
Molecular Mechanisms of Muscle Fatigue


{kind=link}
{kind=link}
Abstract
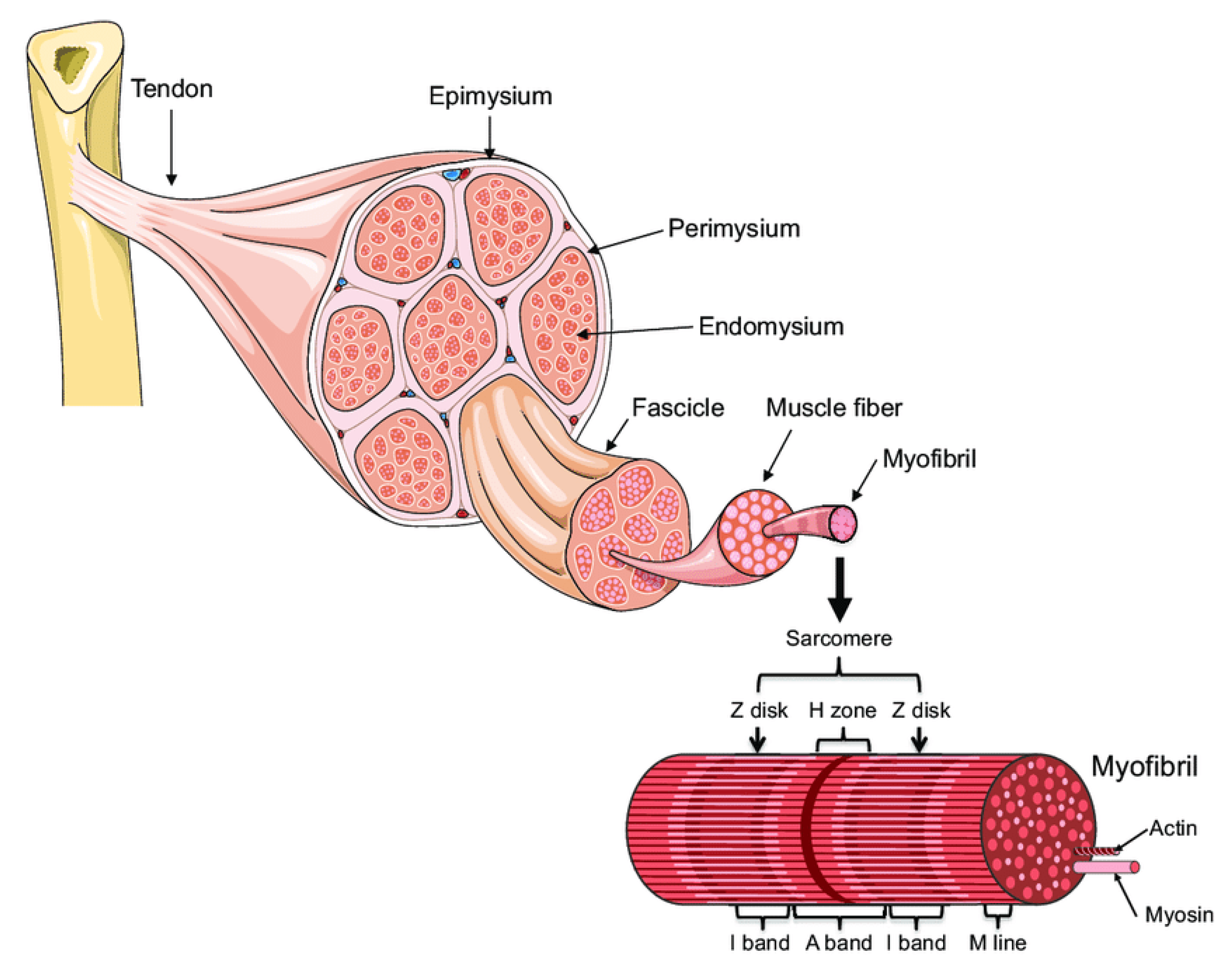
1. Introduction: Muscle Architecture
2. Muscle Fatigue
- (1)
- temporary due to strenuous physical activities and is caused by accumulation in the intracellular space of working muscles with intermediary energy metabolism waste (e.g., lactate) or depletion of their energy-rich compounds (e.g., muscle glycogen store). The time to recover from muscle fatigue will depend on the extent of the intensity and length of the physical task. On average, the individual should be fully recovered within 3 to 5 days. The usual intervention to speed up muscle recovery involves massage, cold compression, and light analgesics’ intake. However, muscle fatigue lasting beyond 2 weeks should require medical attention.
- (2)
- chronic due to either:
- (i)
- muscle atrophies (muscles waste away) due to immobilization, also called disuse atrophy, the presence of chronic inflammation in cardiovascular and respiratory disorders (e.g., heart failure, chronic obstructive pulmonary disease (COPD)), trauma, critical illness, medication (PPAR agonism),
- (ii)
- muscle atrophy with aging (sarcopenia), or
- (iii)
- neurogenic muscle atrophy due to obstructions or interference with different stages of nerve signal propagation from CNS to motor neuron plate due to disease or spinal injury. Depending upon location, it can be divided into central and peripheral [10]. Central fatigue is initiated at the central nervous system (CNS), such as in multiple sclerosis, thereby decreasing the neural drive to the muscle [11,12]. In contrast, peripheral fatigue is generated by changes at or distal to the neuromuscular junction such as in (a) autoimmune diseases caused by abnormal autoimmune reactions targeting neuromuscular synaptic proteins, as in Graves disease, Guillain-Barré syndrome, and myasthenia gravis, or (b) muscular dystrophies (MDs) due to genetic defects. MDs are progressive and debilitating diseases characterized by muscle wasting and progressive weakness. Duchenne (lacks the dystrophin component of the dystrophin-glycoprotein complex), Becker (contains a mutated dystrophin gene), and Limb-girdle type IIA (includes a mutation in the gene coding for calpain 3-P94) are typical examples of MDs.
3. Reduction of Muscle Mass and Function
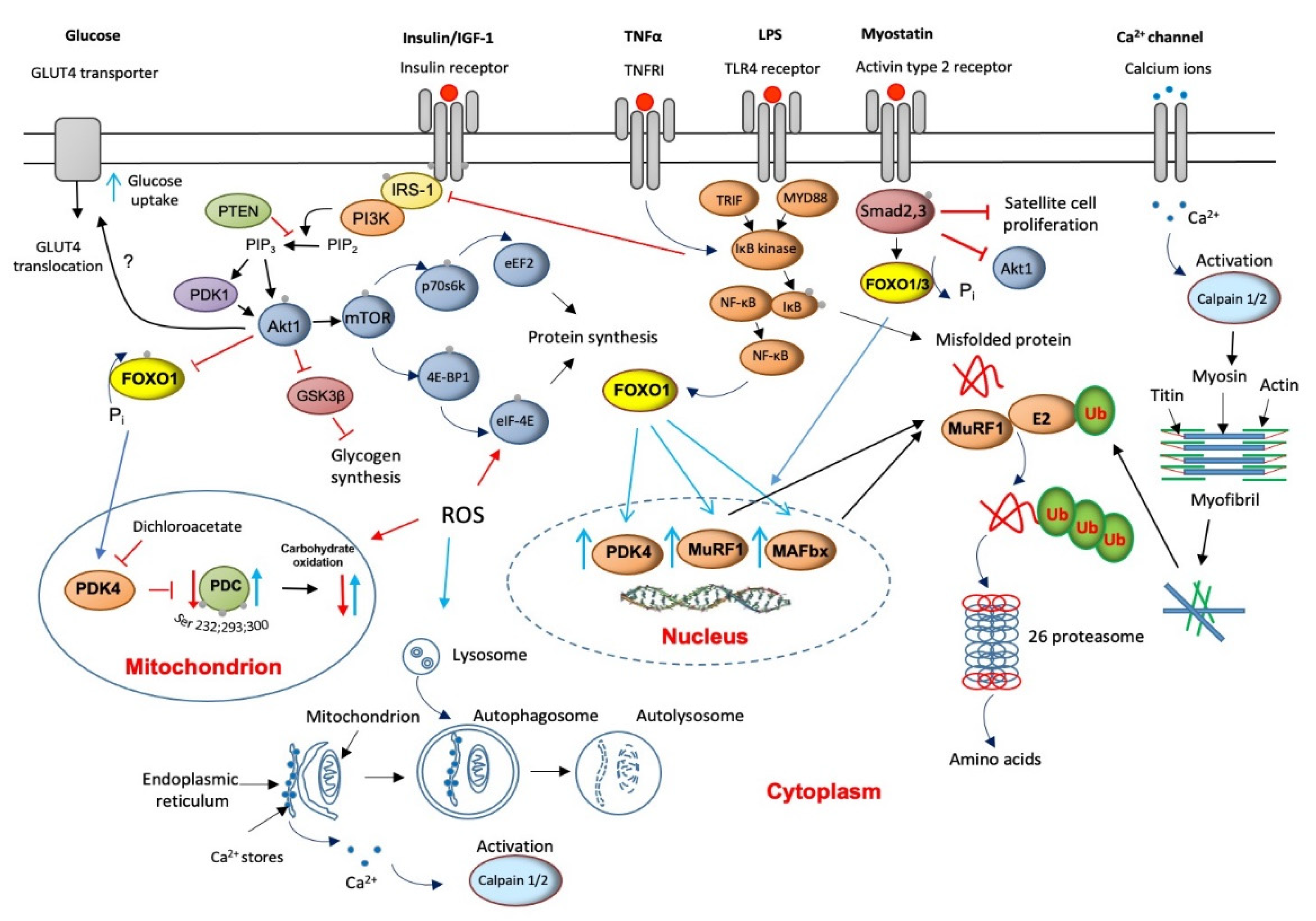
4. Major Molecular Mechanisms of Underlying Muscle Wasting
5. The Role of Lysosomal Autophagy in Muscle Protein Breakdown
6. Muscle Calpains’ Expression Is Increased in Muscle Wasting
7. Muscle Myostatin Expression Is Increased in Muscle Wasting
8. Apoptosis Is Increased in Muscle Wasting
9. Critical Illness Is Associated with Muscle Insulin Resistance
10. Muscle INSULIN Resistance Is Increased with PPAR Agonism and Statins’ Treatment
11. ROS Involvement in Muscle Wasting
12. Protein Synthesis Is Inhibited in Critical Illness but Is Accompanied by the Initiation of an Anabolic Restoration Program
13. Muscle Wasting with Ageing Sarcopenia
14. Muscle Wasting with Chronic Chemical Exposure
15. Muscle Fatigue Associated with Neurogenic Muscle Atrophy Induced by Viruses
16. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation List
| 4E-BP1 | eukaryotic translation initiation factor 4E-binding protein 1 |
| eIF-2B | eukaryotic translation initiation factor 2B |
| eIF-4F | eukaryotic translation initiation factor 4F |
| eIF-4E | eukaryotic translation initiation factor 4E |
| Akt1 | Protein kinase B, |
| AMPK | 5’ AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| Bax | Apoptosis regulator BAX |
| Bcl-2 | B-cell lymphoma 2 |
| CHO | Carbohydrates |
| CK | Creatine kinase |
| CNS | Central nervous system |
| COPD | Chronic obstructive pulmonary disease |
| COVID-19 | coronavirus disease 2019 |
| DCA | Dichloroacetate |
| E3-ligases. | ubiquitin ligase |
| eIF-2B | eukaryotic translation initiation factor 2B |
| eNOS endothelial | Endothelial nitric oxide synthase |
| FOXO1 | Forkhead box protein O1 |
| FoxO3a | Forkhead box O3 |
| GDF8 | myostatin or growth differentiation factor 8 |
| GSK3β | Glycogen synthase kinase-3 |
| GWI | Gulf War illness |
| HERV-W | Human Endogenous Retrovirus-W |
| HERVs | human endogenous retroviruses |
| HSP70 | heat shock protein 70kDa |
| IL-6 | Interleukin 6 |
| iNOS inducible | Inducible nitric oxide synthase |
| IRS-1 | Insulin receptor substrate-1 |
| MAFbx | F-box only protein 32 |
| MAPK | Mitogen-activated protein kinase |
| MEF-2 | myocyte enhancer factor-2 |
| MF | Muscle fatigue |
| MnSOD | Mitochondrial isoform of superoxide dismutase |
| MS | Multiple Sclerosis |
| MSRV | MS-associated retrovirus |
| mTOR | mechanistic target of rapamycin |
| MuRF1 | muscle RING-finger protein-1 |
| Myf5 | Myogenic factor 5 |
| MyoD | myoblast determination protein 1 |
| MyoD88 | myeloid differentiation primary response 88 |
| Myogenin | Muscle-specific basic helix-loop-helix (bHLH) transcription factor |
| Myonuclei | nuclei of a muscle fiber located in the space between myofibrils and sarcolemma |
| GDF8 | Myostatin or growth differentiation factor 8 |
| NADPH oxidase | Nicotinamide adenine dinucleotide phosphate reduced oxidase |
| nNOS neuronal | Neuronal nitric oxide synthase |
| NO | nitric oxide |
| p70s6k | Ribosomal protein S6 kinase beta-1 |
| PDC | Pyruvate dehydrogenase complex |
| PDK4 | Pyruvate dehydrogenase kinase isoform 4 |
| PI3k | Phosphoinositide 3-kinases |
| PPARδ | Peroxisome Proliferator-Activated Receptor Delta |
| RhoA | Transforming protein RhoA/Ras homolog family member actor |
| ROS | reactive oxygen species |
| SAPK/JNK | Stress-activated protein kinase |
| Smac/DIABLO | Second mitochondria-derived activator of caspase |
| Th17 | T helper 17 cell |
| TLR2 | toll-like receptor 2 |
| TLR4 | Toll-like receptor 4 |
| TNF-α | tumor necrosis factor-alpha |
| Tregs | regulatory T cells |
| UPP | Ubiquitin ATP-dependent proteasome |
References
- Dave, H.D.; Shook, M.; Varacallo, M. Anatomy, Skeletal Muscle; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Staron, R.S.; Hagerman, F.C.; Hikida, R.S.; Murray, T.F.; Hostler, D.P.; Crill, M.T.; Ragg, K.E.; Toma, K. Fiber type composition of the vastus lateralis muscle of young men and women. J. Histochem. Cytochem. 2000, 48, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Bonewald, L.F. Bone and muscle. In Basic and Applied Bone Biology; Academic Press: Cambridge, MA, USA, 2019; pp. 317–332. [Google Scholar]
- Schiaffino, S. Muscle fiber type diversity revealed by anti-myosin heavy chain antibodies. FEBS J. 2018, 285, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, D.L.; Roberts, M.D.; Haun, C.T.; Schoenfeld, B.J. Muscle Fiber Type Transitions with Exercise Training: Shifting Perspectives. Sports 2021, 9, 127. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C.; Akimoto, T.; Blaauw, B. Molecular Mechanisms of Skeletal Muscle Hypertrophy. J. Neuromuscul. Dis. 2021, 8, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.H. Human muscle function and fatigue. Ciba. Found Symp. 1981, 82, 1–18. [Google Scholar]
- Fitts, R.H. Cellular mechanisms of muscle fatigue. Physiol. Rev. 1994, 74, 49–94. [Google Scholar] [CrossRef]
- Mills, R.J.; Young, C.A. A medical definition of fatigue in multiple sclerosis. QJM 2008, 101, 49–60. [Google Scholar] [CrossRef]
- Wan, J.J.; Qin, Z.; Wang, P.Y.; Sun, Y.; Liu, X. Muscle fatigue: General understanding and treatment. Exp. Mol. Med. 2017, 49, e384. [Google Scholar] [CrossRef]
- Gandevia, S.C. Spinal and supraspinal factors in human muscle fatigue. Physiol. Rev. 2001, 81, 1725–1789. [Google Scholar] [CrossRef]
- Bigland-Ritchie, B.; Jones, D.A.; Hosking, G.P.; Edwards, R.H. Central and peripheral fatigue in sustained maximum voluntary contractions of human quadriceps muscle. Clin. Sci. Mol. Med. 1978, 54, 609–614. [Google Scholar] [CrossRef]
- Ali, N.A.; O’Brien, J.M., Jr.; Hoffmann, S.P.; Phillips, G.; Garland, A.; Finley, J.C.; Almoosa, K.; Hejal, R.; Wolf, K.M.; Lemeshow, S.; et al. Acquired weakness, handgrip strength, and mortality in critically ill patients. Am. J. Respir. Crit. Care. Med. 2008, 178, 261–268. [Google Scholar] [CrossRef]
- Choi, Y.; Oh, D.Y.; Kim, T.Y.; Lee, K.H.; Han, S.W.; Im, S.A.; Kim, T.Y.; Bang, Y.J. Skeletal Muscle Depletion Predicts the Prognosis of Patients with Advanced Pancreatic Cancer Undergoing Palliative Chemotherapy, Independent of Body Mass Index. PLoS ONE 2015, 10, e0139749. [Google Scholar] [CrossRef]
- Srikanthan, P.; Karlamangla, A.S. Muscle mass index as a predictor of longevity in older adults. Am. J. Med. 2014, 127, 547–553. [Google Scholar] [CrossRef]
- Wang, H.; Hai, S.; Liu, Y.; Liu, Y.; Dong, B. Skeletal Muscle Mass as a Mortality Predictor among Nonagenarians and Centenarians: A Prospective Cohort Study. Sci. Rep. 2019, 9, 2420. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.B.; Kupelian, V.; Visser, M.; Simonsick, E.M.; Goodpaster, B.H.; Kritchevsky, S.B.; Tylavsky, F.A.; Rubin, S.M.; Harris, T.B. Strength, but not muscle mass, is associated with mortality in the health, aging and body composition study cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 72–77. [Google Scholar] [CrossRef]
- Metter, E.J.; Talbot, L.A.; Schrager, M.; Conwit, R. Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, B359–B365. [Google Scholar] [CrossRef] [PubMed]
- Gamrin, L.; Essen, P.; Forsberg, A.M.; Hultman, E.; Wernerman, J. A descriptive study of skeletal muscle metabolism in critically ill patients: Free amino acids, energy-rich phosphates, protein, nucleic acids, fat, water, and electrolytes. Crit. Care. Med. 1996, 24, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Alkner, B.A.; Tesch, P.A. Knee extensor and plantar flexor muscle size and function following 90 days of bed rest with or without resistance exercise. Eur. J. Appl. Physiol. 2004, 93, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Reid, W.D.; MacGowan, N.A. Respiratory muscle injury in animal models and humans. Mol. Cell. Biochem. 1998, 179, 63–80. [Google Scholar] [CrossRef]
- Constantin, D.; Menon, M.K.; Houchen-Wolloff, L.; Morgan, M.D.; Singh, S.J.; Greenhaff, P.; Steiner, M.C. Skeletal muscle molecular responses to resistance training and dietary supplementation in COPD. Thorax 2013, 68, 625–633. [Google Scholar] [CrossRef]
- Iannuzzi-Sucich, M.; Prestwood, K.M.; Kenny, A.M. Prevalence of sarcopenia and predictors of skeletal muscle mass in healthy, older men and women. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, M772–M777. [Google Scholar] [CrossRef]
- Du, Y.; Wang, X.; Xie, H.; Zheng, S.; Wu, X.; Zhu, X.; Zhang, X.; Xue, S.; Li, H.; Hong, W.; et al. Sex differences in the prevalence and adverse outcomes of sarcopenia and sarcopenic obesity in community dwelling elderly in East China using the AWGS criteria. BMC Endocr. Disord. 2019, 19, 109. [Google Scholar] [CrossRef]
- Jagoe, R.T.; Goldberg, A.L. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 183–190. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Rabuel, C.; Renaud, E.; Brealey, D.; Ratajczak, P.; Damy, T.; Alves, A.; Habib, A.; Singer, M.; Payen, D.; Mebazaa, A. Human septic myopathy: Induction of cyclooxygenase, heme oxygenase and activation of the ubiquitin proteolytic pathway. Anesthesiology 2004, 101, 583–590. [Google Scholar] [CrossRef]
- Constantin, D.; McCullough, J.; Mahajan, R.P.; Greenhaff, P.L. Novel events in the molecular regulation of muscle mass in critically ill patients. J. Physiol. 2011, 589, 3883–3895. [Google Scholar] [CrossRef] [PubMed]
- Klaude, M.; Mori, M.; Tjader, I.; Gustafsson, T.; Wernerman, J.; Rooyackers, O. Protein metabolism and gene expression in skeletal muscle of critically ill patients with sepsis. Clin. Sci. 2012, 122, 133–142. [Google Scholar] [CrossRef] [PubMed]
- McKeran, R.O.; Halliday, D.; Purkiss, P.; Royston, P. 3-Methylhistidine excretion as an index of myofibrillar protein catabolism in neuromuscular disease. J. Neurol. Neurosurg. Psychiatry 1979, 42, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, J.G.; Nedergaard, A.; Reitelseder, S.; Mikkelsen, U.R.; Dideriksen, K.J.; Agergaard, J.; Kreiner, F.; Pott, F.C.; Schjerling, P.; Kjaer, M. Activated protein synthesis and suppressed protein breakdown signaling in skeletal muscle of critically ill patients. PLoS ONE 2011, 6, e18090. [Google Scholar] [CrossRef] [PubMed]
- Winkelman, C. Inactivity and inflammation in the critically ill patient. Crit. Care Clin. 2007, 23, 21–34. [Google Scholar] [CrossRef]
- Riedemann, N.C.; Guo, R.F.; Ward, P.A. The enigma of sepsis. J. Clin. Investig. 2003, 112, 460–467. [Google Scholar] [CrossRef]
- Rui, L.; Aguirre, V.; Kim, J.K.; Shulman, G.I.; Lee, A.; Corbould, A.; Dunaif, A.; White, M.F. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Investig. 2001, 107, 181–189. [Google Scholar] [CrossRef]
- Latres, E.; Amini, A.R.; Amini, A.A.; Griffiths, J.; Martin, F.J.; Wei, Y.; Lin, H.C.; Yancopoulos, G.D.; Glass, D.J. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J. Biol. Chem. 2005, 280, 2737–2744. [Google Scholar] [CrossRef]
- Medina, E.A.; Afsari, R.R.; Ravid, T.; Castillo, S.S.; Erickson, K.L.; Goldkorn, T. Tumor necrosis factor-{alpha} decreases Akt protein levels in 3T3-L1 adipocytes via the caspase-dependent ubiquitination of Akt. Endocrinology 2005, 146, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Powers, S.K.; Lynch, G.S.; Murphy, K.T.; Reid, M.B.; Zijdewind, I. Disease-Induced Skeletal Muscle Atrophy and Fatigue. Med. Sci. Sports Exerc. 2016, 48, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Kasper, C.E.; Talbot, L.A.; Gaines, J.M. Skeletal muscle damage and recovery. AACN Clin. Issues 2002, 13, 237–247. [Google Scholar] [CrossRef]
- Jones, S.W.; Hill, R.J.; Krasney, P.A.; O’Conner, B.; Peirce, N.; Greenhaff, P.L. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J. 2004, 18, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.N.; Hocker, A.D.; Vermillion, B.R.; Smolkowski, K.; Shah, S.N.; Jewett, B.A.; Dreyer, H.C. MAFbx, MuRF1, and the stress-activated protein kinases are upregulated in muscle cells during total knee arthroplasty. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R376–R386. [Google Scholar] [CrossRef]
- Baracos, V.E.; DeVivo, C.; Hoyle, D.H.; Goldberg, A.L. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am. J. Physiol. 1995, 268 Pt 1, E996–E1006. [Google Scholar] [CrossRef]
- Guarnieri, G.; Toigo, G.; Situlin, R.; Del Bianco, M.A.; Crapesi, L. Cathepsin B and D activity in human skeletal muscle in disease states. Adv. Exp. Med. Biol. 1988, 240, 243–256. [Google Scholar] [PubMed]
- Wagenmakers, A.J. Muscle function in critically ill patients. Clin. Nutr. 2001, 20, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Reffo, P.; Penna, F.; Autelli, R.; Bonelli, G.; Baccino, F.M. Ca(2+)-dependent proteolysis in muscle wasting. Int. J. Biochem. Cell. Biol. 2005, 37, 2134–2146. [Google Scholar] [CrossRef]
- Lee, S.J. Regulation of muscle mass by myostatin. Annu. Rev. Cell. Dev. Biol. 2004, 20, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.R.; Cook, S.A.; Buranasombati, C.; Rosenberg, M.A.; Rosenzweig, A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am. J. Physiol. Cell Physiol. 2009, 297, C1124–C1132. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, C.; Plummer, E.; Thomas, M.; Hennebry, A.; Ashby, M.; Ling, N.; Smith, H.; Sharma, M.; Kambadur, R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J. Cell Physiol. 2006, 209, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Siu, P.M.; Alway, S.E. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 2005, 565 Pt 1, 309–323. [Google Scholar] [CrossRef]
- Schwartz, L.M. Skeletal Muscles Do Not Undergo Apoptosis During Either Atrophy or Programmed Cell Death-Revisiting the Myonuclear Domain Hypothesis. Front. Physiol. 2018, 9, 1887. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Kitayama, K.; Yamashita, H.; Mori, N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 2003, 375 Pt 2, 365–371. [Google Scholar] [CrossRef]
- Constantin-Teodosiu, D. Regulation of muscle pyruvate dehydrogenase complex in insulin resistance: Effects of exercise and dichloroacetate. Diabetes. Metab. J. 2013, 37, 301–314. [Google Scholar] [CrossRef]
- Investigators, N.-S.S.; Finfer, S.; Liu, B.; Chittock, D.R.; Norton, R.; Myburgh, J.A.; McArthur, C.; Mitchell, I.; Foster, D.; Dhingra, V.; et al. Hypoglycemia and risk of death in critically ill patients. N. Engl. J. Med. 2012, 367, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Constantin, D.; Constantin-Teodosiu, D.; Layfield, R.; Tsintzas, K.; Bennett, A.J.; Greenhaff, P.L. PPARdelta agonism induces a change in fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function in rat skeletal muscle. J. Physiol. 2007, 583 Pt 1, 381–390. [Google Scholar] [CrossRef]
- Mallinson, J.E.; Constantin-Teodosiu, D.; Sidaway, J.; Westwood, F.R.; Greenhaff, P.L. Blunted Akt/FOXO signalling and activation of genes controlling atrophy and fuel use in statin myopathy. J. Physiol. 2009, 587, 219–230. [Google Scholar] [CrossRef]
- Constantin-Teodosiu, D.; Baker, D.J.; Constantin, D.; Greenhaff, P.L. PPARdelta agonism inhibits skeletal muscle PDC activity, mitochondrial ATP production and force generation during prolonged contraction. J. Physiol. 2009, 587, 231–239. [Google Scholar] [CrossRef]
- Thompson, P.D.; Clarkson, P.; Karas, R.H. Statin-associated myopathy. JAMA 2003, 289, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Constantin-Teodosiu, D.; Constantin, D.; Stephens, F.; Laithwaite, D.; Greenhaff, P.L. The role of FOXO and PPAR transcription factors in diet-mediated inhibition of PDC activation and carbohydrate oxidation during exercise in humans and the role of pharmacological activation of PDC in overriding these changes. Diabetes 2012, 61, 1017–1024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mallinson, J.E.; Constantin-Teodosiu, D.; Glaves, P.D.; Martin, E.A.; Davies, W.J.; Westwood, F.R.; Sidaway, J.E.; Greenhaff, P.L. Pharmacological activation of the pyruvate dehydrogenase complex reduces statin-mediated upregulation of FOXO gene targets and protects against statin myopathy in rodents. J. Physiol. 2012, 590, 6389–6402. [Google Scholar] [CrossRef] [PubMed]
- Crossland, H.; Constantin-Teodosiu, D.; Gardiner, S.M.; Constantin, D.; Greenhaff, P.L. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J. Physiol. 2008, 586, 5589–5600. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Kavazis, A.N.; McClung, J.M. Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. (1985) 2007, 102, 2389–2397. [Google Scholar] [CrossRef]
- Powers, S.K.; Smuder, A.J.; Criswell, D.S. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid. Redox Signal 2011, 15, 2519–2528. [Google Scholar] [CrossRef]
- Abrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of Oxidative Stress as Key Regulator of Muscle Wasting during Cachexia. Oxid. Med. Cell Longev. 2018, 2018, 2063179. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Brocca, L.; Pellegrino, M.A.; Desaphy, J.F.; Pierno, S.; Camerino, D.C.; Bottinelli, R. Is oxidative stress a cause or consequence of disuse muscle atrophy in mice? A proteomic approach in hindlimb-unloaded mice. Exp. Physiol. 2010, 95, 331–350. [Google Scholar] [CrossRef]
- Fredriksson, K.; Tjader, I.; Keller, P.; Petrovic, N.; Ahlman, B.; Scheele, C.; Wernerman, J.; Timmons, J.A.; Rooyackers, O. Dysregulation of mitochondrial dynamics and the muscle transcriptome in ICU patients suffering from sepsis induced multiple organ failure. PLoS ONE 2008, 3, e3686. [Google Scholar] [CrossRef]
- Essen, P.; McNurlan, M.A.; Gamrin, L.; Hunter, K.; Calder, G.; Garlick, P.J.; Wernerman, J. Tissue protein synthesis rates in critically ill patients. Crit. Care Med. 1998, 26, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Tjader, I.; Essen, P.; Garlick, P.J.; McMnurlan, M.A.; Rooyackers, O.; Wernerman, J. Impact of surgical trauma on human skeletal muscle protein synthesis. Clin. Sci. 2004, 107, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Frost, R.A.; Vary, T.C. Regulation of muscle protein synthesis during sepsis and inflammation. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E453–E459. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar] [CrossRef]
- Hardt, S.E.; Sadoshima, J. Glycogen synthase kinase-3beta: A novel regulator of cardiac hypertrophy and development. Circ. Res. 2002, 90, 1055–1063. [Google Scholar] [CrossRef]
- Wang, H.; Brown, J.; Gu, Z.; Garcia, C.A.; Liang, R.; Alard, P.; Beurel, E.; Jope, R.S.; Greenway, T.; Martin, M. Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-beta-signaling pathways regulates the innate inflammatory response. J. Immunol. 2011, 186, 5217–5226. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Streeper, T.; Cawthon, P.; Baldwin, K.; Taaffe, D.R.; Harris, T.B. Sarcopenia: Etiology, clinical consequences, intervention, and assessment. Osteoporos. Int. 2010, 21, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.V.; Faulkner, J.A. Skeletal muscle weakness in old age: Underlying mechanisms. Med. Sci. Sports Exerc. 1994, 26, 432–439. [Google Scholar] [CrossRef]
- Chen, W.; Datzkiw, D.; Rudnicki, M.A. Satellite cells in ageing: Use it or lose it. Open. Biol. 2020, 10, 200048. [Google Scholar] [CrossRef]
- Verdijk, L.B.; Koopman, R.; Schaart, G.; Meijer, K.; Savelberg, H.H.; van Loon, L.J. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E151–E157. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Schertzer, J.D.; Lynch, G.S. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 2008, 9, 213–228. [Google Scholar] [CrossRef]
- Lexell, J.; Taylor, C.C. Variability in muscle fibre areas in whole human quadriceps muscle: Effects of increasing age. J. Anat. 1991, 174, 239–249. [Google Scholar]
- Frontera, W.R.; Suh, D.; Krivickas, L.S.; Hughes, V.A.; Goldstein, R.; Roubenoff, R. Skeletal muscle fiber quality in older men and women. Am. J. Physiol. Cell Physiol. 2000, 279, C611–C618. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylavsky, F.A.; Visser, M.; Newman, A.B. The loss of skeletal muscle strength, mass, and quality in older adults: The health, aging and body composition study. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 1059–1064. [Google Scholar] [CrossRef]
- Conboy, I.M.; Conboy, M.J.; Wagers, A.J.; Girma, E.R.; Weissman, I.L.; Rando, T.A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005, 433, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Degens, H. The role of systemic inflammation in age-related muscle weakness and wasting. Scand. J. Med. Sci. Sports 2010, 20, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Ralston, A.; Shaw, K. Gene Expression Regulates Cell Differentiation. Nature Educ. 2008, 1, 27. [Google Scholar]
- Mawson, A.R.; Croft, A.M. Gulf War Illness: Unifying Hypothesis for a Continuing Health Problem. Int. J. Environ. Res. Public Health 2019, 16, 111. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Sanchez, I.; Navarrete-Yanez, V.; Garate-Carrillo, A.; Loredo, M.; Lira-Romero, E.; Estrada-Mena, J.; Campeau, A.; Gonzalez, D.; Carrillo-Terrazas, M.; Moreno-Ulloa, A.; et al. Development of muscle atrophy and loss of function in a Gulf-War illness model: Underlying mechanisms. Sci. Rep. 2020, 10, 14526. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.M.; Parry, G.J. Neuropathies associated with excessive exposure to lead. Muscle Nerve 2006, 33, 732–741. [Google Scholar] [CrossRef]
- Nyirenda, M.H.; Morandi, E.; Vinkemeier, U.; Constantin-Teodosiu, D.; Drinkwater, S.; Mee, M.; King, L.; Podda, G.; Zhang, G.X.; Ghaemmaghami, A.; et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: A novel mechanism of reduced regulatory T cell function in multiple sclerosis. J. Immunol. 2015, 194, 5761–5774. [Google Scholar] [CrossRef] [PubMed]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantin-Teodosiu, D.; Gran, B. Do Antiretroviral Drugs Protect From Multiple Sclerosis by Inhibiting Expression of MS-Associated Retrovirus? Front. Immunol. 2018, 9, 3092. [Google Scholar] [CrossRef]
- Mendelson, M.; Nel, J.; Blumberg, L.; Madhi, S.A.; Dryden, M.; Stevens, W.; Venter, F.W.D. Long-COVID: An evolving problem with an extensive impact. S. Afr. Med. J. 2020, 111, 10–12. [Google Scholar] [CrossRef]
- Ma, K.; Mallidis, C.; Bhasin, S.; Mahabadi, V.; Artaza, J.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E363–E371. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Constantin-Teodosiu, D.; Constantin, D. Molecular Mechanisms of Muscle Fatigue. Int. J. Mol. Sci. 2021, 22, 11587. https://doi.org/10.3390/ijms222111587
Constantin-Teodosiu D, Constantin D. Molecular Mechanisms of Muscle Fatigue. International Journal of Molecular Sciences. 2021; 22(21):11587. https://doi.org/10.3390/ijms222111587
Chicago/Turabian StyleConstantin-Teodosiu, Dumitru, and Despina Constantin. 2021. "Molecular Mechanisms of Muscle Fatigue" International Journal of Molecular Sciences 22, no. 21: 11587. https://doi.org/10.3390/ijms222111587
APA StyleConstantin-Teodosiu, D., & Constantin, D. (2021). Molecular Mechanisms of Muscle Fatigue. International Journal of Molecular Sciences, 22(21), 11587. https://doi.org/10.3390/ijms222111587