
Whey-Derived Peptides at the Heart of the COVID-19 Pandemic


Abstract
1. Introduction
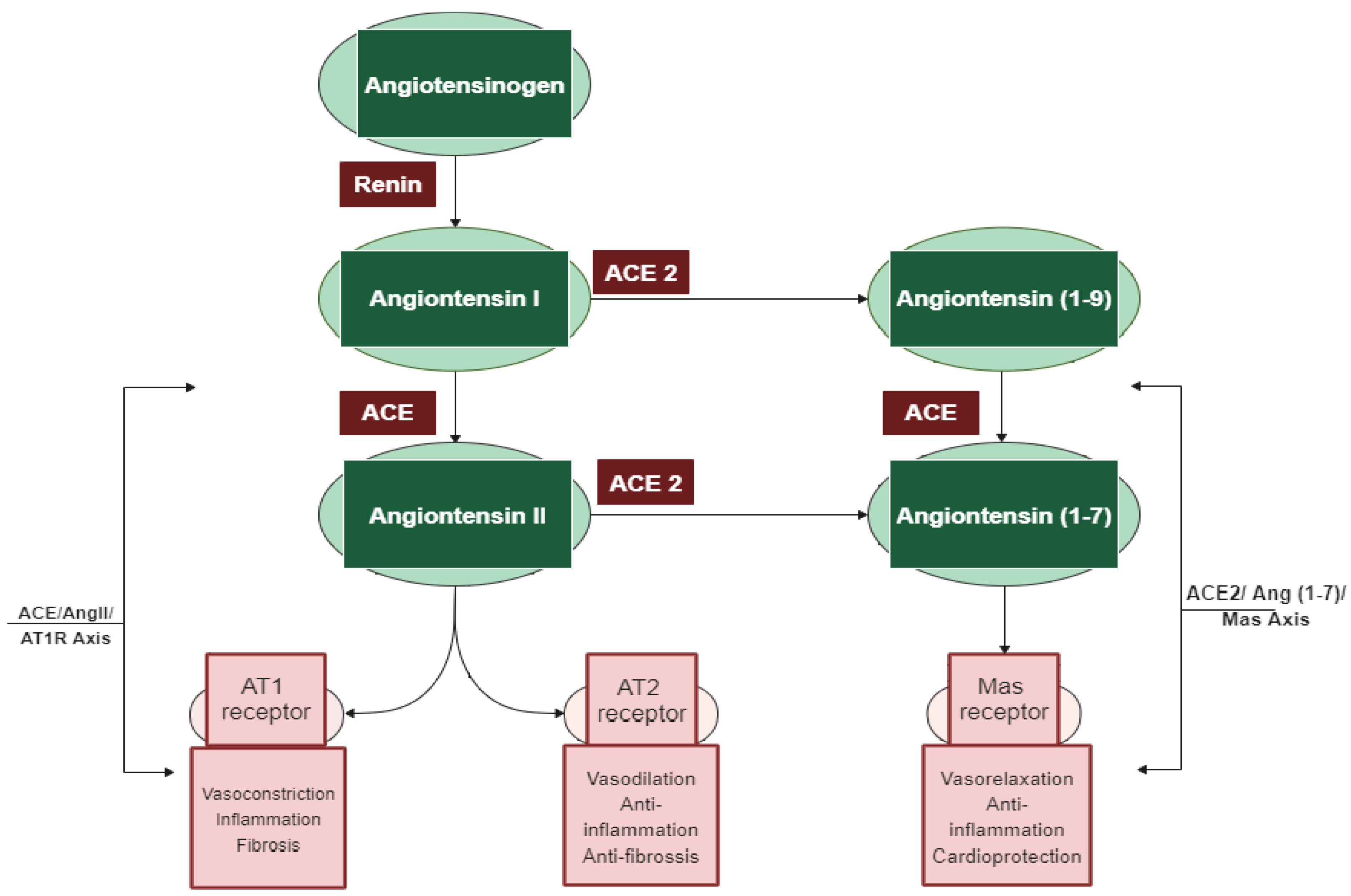
1.1. The Classical and Counter-Regulatory Renin–Angiotensin System (RAS) Pathways
1.2. Characteristics of SARS-CoV-2
1.3. Controversies Regarding the Role of ACE2 in COVID-19
1.4. Whey-Derived Peptides as Promising Therapeutic Candidates
2. Results and Discussion
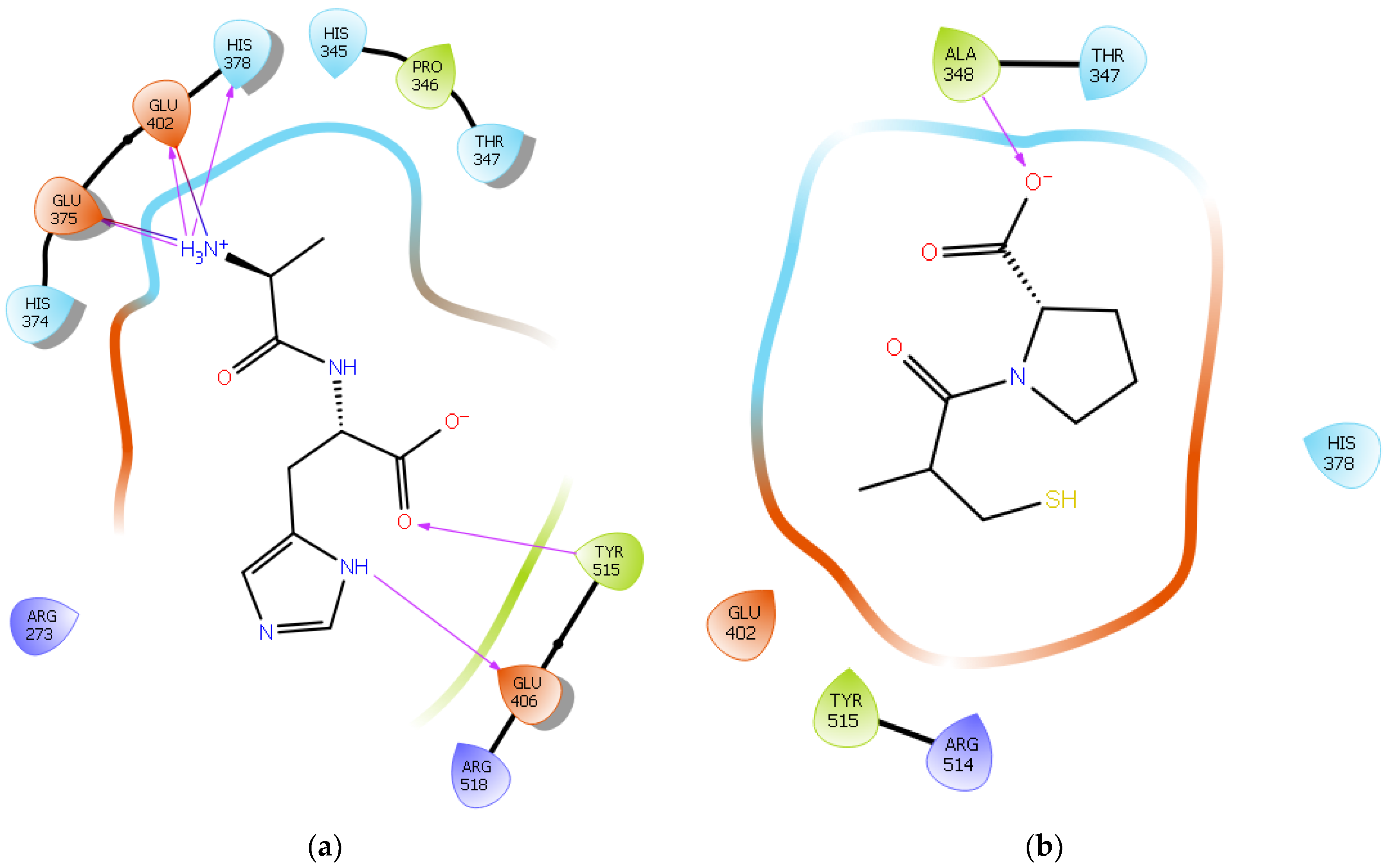
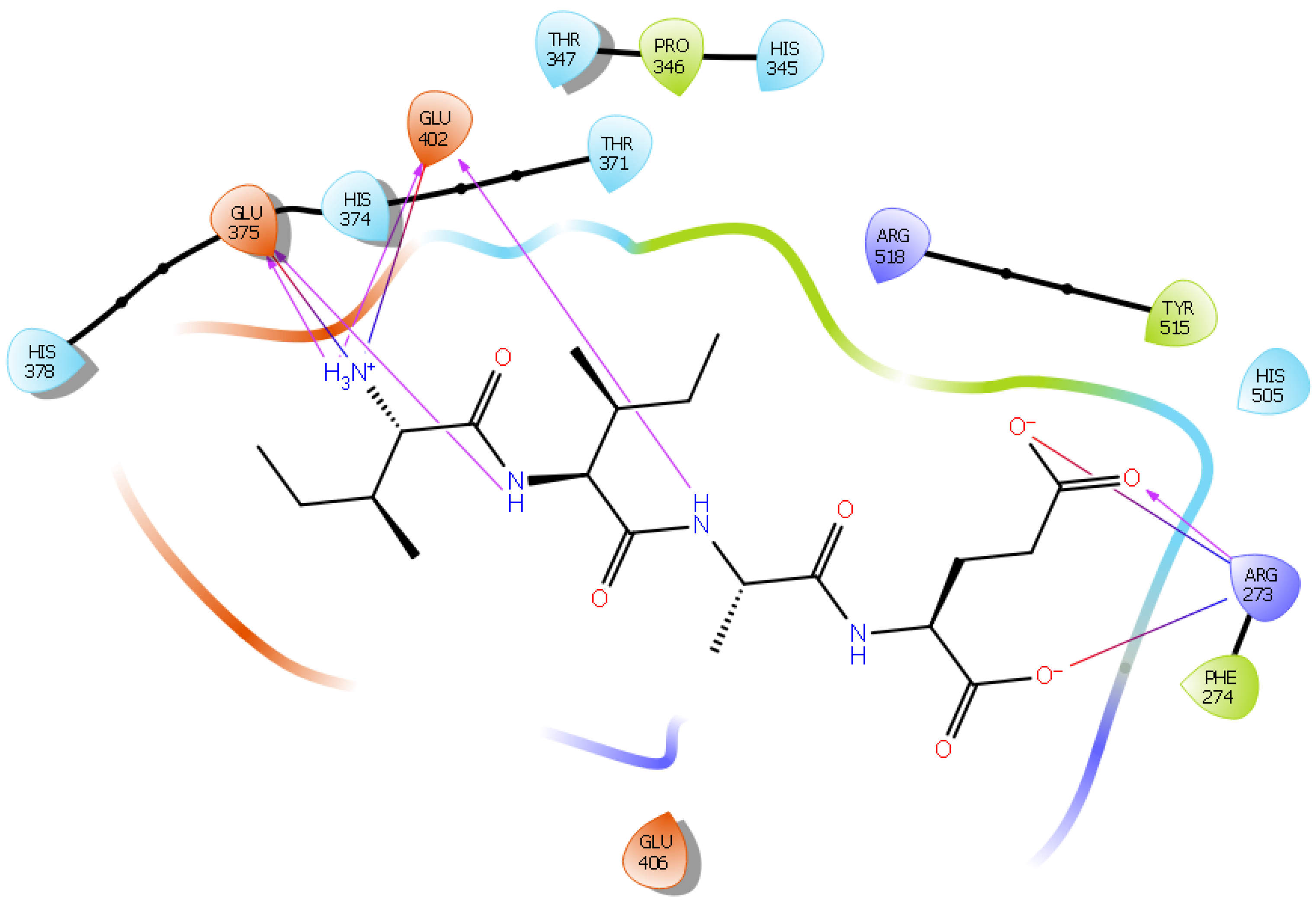
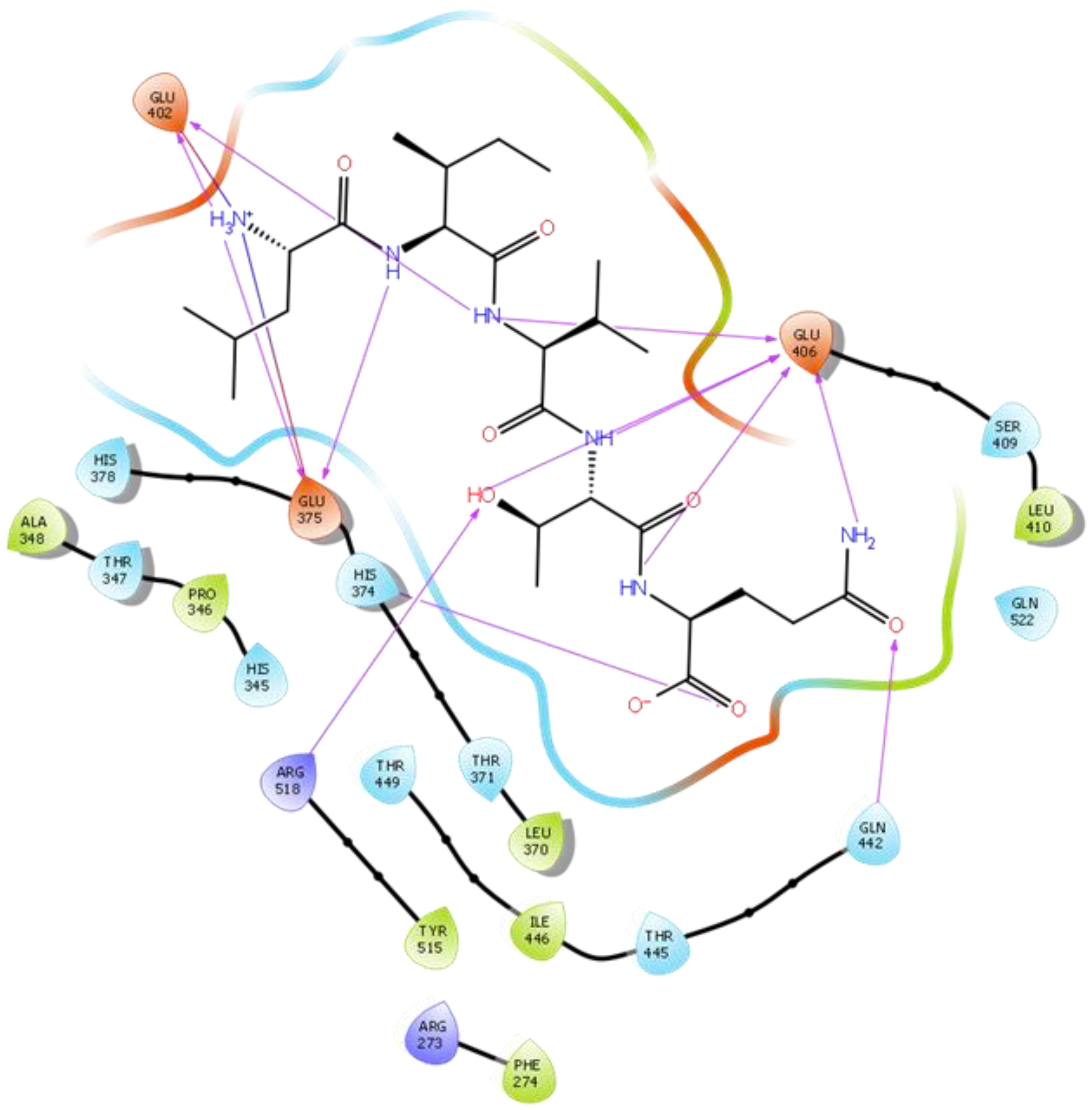
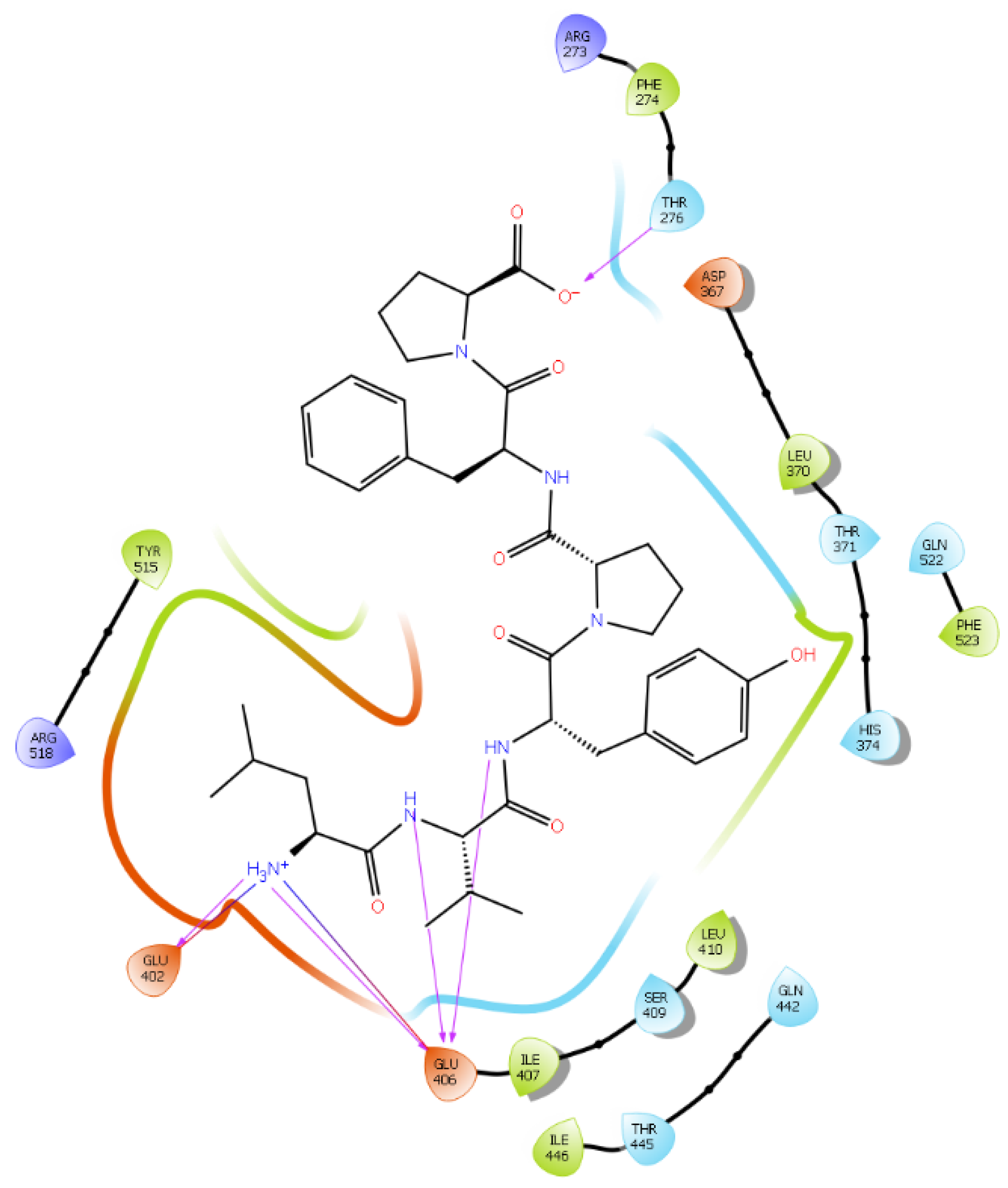
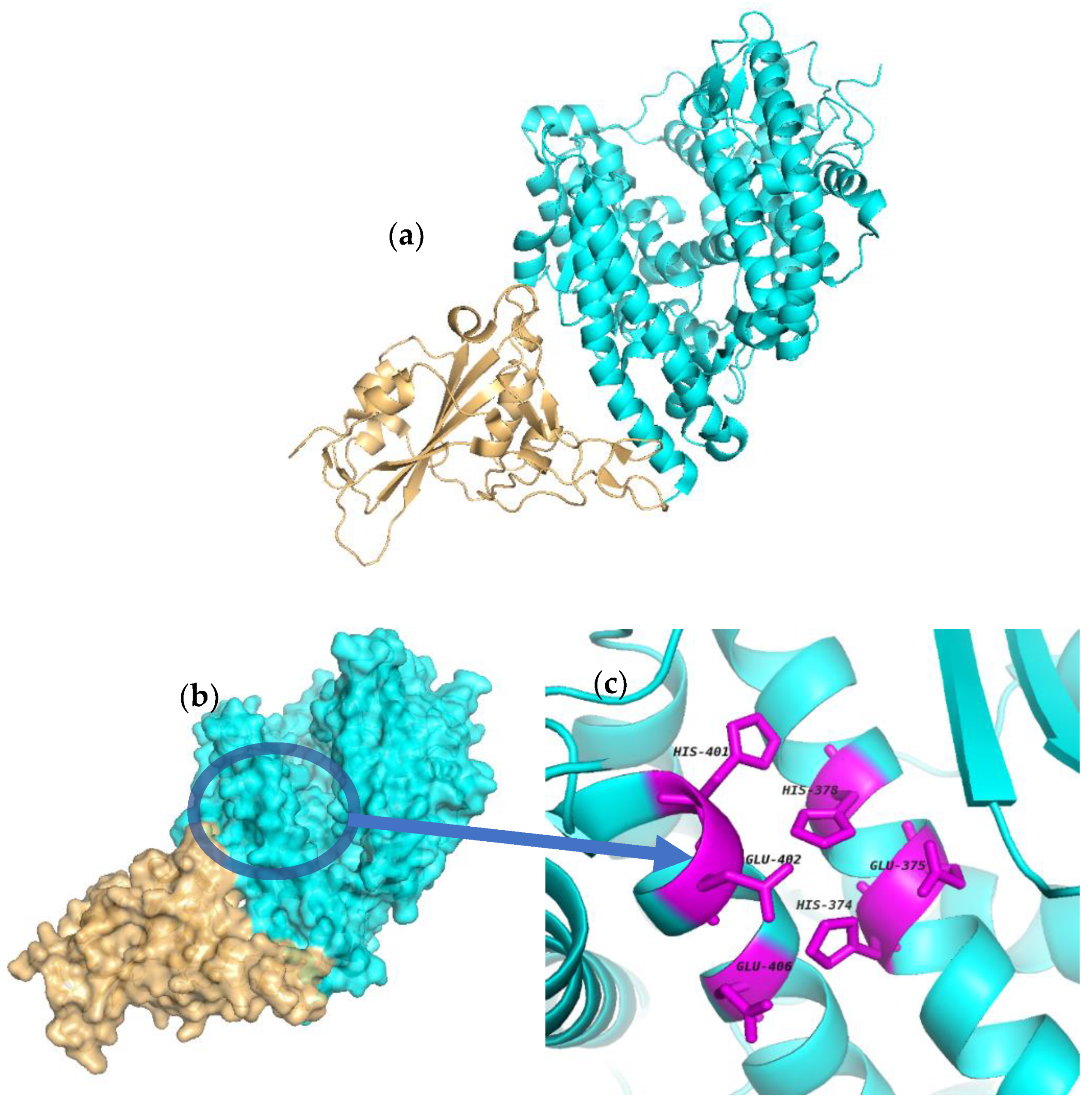
2.1. Molecular Docking
2.2. ACE and ACE2
2.3. Potential Use of ACE Inhibitors in the Treatment of COVID-19
3. Materials and Methods
3.1. Structure Similarity between 1RL4 and 6M0J
3.2. Docking Procedure
3.3. Docking Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization (WHO). Cardiovascular Diseases (CVDs): Key Facts. Available online: https://www.who.int/news-room/factsheets/detail/cardiovascular-diseases-(cvds) (accessed on 20 June 2021).
- Townsend, N.W.J.; Bhatnagar, P.; Wickramasinghe, K.; Rayner, M. Cardiovascular Disease Statistics, 2014; British Heart Foundation: London, UK, 2014. [Google Scholar]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef]
- Borghi, C.; Force, S.T.; Rossi, F.; Force, S.T. Role of the renin-angiotensin-aldosterone system and its pharmacological inhibitors in cardiovascular diseases: Complex and critical issues. High Blood Press. Cardiovasc. Prev. 2015, 22, 429–444. [Google Scholar] [CrossRef]
- Chappell, M.C. Biochemical evaluation of the renin-angiotensin system: The good, bad, and absolute? Am. J. Physiol. Circ. Physiol. 2016, 310, H137–H152. [Google Scholar] [CrossRef]
- Griendling, K.; Murphy, T.J.; Alexander, R.W. Molecular biology of the renin-angiotensin system. Circulation 1993, 87, 1816–1828. [Google Scholar] [CrossRef]
- Acharya, K.R.; Sturrock, E.D.; Riordan, J.F.; Ehlers, M.R.W. Ace revisited: A new target for structure-based drug design. Nat. Rev. Drug Discov. 2003, 2, 891–902. [Google Scholar] [CrossRef]
- Li, G.H.; Le, G.W.; Shi, Y.H.; Shrestha, S. Angiotensin I–converting enzyme inhibitory peptides derived from food proteins and their physiological and pharmacological effects. Nutr. Res. 2004, 24, 469–486. [Google Scholar] [CrossRef]
- Wei, L.; Alhenc-Gelas, F.; Corvol, P.; Clauser, E. The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem. 1991, 266, 9002–9008. [Google Scholar] [CrossRef]
- Sturrock, E.D.; Natesh, R.; Van Rooyen, J.M.; Acharya, K.R. Structure of angiotensin I-converting enzyme. Cell. Mol. Life Sci. 2004, 61, 2677–2686. [Google Scholar] [CrossRef]
- Carey, R.M.; Siragy, H.M. Newly Recognized Components of the Renin-Angiotensin System: Potential Roles in Cardiovascular and Renal Regulation. Endocr. Rev. 2003, 24, 261–271. [Google Scholar] [CrossRef]
- Natesh, R.; Schwager, S.L.U.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nat. Cell Biol. 2003, 421, 551–554. [Google Scholar] [CrossRef]
- Tzakos, A.G.; Galanis, A.S.; Spyroulias, G.A.; Cordopatis, P.; Manessi-Zoupa, E.; Gerothanassis, I.P. Structure–function discrimination of the N-and C-catalytic domains of human angiotensin-converting enzyme: Implications for Cl–activation and peptide hydrolysis mechanisms. Protein Eng. 2003, 16, 993–1003. [Google Scholar] [CrossRef][Green Version]
- Cat, A.N.D.; Touyz, R.M. Cell Signaling of Angiotensin II on Vascular Tone: Novel Mechanisms. Curr. Hypertens. Rep. 2011, 13, 122–128. [Google Scholar] [CrossRef]
- Padia, S.H.; Carey, R.M. AT2 receptors: Beneficial counter-regulatory role in cardiovascular and renal function. Pflügers Archiv-Eur. J. Physiol. 2013, 465, 99–110. [Google Scholar] [CrossRef]
- Santos, R.A. Angiotensin-(1–7). Hypertension 2014, 63, 1138–1147. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A Novel Angiotensin-Converting Enzyme–Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Acton, S. Hy-Drolysis of Biological Peptides by Human Angiotensin-Converting Enzyme-Related Carboxypep-Tidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Simoes e Silva, A.C.S.; Maric, C.; Silva, D.M.R.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef]
- Ferreira, A.; Santos, R.; Almeida, A. Angiotensin-(1–7) improves the post-ischemic function in isolated perfused rat hearts. Braz. J. Med. Biol. Res. 2002, 35, 1083–1090. [Google Scholar] [CrossRef]
- D’Ardes, D.; Boccatonda, A.; Rossi, I.; Guagnano, M.T.; Santilli, F.; Cipollone, F.; Bucci, M. COVID-19 and RAS: Unravelling an Unclear Relationship. Int. J. Mol. Sci. 2020, 21, 3003. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.J.; Santos, R.A.S. Cardiovascular actions of angiotensin-(1-7). Braz. J. Med. Biol. Res. 2005, 38, 499–507. [Google Scholar] [CrossRef]
- World Health Organization. Coronavirus Disease (COVID-19) Outbreak Situation. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 14 October 2021).
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Bi, Y. Genomic Characterisa-Tion and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Wei, Q.; National Pathogen Resource Center, Chinese Center for Disease Control and Prevention; Wang, Y.; Ma, J.; Han, J.; Jiang, M.; Zhao, L.; Ye, F.; Song, J.; Liu, B.; et al. Description of the First Strain of 2019-NCoV, C-Tan-NCoV Wuhan Strain—National Pathogen Resource Center, China, 2020. China CDC Wkly. 2020, 2, 81–82. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Hu, Y.; Song, Z.-G.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. Complete genome characterisation of a novel coronavirus associated with severe human respiratory disease in Wuhan, China. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Veesler, D. Structural Insights into Coronavirus Entry; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 105, pp. 93–116. [Google Scholar]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; McLel-lan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Confor-Mation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 Productively Infects Human Gut Enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Remuzzi, G. Should COVID-19 Concern Nephrologists? Why and to What Extent? The Emerging Impasse of Angiotensin Blockade. Nephron 2020, 144, 213–221. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. A hypothesis for pathobiology and treatment of COVID-19: The centrality of ACE1 / ACE2 imbalance. Br. J. Pharmacol. 2020, 177, 4825–4844. [Google Scholar] [CrossRef]
- Saadah, L.M.; Abu Deiab, G.I.; Al-Balas, Q.; Basheti, I.A. Carnosine to Combat Novel Coronavirus (nCoV): Molecular Docking and Modeling to Cocrystallized Host Angiotensin-Converting Enzyme 2 (ACE2) and Viral Spike Protein. Molecules 2020, 25, 5605. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.F.; Lopes, F.E.; Amaral, J.L.; Freitas, C.D.; Oliveira, J.T. A molecular docking study revealed that synthetic peptides induced conformational changes in the structure of SARS-CoV-2 spike glycoprotein, disrupting the interaction with human ACE2 receptor. Int. J. Biol. Macromol. 2020, 164, 66–76. [Google Scholar] [CrossRef]
- Srivastava, N.; Garg, P.; Srivastava, P.; Seth, P.K. A Molecular Dynamics Simula-Tion Study of the ACE2 Receptor with Screened Natural Inhibitors to Identify Novel Drug Candi-Date against COVID-19. PeerJ 2021, 9, 11171. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Godoy, I.; Jalil, J.E.; Varas, M.; Collantes, P.; Pinto, M.; Roman, M.; Ramirez, C.; Copaja, M.; Diaz-Araya, G.; et al. Enalapril Attenuates Downregulation of Angiotensin-Converting Enzyme 2 in the Late Phase of Ventricular Dysfunction in Myocardial Infarcted Rat. Hypertension 2006, 48, 572–578. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Yuen, K.Y.; Osterhaus, A.D.M.E.; Stöhr, K. The Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 349, 2431–2441. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Sun, M.L.; Yang, J.M.; Sun, Y.P.; Su, G. Inhibitors of RAS might be a good choice for the therapy of COVID-19 pneumonia. Chin. J. Tuberc. Respir. Dis. 2020, 43, E014. [Google Scholar]
- Vuille-dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; et al. Human Intestine Luminal ACE2 and Amino Acid Trans-Porter Expression Increased by ACE-Inhibitors. Amino Acids 2015, 47, 693–705. [Google Scholar] [CrossRef]
- Zheng, Y.-Y.; Ma, Y.-T.; Zhang, J.-Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Kuster, G.M.; Pfister, O.; Burkard, T.; Zhou, Q.; Twerenbold, R.; Haaf, P.; Widmer, A.F.; Osswald, S. SARS-CoV2: Should Inhibitors of the Renin-Angiotensin System Be Withdrawn in Patients with COVID-19? Eur. Heart J. 2020, 41, 1801–1803. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, D.; Italia, L.; Adamo, M.; Inciardi, R.M.; Lombardi, C.M.; Solomon, S.D.; Metra, M. COVID- 19 and heart failure: From infection to inflammation and angiotensin II stimulation. Searching for evidence from a new disease. Eur. J. Heart Fail. 2020, 22, 957–966. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Karakiulakis, G.; Roth, M. Are Patients with Hypertension and Diabetes Mellitus at Increased Risk for COVID-19 Infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Circ. Physiol. 2020, 318, H1084–H1090. [Google Scholar] [CrossRef]
- Zambelli, V.; Bellani, G.; Borsa, R.; Pozzi, F.; Grassi, A.; Scanziani, M.; Castiglioni, V.; Masson, S.; Decio, A.; Laffey, J.G.; et al. Angiotensin-(1–7) improves oxygenation, while reducing cellular infiltrate and fibrosis in experimental Acute Respiratory Distress Syndrome. Intensiv. Care Med. Exp. 2015, 3, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Kalra, A.; Nowacki, A.S.; Anjewierden, S.; Han, Z.; Bhat, P.; Carmona-Rubio, A.E.; Jacob, M.; Procop, G.W.; Harrington, S.; et al. Association of use of angiotensin-converting enzyme inhibitors and angiotensin IIreceptor blockers with testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.-J.; Xie, J.; Liu, Y.-M.; Zhao, Y.-C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Young, D.; Coupland, C.; Channon, K.M.; Tan, P.S.; A Harrison, D.; Rowan, K.; Aveyard, P.; Pavord, I.D.; Watkinson, P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: Cohort study including 8.3 million people. Heart 2020, 106, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, M.L.; Duan, Z.L.; Liu, F.L.; Jin, L.; Long, C.B.; Lai, R. Dalbavancin Binds ACE2 to Block Its Interaction with SARS-CoV-2 Spike Protein and Is Effec-Tive in Inhibiting SARS-CoV-2 Infection in Animal Models. Cell Res. 2021, 31, 17–24. [Google Scholar] [CrossRef]
- Mouffouk, C.; Mouffouk, S.; Mouffouk, S.; Hambaba, L.; Haba, H. Flavonols as Potential Antiviral Drugs Targeting SARS-CoV-2 Proteases (3CLpro and PLpro), Spike Protein, RNA-Dependent RNA Polymerase (RdRp) and Angiotensin-Converting Enzyme II Receptor (ACE2). Eur. J. Pharmacol. 2021, 891, 173759. [Google Scholar] [CrossRef]
- Brauer, F.; Schmidt, K.; Zahn, R.C.; Richter, C.; Radeke, H.H.; Schmitz, J.E.; Egerer, L. A Rationally Engineered Anti-HIV Peptide Fusion Inhibitor with Greatly Reduced Immu-Nogenicity. Antimicrob. Agents Chemother. 2013, 57, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and Peptide-Based Inhibitors of SARS-CoV-2 Entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of Critical Determinants on ACE2 for SARS-CoV Entry and Development of a Potent Entry Inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Maqueda, D.; Miralles, B.; Recio, I.; Hernández-Ledesma, B. Antihypertensive peptides from food proteins: A review. Food Funct. 2012, 3, 350–361. [Google Scholar] [CrossRef]
- Giromini, C.; Fekete, Á.A.; Givens, D.I.; Baldi, A.; Lovegrove, J.A. Short-Communication: A Comparison of the In Vitro Angiotensin-1-Converting Enzyme Inhibitory Capacity of Dairy and Plant Protein Supplements. Nutrients 2017, 9, 1352. [Google Scholar] [CrossRef]
- Fekete, A.A.; Giromini, C.; Chatzidiakou, Y.; Givens, D.I.; Lovegrove, J.A. Whey Protein Lowers Blood Pressure and Improves Endothelial Function and Lipid Biomarkers in Adults with Prehypertension and Mild Hypertension: Results from the Chronic Whey2Go Ran-Domized Controlled Trial. Am. J. Clin. Nutr. 2016, 104, 1534–1544. [Google Scholar] [CrossRef]
- Fekete, A.A.; Givens, D.I.; Lovegrove, J.A. The Impact of Milk Proteins and Pep-Tides on Blood Pressure and Vascular Function: A Review of Evidence from Human Intervention Studies. Nutr. Res. Rev. 2013, 26, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Welderufael, F.T.; Gibson, T.; Methven, L.; Jauregi, P. Chemical characterisation and determination of sensory attributes of hydrolysates produced by enzymatic hydrolysis of whey proteins following a novel integrative process. Food Chem. 2012, 134, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Chamata, Y.; Watson, K.A.; Jauregi, P. Whey-Derived Peptides Interactions with ACE by Molecular Docking as a Potential Predictive Tool of Natural ACE Inhibitors. Int. J. Mol. Sci. 2020, 21, 864. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]
- Guy, J.L.; Jackson, R.M.; Acharya, K.R.; Sturrock, E.D.; Hooper, A.N.M.; Turner, A.J. Angiotensin-Converting Enzyme-2 (ACE2): Comparative Modeling of the Active Site, Specificity Requirements, and Chloride Dependence. Biochemistry 2003, 42, 13185–13192. [Google Scholar] [CrossRef] [PubMed]
- Teralı, K.; Baddal, B.; Gülcan, H.O. Prioritizing potential ACE2 inhibitors in the COVID-19 pandemic: Insights from a molecular mechanics-assisted structure-based virtual screening experiment. J. Mol. Graph. Model. 2020, 100, 107697. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.L.; Jackson, R.M.; Jensen, H.A.; Hooper, N.; Turner, A.J. Identification of critical active-site residues in angiotensin-converting enzyme-2 (ACE2) by site-directed mutagenesis. FEBS J. 2005, 272, 3512–3520. [Google Scholar] [CrossRef]
- Upreti, S.; Prusty, J.S.; Pandey, S.C.; Kumar, A.; Samant, M. Identification of novel inhibitors of angiotensin-converting enzyme 2 (ACE-2) receptor from Urtica dioica to combat coronavirus disease 2019 (COVID-19). Mol. Divers. 2021, 25, 1795–1809. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yamamoto, N.; Sakai, K.; Okubo, A.; Yamazaki, S.; Takano, T. Purification and Characterization of Angiotensin I-Converting Enzyme Inhibitors from Sour Milk. J. Dairy Sci. 1995, 78, 777–783. [Google Scholar] [CrossRef]
- Bimonte, S.; Crispo, A.; Amore, A.; Celentano, E.; Cuomo, A.; Cascella, M. Potential Antiviral Drugs for SARS-Cov-2 Treatment: Preclinical Findings and Ongoing Clinical Research. In Vivo 2020, 34, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Protection, Largest CDC COVID-19 Vaccine Effectiveness Study in Health Workers Shows mRNA Vaccines 94% Effective. Available online: https://www.cdc.gov/media/releases/2021/p0514-covid-19-vaccine-effectiveness.htmlhttps://www.cdc.gov/media/releases/2021/p0514-covid-19-vaccine-effectiveness.html (accessed on 24 July 2021).
- Dror, A.A.; Eisenbach, N.; Taiber, S.; Morozov, N.G.; Mizrachi, M.; Zigron, A.; Srouji, S.; Sela, E. Vaccine hesitancy: The next challenge in the fight against COVID-19. Eur. J. Epidemiol. 2020, 35, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.M.; Alothman, A.; Balkhy, H.H.; Al-Dawood, A.; Aljohani, S.; Al Harbi, S.; Kojan, S.; Aljeraisy, M.; Deeb, A.M.; Assiri, A.M.; et al. Treatment of Middle East Respiratory Syndrome with a combination of lopinavir-ritonavir and interferon-β1b (MIRACLE trial): Study protocol for a randomized controlled trial. Trials 2018, 19, 81. [Google Scholar] [CrossRef]
- COVID-19 Mythbusters—World Health Organization. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/advice-for-public/myth-busters (accessed on 24 July 2021).
- Mohanty, D.; Mohapatra, S.; Misra, S.; Sahu, P. Milk derived bioactive peptides and their impact on human health—A review. Saudi J. Biol. Sci. 2016, 23, 577–583. [Google Scholar] [CrossRef]
- Pihlanto-Leppälä, A. Bioactive peptides derived from bovine whey proteins: Opioid and ace-inhibitory peptides. Trends Food Sci. Technol. 2000, 11, 347–356. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Barochiner, J.; Martínez, R. Use of Inhibitors of the Renin-Angiotensin System in Hypertensive Patients and COVID-19 Severity: A Systematic Review and Meta-Analysis. J. Clin. Pharm. Ther. 2020, 45, 1244–1252. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef]
- Sommerstein, R.; Kochen, M.M.; Messerli, F.H.; Gräni, C. Coronavirus Disease 2019 (COVID-19): Do Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers Have a Biphasic Effect? J. Am. Heart Assoc. 2020, 9, e016509. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Han, B.; Mura, M.; Xia, S.; Wang, S.; Ma, T.; Liu, M.; Liu, Z. Angiotensin-converting enzyme inhibitor captopril prevents oleic acid-induced severe acute lung injury in rats. Shock 2007, 28, 106–111. [Google Scholar] [CrossRef]
- Danser, A.J.; Epstein, M.; Batlle, D. Renin-Angiotensin System Blockers and the COVID-19 Pandemic: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers. Hypertension 2020, 75, 1382–1385. [Google Scholar] [CrossRef]
- Lukkarinen, H.P.; Laine, J.; Aho, H.; Zagariya, A.; Vidyasagar, D.; Kääpä, P.O. Angiotensin II Receptor Inhibition Prevents Pneumocyte Apoptosis in Surfactant-Depleted Rat Lungs: Apoptosis in Surfactant Depleted-Lungs. Pediatr. Pulmonol. 2005, 39, 349–358. [Google Scholar] [CrossRef]
- Medhora, M.; Gao, F.; Jacobs, E.R.; Moulder, J.E. Radiation Damage to the Lung: Mitigation by Angiotensin-Converting Enzyme (ACE) Inhibitors: Radiation Lung Damage and Mitigation. Respirology 2012, 17, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.P.; Bedi, M.; Irving, A.A.; Jacobs, E.; Tomic, R.; Klein, J.; Lawton, C.A.; Moulder, J.E. Mitigation of Late Renal and Pulmonary Injury After Hematopoietic Stem Cell Transplantation. Int. J. Radiat. Oncol. 2012, 83, 292–296. [Google Scholar] [CrossRef]
- Caldeira, D.; Alarcão, J.; Carneiro, A.V.; Costa, J. Risk of pneumonia associated with use of angiotensin converting enzyme inhibitors and angiotensin receptor blockers: Systematic review and meta-analysis. BMJ 2012, 345, e4260. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, E.M.; Nakashima, B.; Cornell, J.; Copeland, L.A.; Pugh, M.J.; Anzueto, A.; Good, C.; Restrepo, M.I.; Downs, J.R.; Frei, C.R.; et al. Population-Based Study of Statins, Angiotensin II Receptor Blockers, and Angiotensin-Converting Enzyme Inhibitors on Pneumonia-Related Outcomes. Clin. Infect. Dis. 2012, 55, 1466–1473. [Google Scholar] [CrossRef]
- Shrikrishna, D.; Astin, R.; Kemp, P.; Hopkinson, N. Renin–angiotensin system blockade: A novel therapeutic approach in chronic obstructive pulmonary disease. Clin. Sci. 2012, 123, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.M.; Park, H.S.; Nath, S.K.; Mancini, B.R.; Decker, R.H. Angiotensin-converting enzyme inhibitors decrease the risk of radiation pneumonitis after stereotactic body radiation therapy. Pr. Radiat. Oncol. 2015, 5, e643–e649. [Google Scholar] [CrossRef] [PubMed]
- ESH STATEMENT ON COVID-19|European Society of Hypertension. Available online: https://www.eshonline.org/esh-content/uploads/2020/06/Statement-ESH-on-Hypertension-RAS-Blockers-and-COVID-19-Update-April-15-2020.pdf (accessed on 20 May 2021).
- Position Statement of the ESC Council on Hypertension on ACE-Inhibitors and Angiotensin Receptor Blockers. Available online: https://www.sphta.org.pt/files/european_society_of_hypertension_-_statement_on_covid-19.pdf (accessed on 20 May 2021).
- Bozkurt, B.; Kovacs, R.; Harrington, B. Joint HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19. J. Card. Fail. 2020, 26, 370. [Google Scholar] [CrossRef]
- Henry, C.; Zaizafoun, M.; Stock, E.; Ghamande, S.; Arroliga, A.C.; White, H.D. Impact of angiotensin-converting enzyme inhibitors and statins on viral pneumonia. Bayl. Univ. Med. Cent. Proc. 2018, 31, 419–423. [Google Scholar] [CrossRef]
- Yang, G. Effects of ARBs and ACEIs on Virus Infection, Inflammatory Status and Clinical Outcomes in COVID-19 Patients with Hypertension: A Single Center Retrospective Study. Hypertension 2020, 76, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Gu, H.; Xie, Z.; Li, T.; Zhang, S.; Lai, C.; Zhu, P.; Wang, K.; Han, L.; Duan, Y.; Zhao, Z.; et al. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci. Rep. 2016, 6, 19840. [Google Scholar] [CrossRef]
- Li, X.; Molina-Molina, M.; Abdul-Hafez, A.; Uhal, V.; Xaubet, A.; Uhal, B.D. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L178–L185. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.; Penninger, J.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Watkins, J. Preventing a covid-19 pandemic. BMJ 2020, 368, m810. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of Angiotensin-Converting Enzyme 2 After Myocardial Infarction by Blockade of Angiotensin II Receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef]
- Epelman, S.; Tang, W.W.; Chen, S.Y.; Van Lente, F.; Francis, G.S.; Sen, S. Detection of Soluble Angiotensin-Converting Enzyme 2 in Heart Failure: Insights Into the Endogenous Counter-Regulatory Pathway of the Renin-Angiotensin-Aldosterone System. J. Am. Coll. Cardiol. 2008, 52, 750–754. [Google Scholar] [CrossRef]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary Angiotensin-Converting Enzyme 2 in Hypertensive Patients May Be Increased by Olmesartan, an Angiotensin II Receptor Blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Oh, Y.-S.; Kim, J.-H.; Chung, W.-B.; Oh, S.-S.; Lee, D.-H.; Choi, Y.-S.; Shin, W.-S.; Park, C.-S.; Youn, H.-J.; et al. Effect of Angiotensin Converting Enzyme Inhibitors and Angiotensin Receptor Blockers on Patients Following Ablation of Atrial Fibrillation. Korean Circ. J. 2009, 39, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Lely, A.T.; Hamming, I.; van Goor, H.; Navis, G.J. Renal ACE2 expression in human kidney disease. J. Pathol. 2004, 204, 587–593. [Google Scholar] [CrossRef]
- Burrell, L.M.; Cooper, M.E.; Johnston, C.I. Myocardial infarction increases ACE2 expression in rat and humans: Reply. Eur. Heart J. 2005, 26, 1142–1143. [Google Scholar] [CrossRef]
- Hampl, V.; Herget, J.; Bíbová, J.; Baňasová, A.; Husková, Z.; Vaňourková, Z.; Jíchová, Š.; Kujal, P.; Vernerová, Z.; Sadowski, J.; et al. Intrapulmonary Activation of the Angiotensin-Converting Enzyme Type 2/Angiotensin 1–7/G-Protein-Coupled Mas Receptor Axis Attenuates Pulmonary Hypertension in Ren-2 Transgenic Rats Exposed to Chronic Hypoxia. Physiol. Res. 2015, 64, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.E.; Kane, C.; Raff, H. Peripheral Chemoreceptor Control of Fetal Renin Re-Sponses to Hypoxia and Hypercapnia. Circ. Res. 1990, 67, 722–732. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Suzuki, K.; Naoki, K.; Nishio, K.; Sato, N.; Takeshita, K.; Kudo, H.; Aoki, T.; Suzuki, Y.; Miyata, A.; et al. Response of Intra-acinar Pulmonary Microvessels to Hypoxia, Hypercapnic Acidosis, and Isocapnic Acidosis. Circ. Res. 1998, 82, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Wang, L.; Yang, C.; He, J.; Wang, X.; Guo, R.; Lan, A.; Dong, X.; Yang, Z.; Wang, H. Cyclooxygenase Mediates Cardioprotection of Angiotensin-(1–7) against Ische-Mia/Reperfusion-Induced Injury through the Inhibition of Oxidative Stress. Mol. Med. Rep. 2011, 4, 1145–1150. [Google Scholar] [PubMed]
- Zamai, L. The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients. Cells 2020, 9, 1704. [Google Scholar] [CrossRef] [PubMed]
- Wakahara, S.; Konoshita, T.; Mizuno, S.; Motomura, M.; Aoyama, C.; Makino, Y.; Kato, N.; Koni, I.; Miyamori, I. Synergistic Expression of Angiotensin-Converting Enzyme (ACE) and ACE2 in Human Renal Tissue and Confounding Effects of Hypertension on the ACE to ACE2 Ratio. Endocrinology 2007, 148, 2453–2457. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B. Ectodomain Shedding of Angiotensin Converting Enzyme 2 in Hu-Man Airway Epithelia. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-α-Converting Enzyme by the Spike Protein of SARS-CoV and ACE2 Induces TNF-α Production and Facilitates Viral Entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.E.; Belyaev, N.D.; Lambert, D.W.; Turner, A.J. Epigenetic Regulation of Angioten-Sin-Converting Enzyme 2 (ACE2) by SIRT1 under Conditions of Cell Energy Stress. Clin. Sci. 2013, 126, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Sharma, R.; Dhingra, N.; Patil, S. CoMFA, CoMSIA, HQSAR and molecular docking analysis of ionone-based chalcone derivatives as antiprostate cancer activity. Indian J. Pharm. Sci. 2016, 78, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Wang, S.-T.; Sun, P.-H.; Song, F.-J. Combined 3D-QSAR Modeling and Molecular Docking Studies on Pyrrole-Indolin-2-ones as Aurora A Kinase Inhibitors. Int. J. Mol. Sci. 2011, 12, 1605–1624. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Chen, W.N.; Chen, W.M. Molecular modeling studies on imidazo [4, 5-b] pyridine derivatives as Aurora A kinase inhibitors using 3D-QSAR and docking approaches. Eur. J. Med. Chem. 2011, 46, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef]
- Schrodinger LLC. Version 1.8, The PyMOL Molecular Graphics System; Technical Report; Schrödinger LLC: New York, NY, USA, 2015; p. 15. [Google Scholar]
- PDBeFold—Structure Similarity, Embl-Ebi. 2019. Available online: http://www.ebi.ac.uk/msd-srv/ssm/cgi-bin/ssmserver (accessed on 14 October 2021).
- Wang, R.; Lu, Y.; Wang, S. Comparative Evaluation of 11 Scoring Functions for Molecular Docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein 6MOJ | Ligand IPP | ||
|---|---|---|---|
| Residue | Atom Name | Interaction Type | Distance (Å) |
| NH1 Arg 273 | O− (Pro2) | Salt bridge | 3.0 |
| OE1 Glu 375 | NH3+ (Ile) | Salt bridge | 4.1 |
| OE2 Glu 375 | NH3+ (Ile) | Hydrogen bond | 2.0 |
| NE2 His 378 | NH3+ (Ile) | Hydrogen bond | 2.4 |
| OE1 Glu 402 | NH3+ (Ile) | Hydrogen bond | 2.9 |
| OE2 Glu 402 | NH3+ (Ile) | Salt bridge | 3.1 |
| Ligand IIAE | |||
| NH2 Arg 273 | O1 (Glu) | Salt bridge | 3.0 |
| NH2 Arg 273 | OE1 (Glu) | Hydrogen bond | 2.9 |
| NH1 Arg 273 | OE2 (Glu) | Salt bridge | 3.0 |
| OE1 Glu 375 | NH3+ (Ile) | Salt bridge | 3.0 |
| OE2 Glu 375 | NH3+ (Ile) | Hydrogen bond | 2.6 |
| OE2 Glu 375 | NH (Ile) | Hydrogen bond | 2.4 |
| OE1 Glu 402 | NH3+ (Ile) | Hydrogen bond | 2.9 |
| OE2 Glu 402 | NH (Ala) | Hydrogen bond | 2.9 |
| CG Glu 402 | NH3+ (Ile) | Salt bridge | 4.3 |
| Ligand LIVTQ | |||
| ND1 His 374 | O (Gln) | Hydrogen bond | 2.8 |
| OE1 Glu 375 | NH3+ (Leu) | Salt bridge | 3.5 |
| OE2 Glu 375 | NH3+ (Leu) | Hydrogen bond | 2.9 |
| OE2 Glu 375 | NH (Ile) | Hydrogen bond | 2.3 |
| OE1 Glu 402 | NH3+ (Leu) | Hydrogen bond | 2.0 |
| OE2 Glu 402 | NH3+ (Leu) | Salt bridge | 4.3 |
| OE2 Glu 402 | NH (Val) | Hydrogen bond | 2.6 |
| OE1 Glu 406 | OH (Thr) | Hydrogen bond | 2.9 |
| OE1 Glu 406 | NH2 (Gln) | Hydrogen bond | 2.9 |
| OE1 Glu 406 | NH (Gln) | Hydrogen bond | 2.9 |
| OE2 Glu 406 | NH (Thr) | Hydrogen bond | 2.9 |
| OE2 Glu 406 | NH (Val) | Hydrogen bond | 2.3 |
| NE2 Gln 442 | O (Gln) | Hydrogen bond | 2.8 |
| NH2 Arg 518 | OH (Thr) | Hydrogen bond | 2.1 |
| Ligand LVYPFP | |||
| CG2 Thr 276 | O− (Pro) | Hydrogen bond | 2.6 |
| OE1 Glu 402 | NH3+ (Leu) | Salt bridge | 3.8 |
| OE2 Glu 402 | NH3+ (Leu) | Hydrogen bond | 2.9 |
| CO Glu 406 | NH3+ (Leu) | Salt bridge | 4.0 |
| OE1 Glu 406 | NH (Val) | Hydrogen bond | 2.8 |
| OE1 Glu 406 | NH (Tyr) | Hydrogen bond | 2.4 |
| OE2 Glu 406 | NH3+ (Leu) | Hydrogen bond | 2.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamata, Y.; Jackson, K.G.; Watson, K.A.; Jauregi, P. Whey-Derived Peptides at the Heart of the COVID-19 Pandemic. Int. J. Mol. Sci. 2021, 22, 11662. https://doi.org/10.3390/ijms222111662
Chamata Y, Jackson KG, Watson KA, Jauregi P. Whey-Derived Peptides at the Heart of the COVID-19 Pandemic. International Journal of Molecular Sciences. 2021; 22(21):11662. https://doi.org/10.3390/ijms222111662
Chicago/Turabian StyleChamata, Yara, Kim G. Jackson, Kimberly A. Watson, and Paula Jauregi. 2021. "Whey-Derived Peptides at the Heart of the COVID-19 Pandemic" International Journal of Molecular Sciences 22, no. 21: 11662. https://doi.org/10.3390/ijms222111662
APA StyleChamata, Y., Jackson, K. G., Watson, K. A., & Jauregi, P. (2021). Whey-Derived Peptides at the Heart of the COVID-19 Pandemic. International Journal of Molecular Sciences, 22(21), 11662. https://doi.org/10.3390/ijms222111662