. Medial region of the amygdala: Involvement in adrenal-steroid-induced salt-appetite. Brain Res. 1993, 600, 20–26. [Google Scholar] [CrossRef]
- Bernard, J.F.; Alden, M.; Besson, J.M. The organization of the efferent projections from the pontine parabrachial area to the amygdaloid complex: A Phaselous Vulgaris leucoagglutinin (PHA-L) study in the rat. J. Comp. Neurol. 1993, 329, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Geerling, G.C.; Loewy, A.D. Aldosterone-sensitive neurons in the nucleus of the solitary tract: Bidirectional connections with the central nucleus of the amygdala. J. Comp. Neurol. 2006, 497, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.R.; McEwen, B.S.; Fluharthy, S.J.; Ma, L.Y. The amygdala: Site of genomic and nongenomic arousal of aldosterone-induced sodium intake. Kidney Int. 2000, 57, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, D.A.; Hostage, G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the rat. Exp. Brain Res. 1978, 32, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Tsumori, T.; Oka, T.; Yokota, S. Amygdaloid axon terminals are in contact with trigeminal premotor neurons in the parvicellular reticular formation of the rat medulla oblongata. Brain Res. 2004, 1016, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.B.; Norgren, R. Afferent projections to the oral motor nuclei in the rat. J. Comp. Neurol. 1983, 220, 280–298. [Google Scholar] [CrossRef] [PubMed]
- Cunnigham, E.T.; Sawchenko, P.E. Dorsal medullary pathways subserving oromotor reflexes in the rat: Implications for the central neural control of swallowing. J. Comp. Neurol. 2000, 417, 448–466. [Google Scholar] [CrossRef]
- Black, S.L.; Mogenson, J.G. The regulation of serum sodium in septal-lesioned rats: A test of two hypotheses. Physiol. Behav. 1973, 10, 379–384. [Google Scholar] [CrossRef]
- Blass, E.M.; Nussbaum, A.I.; Hanson, D.G. Septal hyperdipsia: Specific enhancement of drinking to angiotensin in rats. J. Comp. Physiol. Psychol. 1974, 81, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.N. Neuroendocrinology of thirst and salt appetite. In Frontiers in Neuroendocrinology; Ganong, W.F., Martini, L., Eds.; Raven Press: New York, NY, USA, 1978; Volume 5, pp. 101–134. [Google Scholar]
- Harvey, J.A.; Hunt, H.F. Effect of septal lesions on thirst in the rat as indicated by water consumption and operant response for water reward. J. Comp. Physiol. Psychol. 1965, 59, 49–56. [Google Scholar] [CrossRef]
- Lubar, J.F.; Boyce, B.A.; Schaeffer, C.F. Etiology of polydipsia and polyuria in rats with septal lesions. Physiol. Behav. 1968, 3, 289–292. [Google Scholar] [CrossRef]
- Lubar, J.F.; Schaeffer, C.F.; Boyce, B.A. The role of the septal area in the regulation of water intake and associated motivational behavior. Ann. N. Y. Acad. Sci. 1969, 157, 875–893. [Google Scholar] [CrossRef] [PubMed]
- Iovino, M.; Steardo, L. The role of the septal area in the regulation of drinking behavior and plasma ADH secretion. In The Physiology of Thirst and Sodium Appetite; De Caro, G., Epstein, A.N., Massi, M., Eds.; Plenum Pub. Corp.: New York, NY, USA, 1986; pp. 367–374. [Google Scholar]
- Smardencas, A.; Denton, D.A.; McKinley, M.J. Hyperdipsia in the sheep bearing lesions in the medial septal nucleus. Brain Res. 2021, 1752, 147223. [Google Scholar] [CrossRef] [PubMed]
- Tangapregassom, A.M.; Tangapregassom, A.M.; Soulairac, A. Noyaux septaux et équilibre hydrique: Analyse électrophysique et comportamentale. Ann. Endocrinol. 1974, 35, 153–155. [Google Scholar]
- Lorens, S.A.; Sorensen, J.P.; Harvey, J.A. Lesions in the nuclei accumbens septi of the rat. Behavioral and neurochemical effects. J. Comp. Physiol. Psychol. 1970, 73, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Iovino, M.; Poenaru, S.; Annunziato, L. Basal and thirst-evoked vasopressin secreion in rats with electrolytic lesion of the medio-ventral septal area. Brain Res. 1983, 258, 123–126. [Google Scholar] [CrossRef]
- Tangapregassom, A.M.; Tangapregassom, M.J.; Soulairac, A.; Soulairac, M.L. Effects of septal lesions on the ultrastructure of the supra-optic nucleus. Ann. Endocrinol. 1974, 35, 149–152. [Google Scholar]
- Zaborszky, L.; Leranth, C.; Makara, G.B.; Palkovits, M. Quantitative studies on the supraoptic nucleus in the rat. II. Afferent fiber connections. Exp. Brain Res. 1975, 22, 525–540. [Google Scholar] [PubMed]
- Faraci, W.S.; Yang, B.V.; O’Rouke, D.; Spencer, R.W. Septal projections to nuclei functioning in oxytocin release. Am. J. Anat. 1967, 120, 605–610. [Google Scholar]
- Iovino, M.; Papa, M.; Monteleone, P.; Steardo, L. Changes in magnocellular neurosecretory activity following septal forebrain lesions: Morphological and biochemical data. Neuroendocrinol. Lett. 1989, 11, 361–369. [Google Scholar]
- Sirett, N.E.; McLean, A.S.; Bray, J.J.; Hubbard, J.I. Distribution of angiotensin II receptors in rat brain. Brain Res. 1977, 122, 299–312. [Google Scholar] [CrossRef]
- Landas, S.; Phillips, M.I.; Stamler, J.F.; Raizada, M.K. Visualization of specific angiotensin II binding sites in the brain by florescent microscopy. Science 1980, 210, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Iovino, M.; Steardo, L. Effets des lésions septales sur la reponse de la vasopressine à l’angiotensine II. Ann. Endocrinol. 1985, 46, 113–117. [Google Scholar]
- De Kloet, E.R.; van Acker, S.A.B.E.; Siburg, R.M.; Oitzl, M.S.; Meijer, O.C.; Ramouni, K.; de Jong, W. Brain mineralcorticoid receptors and centrally regulated functions. Kidney Int. 2000, 57, 1329–1336. [Google Scholar] [CrossRef]
- Stricker, E.M. Thirst and sodium appetite after colloid treatment in rats with septal lesions. Behav. Neurosci. 1984, 98, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Antunes, V.R.; Camargo, G.M.P.A.; Saad, R.; Saad, W.A.; Luiz, A.C.; Camargo, L.A.A. Role of angiotensin II and vasopressin receptors within the supraoptic nucleus in water and sodium intake induced by the injection of angiotensin II into the medial septal area. Braz. J. Med. Biol. Res. 1998, 31, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.; Bassi, M.; Nakamura, N.B.; Schoorlemmer, G.H.; Menani, J.; Colombari, E. Hindbrain mineralcorticoid mechanisms on sodium appetite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R252–R259. [Google Scholar] [CrossRef]
- Scallert, T. Adipsia produced by lateral hypothalamic lesions: Facilitation of recovery by preoperative restriction of water intake. J. Comp. Physiol. Psychol. 1982, 96, 604–614. [Google Scholar] [CrossRef]
- Mendelson, J. Lateral hypothalamic stimulation in satiated rats: The rewarding effects of self-induced drinking. Science 1967, 157, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. Effect of dorsolateral hypothalamic lesions on sodium appetite elicited by desoxycorticosterone and by acute hyponatremia. J. Comp. Physiol. Psychol. 1964, 58, 394–402. [Google Scholar] [CrossRef]
- Watts, A.G. Osmotic stimulation differentially affects cellular levels of corticotrpin-releasing hormone and neurotensin/neuromedin N mRNAs in the lateral hypothalamic area and central nucleus of the amygdala. Brain Res. 1992, 581, 208–216. [Google Scholar] [CrossRef]
- Tarjan, E.; Denton, D.A.; Weisinger, R. Effect of CRF, ACTH, and adrenal steroids on sodium intake and excretion of rabbits. Kidney Int. 1992, 37, S97–S101. [Google Scholar]
- Kelly, A.B.; Watts, A.G. The region of the pontine parabrachial nucleus is a major target of dehydration sensitive CRH neurons in the rat lateral hypothalamic area. J. Comp. Neurol. 1998, 394, 48–63. [Google Scholar] [CrossRef]
- Hagan, J.J.; Tonnaer, J.A.; Broekkamp, C.L. Cholinergic stimulation of drinking from the lateral hypothalamus: Indication for M2 muscarinic receptor mediation. Pharmacol. Biochem. Behav. 1987, 26, 771–779. [Google Scholar] [CrossRef]
- Carelli, R.M. The nucleus accumbens an reward: Neurophysiological investigations in behaving animals. Behav. Cogn. Neurosci. Rev. 2002, 1, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Roitman, M.F.; Na, E.; Anderson, G.; James, T.A.; Bernstein, I.L. Induction of a salt appetite alters dendritic morphology in nucleus accumbens and sensitizes rats to amphetamine. J. Neursci. 2002, 22, 225. [Google Scholar] [CrossRef]
- Roitman, M.F.; Patterson, T.A.; Sakai, R.R.; Bernstein, I.L.; Fighwicz, D.P. Sodium depletion an aldosterone decrease dopamine transporter activity in nucleus accumbens but not striatum. Am. J. Physiol. 1999, 276 Pt 2, R1339–R1451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Stellar, E.; Epstein, A.N. Together intracranial angiotensin and systemic mineralcorticoid produce avidity for salt in the rat. Physiol. Behav. 1984, 32, 677–681. [Google Scholar] [CrossRef]
- Grafe, L.A.; Flanagan-Cato, L.M. Differential effects of mineralcorticoid and angiotensin II on incentive and mesolimbic activity. Horm. Behav. 2016, 79, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Prakash, M.R.; Norgren, R. Comparing salt appetites: Induction with intracranial hormones or dietary sodium restriction. Brain Res. Bull. 1991, 27, 397–401. [Google Scholar] [CrossRef]
- Wang, T.; Edwards, G.L. Differential effects of dorsomedial medulla lesion size on ingestive behavior in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997, 273, R1299–R1308. [Google Scholar] [CrossRef] [PubMed]
- Schreihofer, A.M.; Anderson, B.K.; Schiltz, J.C.; Xu, L.; Sved, A.F.; Stricker, M.E. Thirst and salt appetite elicited by hypovolemia in rats with chronic lesions of the nucleus of the solitary tract. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 276, R251–R258. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.W.; Swanson, L.W. Projections from the rhomboid nucleus of the bed nucleus of the stria terminalis. Implications for cerebral hemisphere regulation of ingestive behaviors. J. Comp. Neurol. 2003, 463, 434–472. [Google Scholar] [CrossRef]
- Geerling, J.C.; Engeland, W.C.; Kawata, M.; Loewy, A.D. Aldosterone target neurons in the nucleus tractus solitarius drive sodium appetite. J. Neurosci. 2006, 26, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Cullinam, W.E.; Herman, J.P.; Watson, S.J. Ventral subicular interaction with the hypothalamic paraventricular nucleus: Evidence for a relay in the be nucleus of the stria terminalis. J. Comp. Neurfol. 1993, 332, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sawchenko, P.C.; Swanson, L.W. Immunohistochemical identification of neuron in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J. Comp. Neurol. 1982, 205, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Camacho, A.; Phillips, M.I. Horseradish peroxidase study in rat of the neural connections of the organum vasculosum of the lamina terminalis. Neurosci. Lett. 1981, 25, 201–204. [Google Scholar] [CrossRef]
- Feld, J.; Deutch, A.Y. Anatomical substrates of orexin-dopamine interactions: Lateral hypothalamic projections to the ventral tegmental area. Neuroscience 2002, 11, 287–379. [Google Scholar]
- Hoebel, B.G.; Hernandez, L.; Schwartz, D.H.; Mark, G.P.; Hunter, G.A. Microdialysis studies of brain norepinephrine, serotonin, and dopamine release during ingestive behavior, theoretical and clinical implications. Ann. N. Y. Acad. Sci. 1989, 575, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Geerling, J.C.; Lowy, A.D. Aldosterone in the brain. Am. J. Physiol. Renal. Physiol. 2009, 297, F559–F576. [Google Scholar] [CrossRef] [PubMed]
- Heck, G.L.; Mierson, S.; De Simone, J.A. Salt taste transduction occurs through an amiloride-sensitive sodium transport pathway. Science 1984, 223, 403–405. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iovino, M.; Messana, T.; Lisco, G.; Vanacore, A.; Giagulli, V.A.; Guastamacchia, E.; De Pergola, G.; Triggiani, V. Signal Transduction of Mineralocorticoid and Angiotensin II Receptors in the Central Control of Sodium Appetite: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 11735. https://doi.org/10.3390/ijms222111735
Iovino M, Messana T, Lisco G, Vanacore A, Giagulli VA, Guastamacchia E, De Pergola G, Triggiani V. Signal Transduction of Mineralocorticoid and Angiotensin II Receptors in the Central Control of Sodium Appetite: A Narrative Review. International Journal of Molecular Sciences. 2021; 22(21):11735. https://doi.org/10.3390/ijms222111735
Chicago/Turabian StyleIovino, Michele, Tullio Messana, Giuseppe Lisco, Aldo Vanacore, Vito Angelo Giagulli, Edoardo Guastamacchia, Giovanni De Pergola, and Vincenzo Triggiani. 2021. "Signal Transduction of Mineralocorticoid and Angiotensin II Receptors in the Central Control of Sodium Appetite: A Narrative Review" International Journal of Molecular Sciences 22, no. 21: 11735. https://doi.org/10.3390/ijms222111735
APA StyleIovino, M., Messana, T., Lisco, G., Vanacore, A., Giagulli, V. A., Guastamacchia, E., De Pergola, G., & Triggiani, V. (2021). Signal Transduction of Mineralocorticoid and Angiotensin II Receptors in the Central Control of Sodium Appetite: A Narrative Review. International Journal of Molecular Sciences, 22(21), 11735. https://doi.org/10.3390/ijms222111735