
CCNE1 and E2F1 Partially Suppress G1 Phase Arrest Caused by Spliceostatin A Treatment

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
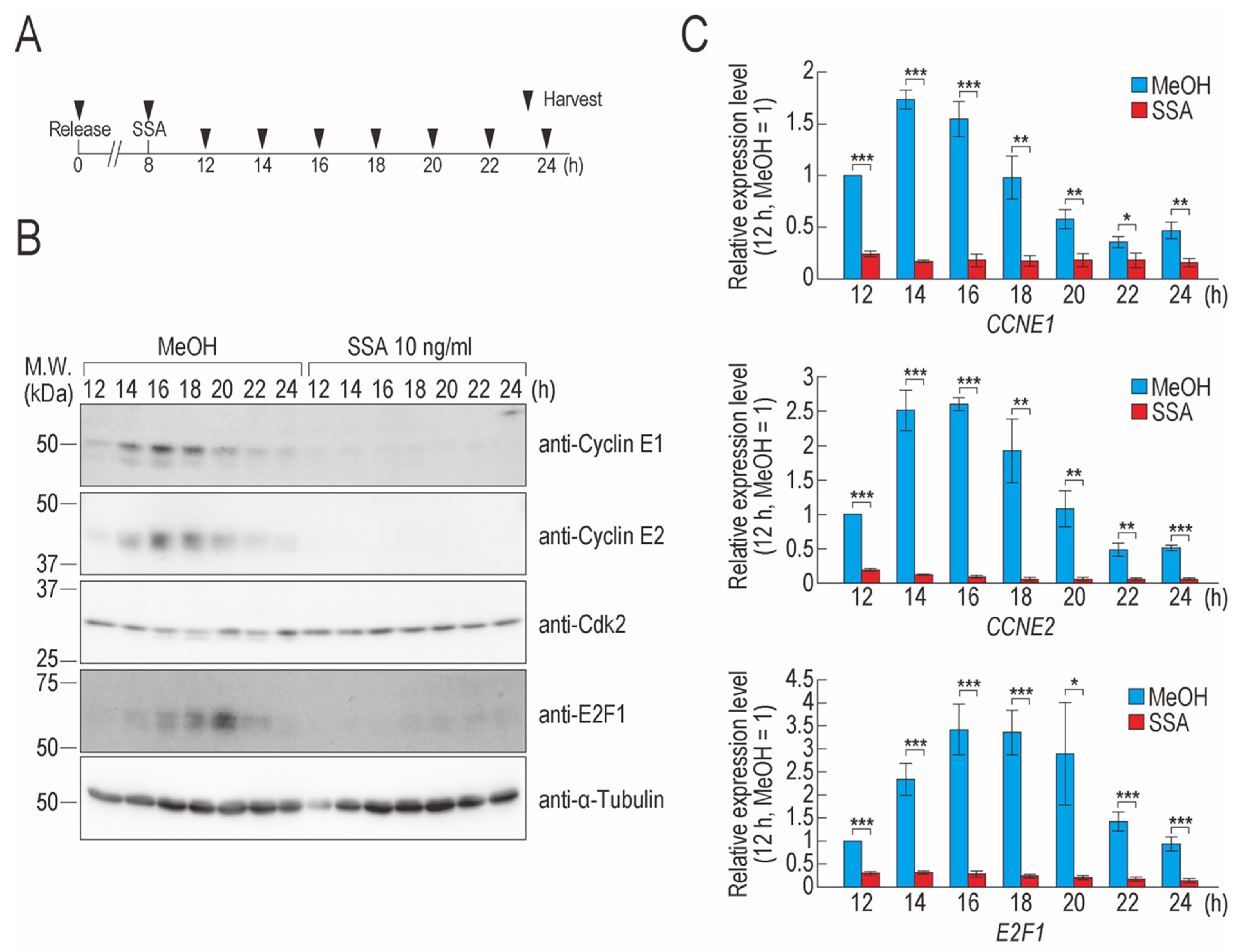
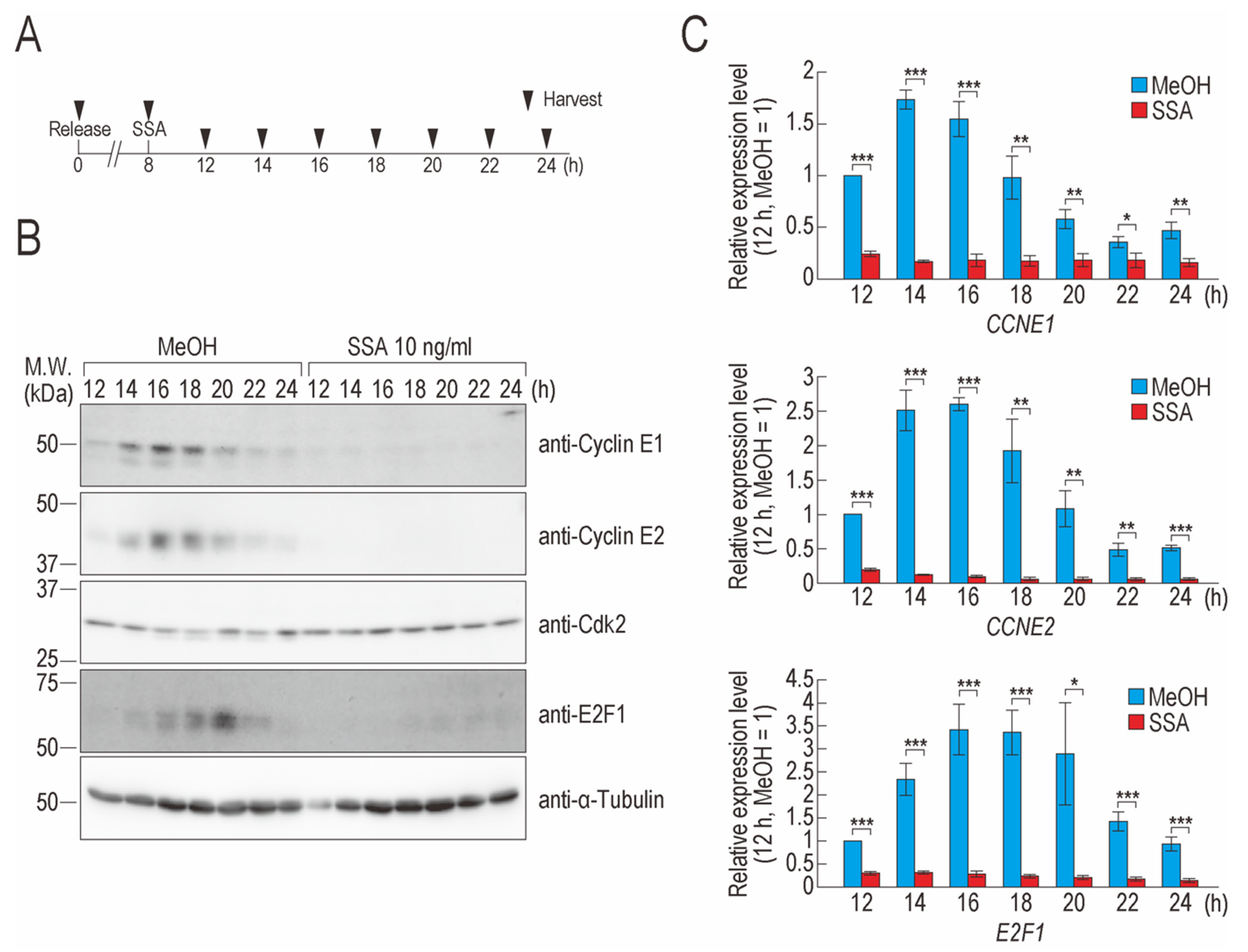
2.1. SSA Treatment Decreases Cyclin E1, Cyclin E2, and E2F1 Levels
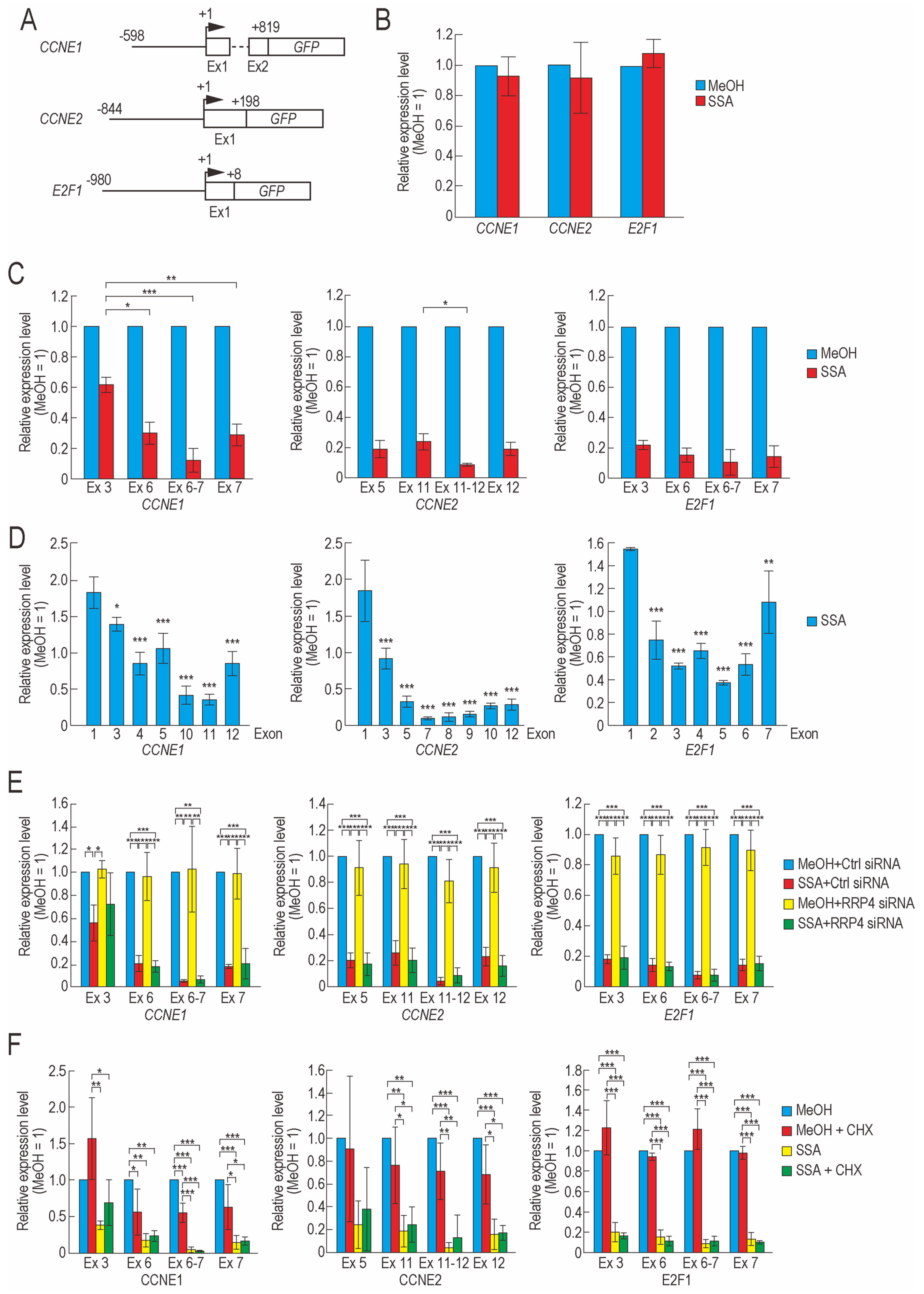
2.2. Transcription Elongation of CCNE1, CCNE2, and E2F1 Is Defective in SSA-Treated Cells
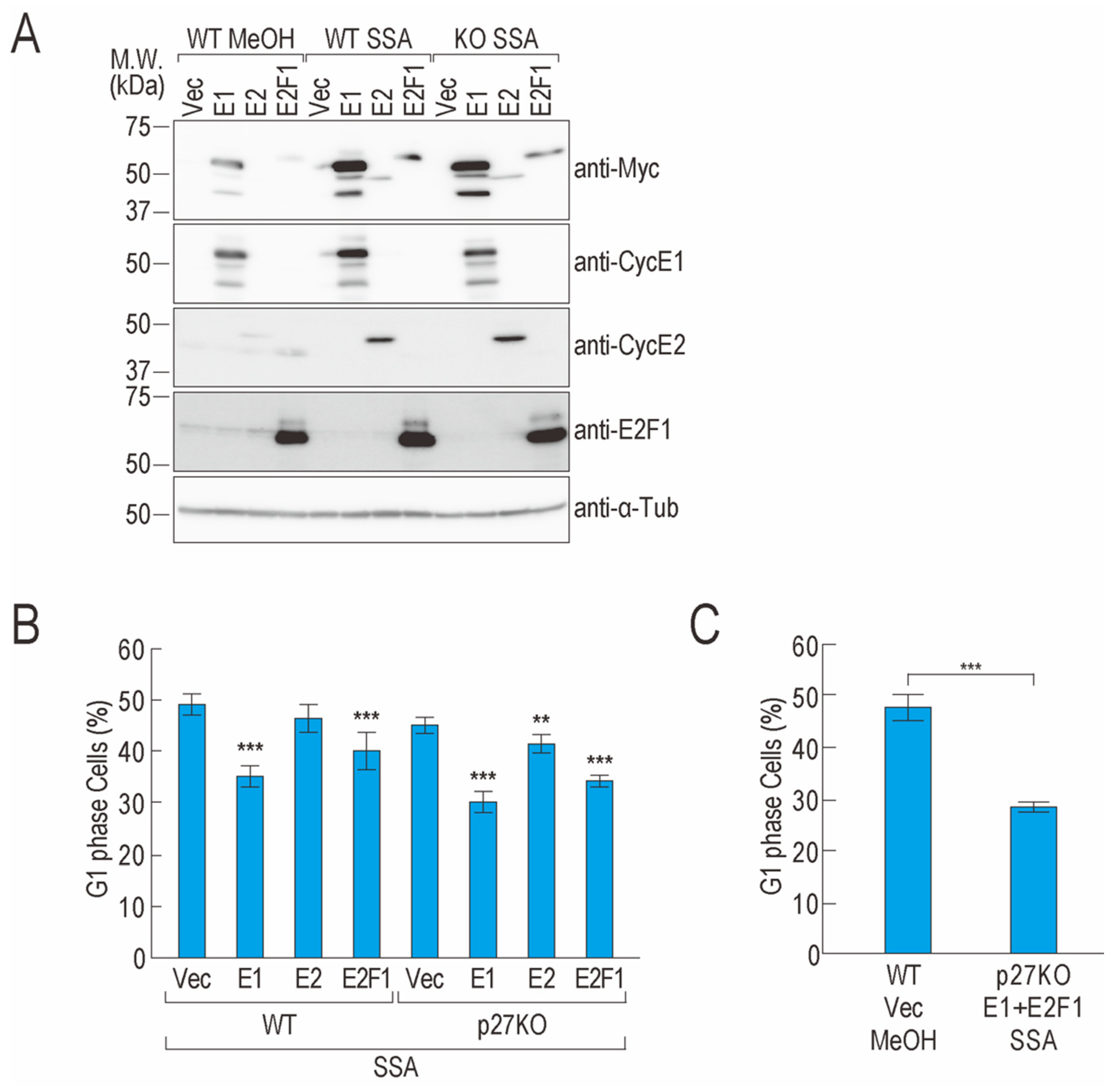
2.3. Overexpression of CCNE1 and E2F1 Partially Suppresses G1 Phase Arrest
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Synchronization, and Reagents
4.2. Antibodies and Immunoblotting
4.3. Cell Cycle Analysis
4.4. siRNA Transfection
4.5. RNA Purification and qRT-PCR
4.6. Plasmid Construction and Transfection
4.7. Establishment of p27KO Cells
4.8. Exon Array Data Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Geng, Y.; Zhou, Y.; Sicinski, P. Cyclin E in normal physiology and disease states. Trends Cell Biol. 2021, 31, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koff, A.; Giordano, A.; Desai, D.; Yamashita, K.; Harper, J.W.; Elledge, S.; Nishimoto, T.; Morgan, D.O.; Franza, B.R.; Roberts, J.M. Formation and Activation of a Cyclin E-cdk2 Complex During the G 1 Phase of the Human Cell Cycle. Science 1992, 257, 1689–1694. [Google Scholar] [CrossRef]
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An integrated view of cyclin E function and regulation. Cell Cycle 2012, 11, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef] [Green Version]
- Hinds, P.W.; Mittnacht, S.; Dulic, V.; Arnold, A.; Reed, S.I.; Weinberg, R.A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992, 70, 993–1006. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Ait-Si-Ali, S.; Ramirez, S.; Barre, F.-X.; Dkhissi, F.; Magnaghi-Jaulin, L.; Girault, J.-A.; Robin, P.; Knibiehler, M.; Pritchard, L.L.; Ducommun, B.; et al. Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nat. Cell Biol. 1998, 396, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Van Tine, B.A.; Wei, Y.; Garrett, M.D.; Nelson, D.; Adams, P.D.; Wang, J.; Qin, J.; Chow, L.T.; Harper, J.W. Cell cycle-regulated phosphorylation of p220NPAT by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000, 14, 2298–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nat. Cell Biol. 2004, 430, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kennedy, B.K.; Lawrence, B.D.; Barbie, D.A.; Matera, A.G.; Fletcher, J.A.; Harlow, E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 2000, 14, 2283–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Lee, M.-H.; Erdjument-Bromage, H.; Koff, A.; Roberts, J.M.; Tempst, P.; Massague, J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 78, 59–66. [Google Scholar] [CrossRef]
- Toyoshima, H.; Hunter, T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef]
- Sheaff, R.J.; Groudine, M.; Gordon, M.; Roberts, J.M.; Clurman, B.E. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997, 11, 1464–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlach, J.; Hennecke, S.; Amati, B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. EMBO J. 1997, 16, 5334–5344. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [Green Version]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D. Spliceostatin A treatment inhibits mitotic clonal expansion and adipogenesis. Biochem. Biophys. Res. Commun. 2019, 514, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kaida, D. Upregulation of p27 cyclin-dependent kinase inhibitor and a C-terminus truncated form of p27 contributes to G1 phase arrest. Sci. Rep. 2016, 6, 27829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli, C.; Skotheim, J.M.; De Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Rosenblatt, J.; Morgan, D. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992, 11, 3995–4005. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Hayashi, M.; Kaida, D. Splicing inhibition decreases phosphorylation level of Ser2 in Pol II CTD. Nucleic Acids Res. 2015, 43, 8258–8267. [Google Scholar] [CrossRef] [Green Version]
- Koga, M.; Satoh, T.; Takasaki, I.; Kawamura, Y.; Yoshida, M.; Kaida, D. U2 snRNP Is Required for Expression of the 3′ End of Genes. PLoS ONE 2014, 9, e98015. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, S.; Fukumura, K.; Hayashi, M.; Kataoka, N.; Mayeda, A.; Kaida, D. Rbm38 Reduces the Transcription Elongation Defect of the SMEK2 Gene Caused by Splicing Deficiency. Int. J. Mol. Sci. 2020, 21, 8799. [Google Scholar] [CrossRef]
- Bousquet-Antonelli, C.; Presutti, C.; Tollervey, D. Identification of a Regulated Pathway for Nuclear Pre-mRNA Turnover. Cell 2000, 102, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Tomecki, R.; Drazkowska, K.; Dziembowski, A. Mechanisms of RNA Degradation by the Eukaryotic Exosome. ChemBioChem 2010, 11, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, Y.; Li, X.; Serin, G.; Maquat, L.E. Evidence for a Pioneer Round of mRNA Translation: mRNAs Subject to Nonsense-Mediated Decay in Mammalian Cells Are Bound by CBP80 and CBP20. Cell 2001, 106, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Mizui, Y.; Sakai, T.; Iwata, M.; Uenaka, T.; Okamoto, K.; Shimizu, H.; Yamori, T.; Yoshimatsu, K.; Asada, M. Pladienolides, New Substances from Culture of Streptomyces platensis Mer-11107 III. In Vitro and In Vivo Antitumor Activities. J. Antibiot. 2004, 57, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, H.; Hori, Y.; Terano, H.; Okuhara, M.; Manda, T.; Matsumoto, S.; Shimomura, K. New Antitumor Substances, FR901463, FR901464 and FR901465. II. Activities against Experimental Tumors in Mice and Mechanism of Action. J. Antibiot. 1996, 49, 1204–1211. [Google Scholar] [CrossRef]
- Nakajima, H.; Sato, B.; Fujita, T.; Takase, S.; Terano, H.; Okuhara, M. New Antitumor Substances, FR901463, FR901464 and FR901465. I. Taxonomy, Fermentation, Isolation, Physico-chemical Properties and Biological Activities. J. Antibiot. 1996, 49, 1196–1203. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kikuchi, K.; Kaida, D. CCNE1 and E2F1 Partially Suppress G1 Phase Arrest Caused by Spliceostatin A Treatment. Int. J. Mol. Sci. 2021, 22, 11623. https://doi.org/10.3390/ijms222111623
Kikuchi K, Kaida D. CCNE1 and E2F1 Partially Suppress G1 Phase Arrest Caused by Spliceostatin A Treatment. International Journal of Molecular Sciences. 2021; 22(21):11623. https://doi.org/10.3390/ijms222111623
Chicago/Turabian StyleKikuchi, Kei, and Daisuke Kaida. 2021. "CCNE1 and E2F1 Partially Suppress G1 Phase Arrest Caused by Spliceostatin A Treatment" International Journal of Molecular Sciences 22, no. 21: 11623. https://doi.org/10.3390/ijms222111623
APA StyleKikuchi, K., & Kaida, D. (2021). CCNE1 and E2F1 Partially Suppress G1 Phase Arrest Caused by Spliceostatin A Treatment. International Journal of Molecular Sciences, 22(21), 11623. https://doi.org/10.3390/ijms222111623