
DUSP9, a Dual-Specificity Phosphatase with a Key Role in Cell Biology and Human Diseases



Abstract
1. General Introduction
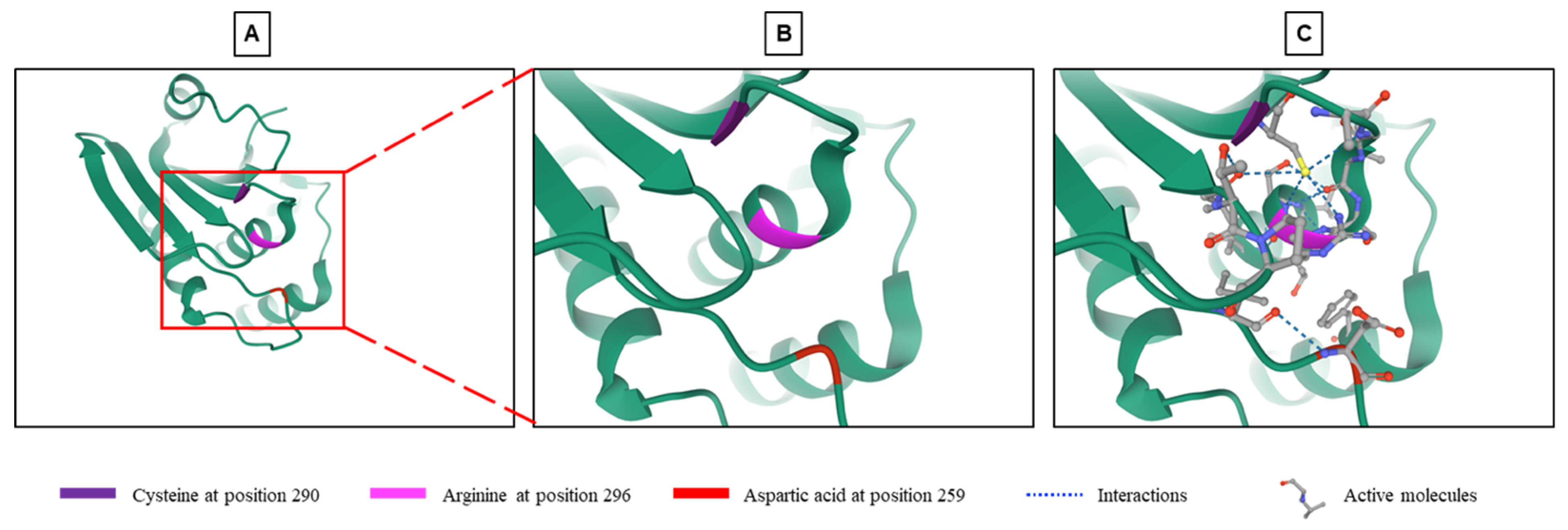
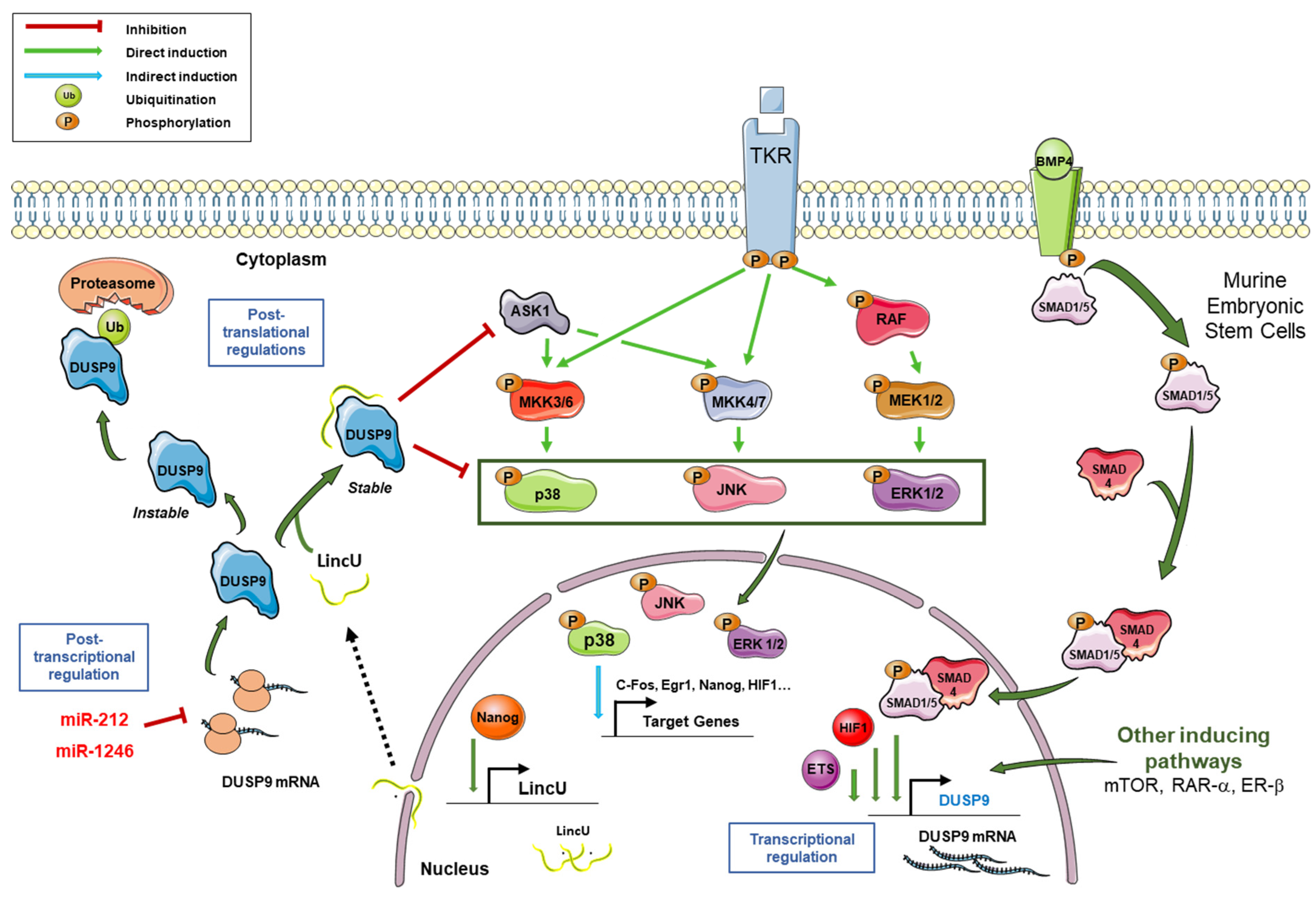
2. General Characteristics of DUSP9 and Mechanisms of Regulation
3. DUSP9 in Embryonic Stem Cell Pluripotency and Sex Differences
4. DUSP9 and Metabolic Diseases
5. DUSP9 and Cardiac Disease
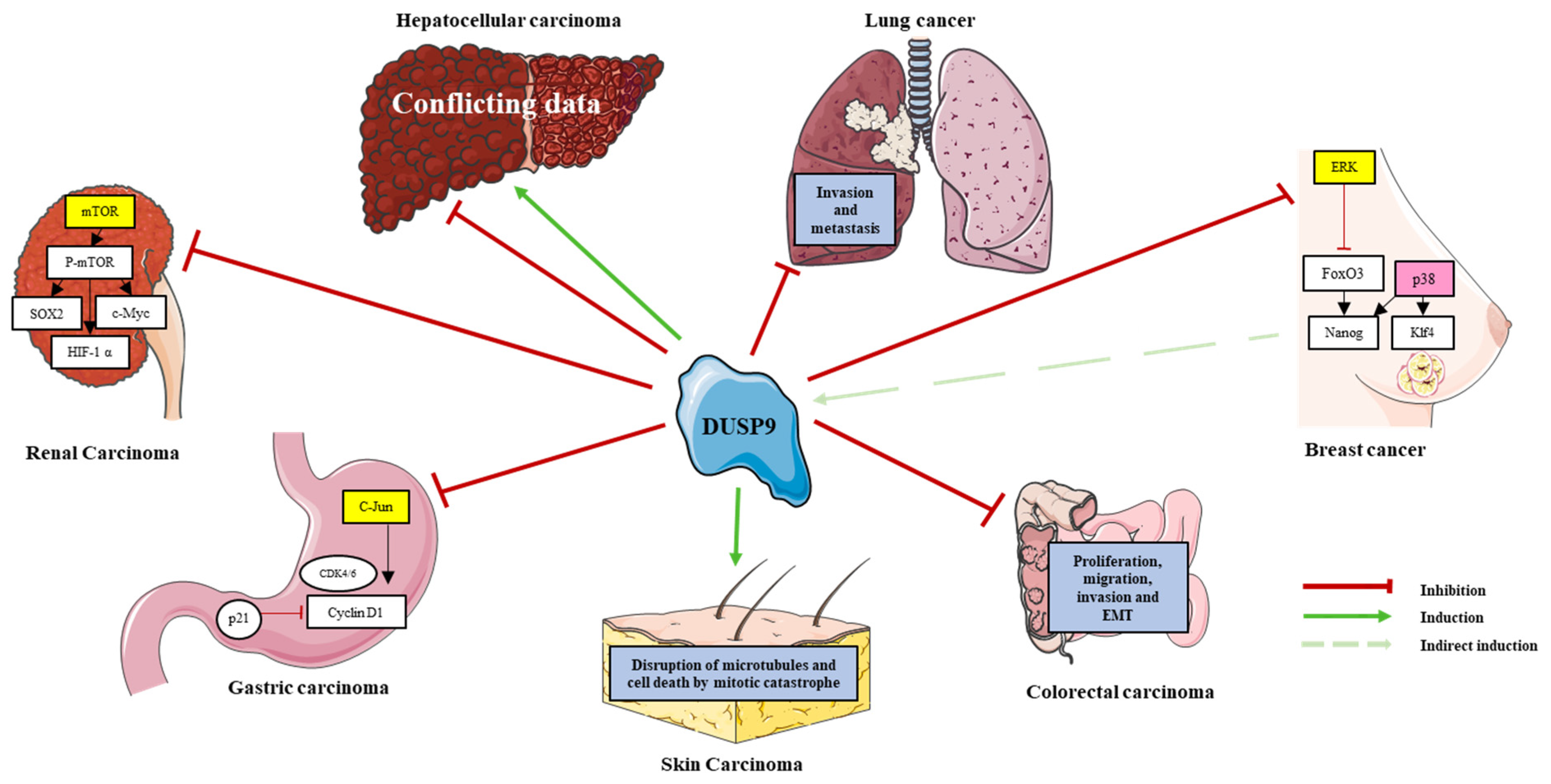
6. DUSP9 in Cancers
6.1. Breast Cancer
6.2. Colorectal Cancer
6.3. Gastric Cancer
6.4. Liver Cancer
6.5. Lung Cancer
6.6. Kidney Cancer
6.7. Skin Cancer
7. DUSP9 Is the Target for Therapy
8. Conclusions
- (1)
- DUSP9 is tightly regulated at transcriptional, post-transcriptional and post-translational levels by transcription factors, promoter methylation, miRNAs, lncRNA and ubiquitination. Its rigorous regulation is therefore necessary to maintain normal cell function and physiological homeostasis.
- (2)
- DUSP9 has a central role in sex differences, metabolic disorders and tumorigenesis. At a pathophysiological level, DUSP9 is strongly involved in the regulation of insulin signaling, and consequently, of the downstream phosphorylation cascades and metabolic processes. Therefore, any therapeutic intervention to increase DUSP9 expression or to control the activity of ERK1/2, JNK, p38 MAPK and/or ASK1 kinases could be beneficial for patients presenting metabolic syndromes such as type 2 diabetes, morbid obesity, liver cirrhosis, NAFLD or its most severe form, NASH.
- (3)
- DUSP9 is central in tumorigenesis and is involved in many adult and pediatric cancers. It clearly acts as a tumor suppressor in kidney cancer, gastric cancer, skin cancer, CRC and lung cancer. Its high expression is associated with poor prognosis in C2A hepatoblastomas and with cancer stemness and drug resistance in breast cancer (Table 2; Figure 3). However, the role of DUSP9 in the adult liver is still a matter of debate and needs further investigations to determine its pro- or anti-tumoral function in hepatic malignancies (Table 2; Figure 3). As NASH can lead to malignant HCC, therapies influencing DUSP9 activity could also be beneficial in patients with liver cancer.
- (4)
- DUSP9 is also an important gene in heart tissue preservation and can counteract the negative effect mediated by pressure overload on cardiac hypertrophy and cardiomyocytes.
- (5)
- Collectively, these data clearly demonstrated the critical role of DUSP9 in both cell physiology and pathologies. It is therefore a promising therapeutic target to fight against the frequent human diseases that are diabetes, heart failure and cancers.
Funding
Conflicts of Interest
Abbreviations
| 3′UTR | 3′-untranslated region |
| ASK1 | MAPKKK apoptosis signal-regulating kinase 1 |
| CRC | colorectal carcinoma |
| DR1 element | inverted direct repeat separated by 1 |
| DUSP | dual specificity phosphatase |
| ERβ | estrogen receptor-beta |
| HCC | hepatocellular carcinoma |
| hESC | human embryonic stem cell |
| HIF1α | hypoxia-inducible factor 1 alpha |
| IHC | immunohistochemistry |
| IRS1 | insulin receptor substrate-1 |
| KIM | kinase-interacting motif |
| KO | knockout |
| MAPK | mitogen-activated protein kinase |
| mEGC | murine embryonic germ cell |
| mESC | mouse embryonic stem cell |
| MKB | MAP kinase-binding motif |
| MKP | MAP kinase phosphatase |
| NASH | non-alcoholic steatohepatitis |
| NAFLD | non-alcoholic fatty liver disease |
| pNPP | para-nitrophenylphosphate |
| RAR | retinoic acid receptor |
References
- Keshet, Y.; Seger, R. The MAP Kinase Signaling Cascades: A System of Hundreds of Components Regulates a Diverse Array of Physiological Functions. In MAP Kinase Signaling Protocols; Seger, R., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 661, pp. 3–38. ISBN 978-1-60761-794-5. [Google Scholar]
- Jiang, L.; Wang, Y.; Liu, G.; Liu, H.; Zhu, F.; Ji, H.; Li, B. C-Phycocyanin Exerts Anti-Cancer Effects via the MAPK Signaling Pathway in MDA-MB-231 Cells. Cancer Cell Int. 2018, 18, 12. [Google Scholar] [CrossRef]
- Chen, H.-F.; Chuang, H.-C.; Tan, T.-H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. P38 MAP-Kinases Pathway Regulation, Function and Role in Human Diseases. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Gaestel, M. MAPK-Activated Protein Kinases (MKs): Novel Insights and Challenges. Front. Cell Dev. Biol. 2016, 3, 88. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The MAPK Pathway across Different Malignancies: A New Perspective: Tissue-Specific MAPK Signaling. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Ghousein, A.; Mosca, N.; Cartier, F.; Charpentier, J.; Dupuy, J.; Raymond, A.; Bioulac-Sage, P.; Grosset, C.F. MiR-4510 Blocks Hepatocellular Carcinoma Development through RAF1 Targeting and RAS/RAF/MEK/ERK Signalling Inactivation. Liver Int. 2020, 40, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Low, H.B.; Zhang, Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016, 16, 85–98. [Google Scholar] [CrossRef]
- Nayak, J.; Gastonguay, A.J.; Talipov, M.R.; Vakeel, P.; Span, E.A.; Kalous, K.S.; Kutty, R.G.; Jensen, D.R.; Pokkuluri, P.R.; Sem, D.S.; et al. Protein Expression, Characterization and Activity Comparisons of Wild Type and Mutant DUSP5 Proteins. BMC Biochem. 2014, 15, 27. [Google Scholar] [CrossRef]
- Kutty, R.G. Dual Specificity Phosphatase 5-Substrate Interaction: A Mechanistic Perspective. Compr. Physiol. 2017, 7, 1449–1461. [Google Scholar] [PubMed]
- Seternes, O.-M.; Kidger, A.M.; Keyse, S.M. Dual-Specificity MAP Kinase Phosphatases in Health and Disease. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2019, 1866, 124–143. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhang, C.; Qu, L.; Lu, C.; Xiao, M.; Ni, R.; Liu, J. MKP-4 Suppresses Hepatocarcinogenesis by Targeting ERK1/2 Pathway. Cancer Cell Int. 2019, 19, 61. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Tan, T.-H. DUSPs, to MAP Kinases and Beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and P38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202. [Google Scholar] [CrossRef]
- Muda, M.; Boschert, U.; Smith, A.; Antonsson, B.; Gillieron, C.; Chabert, C.; Camps, M.; Martinou, I.; Ashworth, A.; Arkinstall, S. Molecular Cloning and Functional Characterization of a Novel Mitogen-Activated Protein Kinase Phosphatase, MKP-4. J. Biol. Chem. 1997, 272, 5141–5151. [Google Scholar] [CrossRef]
- Camps, M.; Nichols, A.; Gillieron, C.; Antonsson, B.; Muda, M.; Chabert, C.; Boschert, U.; Arkinstall, S. Catalytic Activation of the Phosphatase MKP-3 by ERK2 Mitogen-Activated Protein Kinase. Science 1998, 280, 1262–1265. [Google Scholar] [CrossRef]
- Buffet, C. Anomalies Moléculaires de la Voie MAPK et Cancer Papillaire de la Thyroïde: Étude de Deux Phosphatases Spécifiques de ERK, DUSP5 et DUSP6. Ph.D. Thesis, Université René Descartes, Paris, France, 2015. [Google Scholar]
- Chen, K.; Gorgen, A.; Ding, A.; Du, L.; Jiang, K.; Ding, Y.; Sapisochin, G.; Ghanekar, A. Dual-Specificity Phosphatase 9 Regulates Cellular Proliferation and Predicts Recurrence After Surgery in Hepatocellular Carcinoma. Hepatol. Commun. 2021, 5, 1310–1328. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, Y.; Yu, H.; Shen, B.; Liang, Y.; Jin, R.; Liu, X.; Shi, L.; Cai, X. Role of DUSP1/MKP1 in Tumorigenesis, Tumor Progression and Therapy. Cancer Med. 2016, 5, 2061–2068. [Google Scholar] [CrossRef]
- Wei, W.; Jiao, Y.; Postlethwaite, A.; Stuart, J.M.; Wang, Y.; Sun, D.; Gu, W. Dual-Specificity Phosphatases 2: Surprising Positive Effect at the Molecular Level and a Potential Biomarker of Diseases. Genes Immun. 2013, 14, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.-Y.; Lin, Y.-C.; Liao, F.-H.; Chan, Y.-C.; Huang, C.-Y. Dual-Specificity Phosphatase 4 Regulates STAT5 Protein Stability and Helper T Cell Polarization*. PLoS ONE 2015, 10, e0145880. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Budczies, J.; Munkácsy, G.; Esteva, F.J.; Szabó, A.; Miquel, T.P.; Győrffy, B. DUSP4 Is Associated with Increased Resistance against Anti-HER2 Therapy in Breast Cancer. Oncotarget 2017, 8, 77207–77218. [Google Scholar] [CrossRef]
- Seo, H.; Cho, Y.-C.; Ju, A.; Lee, S.; Park, B.C.; Park, S.G.; Kim, J.-H.; Kim, K.; Cho, S. Dual-Specificity Phosphatase 5 Acts as an Anti-Inflammatory Regulator by Inhibiting the ERK and NF-ΚB Signaling Pathways. Sci. Rep. 2017, 7, 17348. [Google Scholar] [CrossRef]
- Muhammad, K.A.; Nur, A.A.; Nurul, H.S.; Narazah, M.Y.; Siti, R.A.R. Dual-Specificity Phosphatase 6 (DUSP6): A Review of Its Molecular Characteristics and Clinical Relevance in Cancer. Cancer Biol. Med. 2018, 15, 14. [Google Scholar] [CrossRef]
- Luan, T.; Zhang, X.; Wang, S.; Song, Y.; Zhou, S.; Lin, J.; An, W.; Yuan, W.; Yang, Y.; Cai, H.; et al. Long Non-Coding RNA MIAT Promotes Breast Cancer Progression and Functions as CeRNA to Regulate DUSP7 Expression by Sponging MiR-155-5p. Oncotarget 2017, 8, 76153–76164. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Zhou, Y.; Long, R.; Chen, C.; Zhao, J.; Cui, P.; Guo, M.; Liang, G.; Xu, L. DUSP8 Phosphatase: Structure, Functions, Expression Regulation and the Role in Human Diseases. Cell Biosci. 2019, 9, 70. [Google Scholar] [CrossRef]
- Jiang, L.; Ren, L.; Guo, X.; Zhao, J.; Zhang, H.; Chen, S.; Le, S.; Liu, H.; Ye, P.; Chen, M.; et al. Dual-Specificity Phosphatase 9 Protects against Cardiac Hypertrophy by Targeting ASK1. Int. J. Biol. Sci. 2021, 17, 2193–2204. [Google Scholar] [CrossRef]
- Li, Z.; Fei, T.; Zhang, J.; Zhu, G.; Wang, L.; Lu, D.; Chi, X.; Teng, Y.; Hou, N.; Yang, X.; et al. BMP4 Signaling Acts via Dual-Specificity Phosphatase 9 to Control ERK Activity in Mouse Embryonic Stem Cells. Cell Stem Cell 2012, 10, 171–182. [Google Scholar] [CrossRef]
- Wei, Q.; Pu, X.; Zhang, L.; Xu, Y.; Duan, M.; Wang, Y. Expression of Dual-Specificity Phosphatase 9 in Placenta and Its Relationship with Gestational Diabetes Mellitus. J. Diabetes Res. 2019, 2019, 1–7. [Google Scholar] [CrossRef]
- Wu, S.; Wang, Y.; Sun, L.; Zhang, Z.; Jiang, Z.; Qin, Z.; Han, H.; Liu, Z.; Li, X.; Tang, A.; et al. Decreased Expression of Dual-Specificity Phosphatase 9 Is Associated with Poor Prognosis in Clear Cell Renal Cell Carcinoma. BMC Cancer 2011, 11, 413. [Google Scholar] [CrossRef]
- Ye, P.; Xiang, M.; Liao, H.; Liu, J.; Luo, H.; Wang, Y.; Huang, L.; Chen, M.; Xia, J. Dual-Specificity Phosphatase 9 Protects Against Nonalcoholic Fatty Liver Disease in Mice through ASK1 Suppression: Steatohepatitis/Metabolic Liver Disease. Hepatology 2019, 69, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, T.; Barrios, A.; Tucker, A.; Collazo, J.; Arias, N.; Fazel, S.; Halim, M.; Huynh, T.; Singh, R.; Pervin, S. DUSP9-Mediated Reduction of PERK1/2 Supports Cancer Stem Cell-like Traits and Promotes Triple Negative Breast Cancer. Am. J. Cancer Res. 2020, 10, 3487–3506. [Google Scholar]
- Wu, Y.-K.; Hu, L.-F.; Lou, D.-S.; Wang, B.-C.; Tan, J. Targeting DUSP16/TAK1 Signaling Alleviates Hepatic Dyslipidemia and Inflammation in High Fat Diet (HFD)-Challenged Mice through Suppressing JNK MAPK. Biochem. Biophys. Res. Commun. 2020, 524, 142–149. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Li, J.-P.; Chiu, L.-L.; Lan, J.-L.; Chen, D.-Y.; Chuang, H.-C.; Huang, C.-Y.; Tan, T.-H. Dual-Specificity Phosphatase 14 (DUSP14/MKP6) Negatively Regulates TCR Signaling by Inhibiting TAB1 Activation. J. Immunol. 2014, 192, 1547–1557. [Google Scholar] [CrossRef]
- Jung, S.; Nah, J.; Han, J.; Choi, S.-G.; Kim, H.; Park, J.; Pyo, H.-K.; Jung, Y.-K. Dual-Specificity Phosphatase 26 (DUSP26) Stimulates Aβ42 Generation by Promoting Amyloid Precursor Protein Axonal Transport during Hypoxia. J. Neurochem. 2016, 137, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M. Dual-Specificity MAP Kinase Phosphatases (MKPs) and Cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef]
- Lang, R.; Hammer, M.; Mages, J. DUSP Meet Immunology: Dual Specificity MAPK Phosphatases in Control of the Inflammatory Response. J. Immunol. 2006, 177, 7497–7504. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Raffi, F.A.M. Dual-Specificity Phosphatases in Immunity and Infection: An Update. Int. J. Mol. Sci. 2019, 20, 2710. [Google Scholar] [CrossRef]
- Theodosiou, A.; Ashworth, A. MAP Kinase Phosphatases. Genome Biol. 2002, 3, REVIEWS3009. [Google Scholar] [CrossRef]
- Jeong, D.G.; Yoon, T.S.; Jung, S.K.; Park, B.C.; Park, H.; Ryu, S.E.; Kim, S.J. Exploring Binding Sites Other than the Catalytic Core in the Crystal Structure of the Catalytic Domain of MKP-4. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 25–31. [Google Scholar] [CrossRef]
- Hong, S.B.; Lubben, T.H.; Dolliver, C.M.; Petrolonis, A.J.; Roy, R.A.; Li, Z.; Parsons, T.F.; Li, P.; Xu, H.; Reilly, R.M.; et al. Expression, Purification, and Enzymatic Characterization of the Dual Specificity Mitogen-Activated Protein Kinase Phosphatase, MKP-4. Bioorg. Chem. 2005, 33, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.-K.; Amini, B.; Andersen, E.; Andersson, A.-C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A Human Protein Atlas for Normal and Cancer Tissues Based on Antibody Proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef]
- Xu, H.; Dembski, M.; Yang, Q.; Yang, D.; Moriarty, A.; Tayber, O.; Chen, H.; Kapeller, R.; Tartaglia, L.A. Dual Specificity Mitogen-Activated Protein (MAP) Kinase Phosphatase-4 Plays a Potential Role in Insulin Resistance. J. Biol. Chem. 2003, 278, 30187–30192. [Google Scholar] [CrossRef] [PubMed]
- Jiapaer, Z.; Li, G.; Ye, D.; Bai, M.; Li, J.; Guo, X.; Du, Y.; Su, D.; Jia, W.; Chen, W.; et al. LincU Preserves Naive Pluripotency by Restricting ERK Activity in Embryonic Stem Cells. Stem Cell Rep. 2018, 11, 395–409. [Google Scholar] [CrossRef]
- Imajo, M.; Kondoh, K.; Yamamoto, T.; Nakayama, K.; Nakajima-Koyama, M.; Nishida, E. Antagonistic Interactions between Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase and Retinoic Acid Receptor Signaling in Colorectal Cancer Cells. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthi, V.P.; Ratri, A.; Masumi, S.; Borosha, S.; Ghosh, S.; Christenson, L.K.; Roby, K.F.; Wolfe, M.W.; Rumi, M.A.K. Granulosa Cell Genes That Regulate Ovarian Follicle Development beyond the Antral Stage: The Role of Estrogen Receptor β. Mol. Cell. Endocrinol. 2021, 528, 111212. [Google Scholar] [CrossRef]
- Trezeguet, V.; Grosset, C.F. Les MicroARN et Leur Potentiel Thérapeutique En Cancérologie: Le Point En 2020. J. Biol. Méd. 2020, 32, 271–277. [Google Scholar]
- Cartier, F.; Indersie, E.; Lesjean, S.; Charpentier, J.; Hooks, K.B.; Ghousein, A.; Desplat, A.; Dugot-Senant, N.; Trézéguet, V.; Sagliocco, F.; et al. New Tumor Suppressor MicroRNAs Target Glypican-3 in Human Liver Cancer. Oncotarget 2017, 8, 41211–41226. [Google Scholar] [CrossRef]
- Indersie, E.; Lesjean, S.; Hooks, K.B.; Sagliocco, F.; Ernault, T.; Cairo, S.; Merched-Sauvage, M.; Rullier, A.; Le Bail, B.; Taque, S.; et al. MicroRNA Therapy Inhibits Hepatoblastoma Growth in Vivo by Targeting Beta-Catenin and Wnt Signaling. Hepatol. Commun. 2017, 1, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Jalvy, S.; Ladeiro, Y.; Combe, C.; Vachet, L.; Sagliocco, F.; Bioulac-Sage, P.; Pitard, V.; Jacquemin-Sablon, H.; Zucman-Rossi, J.; et al. A Functional Screening Identifies Five Micrornas Controlling Glypican-3: Role of Mir-1271 down-Regulation in Hepatocellular Carcinoma. Hepatology 2013, 57, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Xiao, X.; Dong, Y.; Zhou, X.H. Fermented Barley Extracts with Lactobacillus Plantarum Dy-1 Rich in Vanillic Acid Modulate Glucose Consumption in Human HepG2 Cells. Biomed. Environ. Sci. 2018, 31, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Liang, N.; Huang, Q.; Sun, T.; Xue, H.; Xie, T.; Wang, X.; Wang, Q. Downregulation of DUSP9 Promotes Tumor Progression and Contributes to Poor Prognosis in Human Colorectal Cancer. Front. Oncol. 2020, 10, 547011. [Google Scholar] [CrossRef]
- Chang, J.; Huang, L.; Cao, Q.; Liu, F. Identification of Colorectal Cancer-Restricted MicroRNAs and Their Target Genes Based on High-Throughput Sequencing Data. OncoTargets Ther. 2016, 9, 1787–1794. [Google Scholar] [CrossRef]
- Choi, J.; Clement, K.; Huebner, A.J.; Webster, J.; Rose, C.M.; Brumbaugh, J.; Walsh, R.M.; Lee, S.; Savol, A.; Etchegaray, J.P.; et al. DUSP9 Modulates DNA Hypomethylation in Female Mouse Pluripotent Stem Cells. Cell Stem Cell 2017, 20, 706–719.e7. [Google Scholar] [CrossRef]
- Song, J.; Janiszewski, A.; De Geest, N.; Vanheer, L.; Talon, I.; El Bakkali, M.; Oh, T.; Pasque, V. X-Chromosome Dosage Modulates Multiple Molecular and Cellular Properties of Mouse Pluripotent Stem Cells Independently of Global DNA Methylation Levels. Stem Cell Rep. 2019, 12, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Genolet, O.; Monaco, A.A.; Dunkel, I.; Boettcher, M.; Schulz, E.G. Identification of X-Chromosomal Genes That Drive Sex Differences in Embryonic Stem Cells through a Hierarchical CRISPR Screening Approach. Genome Biol. 2021, 22, 110. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxid. Med. Cell. Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef]
- Jung, H.J.; Suh, Y. Regulation of IGF -1 Signaling by MicroRNAs. Front. Genet. 2015, 5. [Google Scholar] [CrossRef]
- Mukherjee, B.; Hossain, C.M.; Mondal, L.; Paul, P.; Ghosh, M.K. Obesity and Insulin Resistance: An Abridged Molecular Correlation. Lipid Insights 2013, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, Insulin Resistance, and the Metabolic Syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef]
- Christie, G.R.; Williams, D.J.; Macisaac, F.; Dickinson, R.J.; Rosewell, I.; Keyse, S.M. The Dual-Specificity Protein Phosphatase DUSP9/MKP-4 Is Essential for Placental Function but Is Not Required for Normal Embryonic Development. Mol. Cell. Biol. 2005, 25, 8323–8333. [Google Scholar] [CrossRef] [PubMed]
- Bazuine, M.; Carlotti, F.; Tafrechi, R.S.; Hoeben, R.C.; Maassen, J.A. Mitogen-Activated Protein Kinase (MAPK) Phosphatase-1 and -4 Attenuate P38 MAPK during Dexamethasone-Induced Insulin Resistance in 3T3-L1 Adipocytes. Mol. Endocrinol. 2004, 18, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, B.; Eberle, D.; Suzuki, R.; Kahn, C.R. Overexpression of the Dual-Specificity Phosphatase MKP-4/DUSP-9 Protects against Stress-Induced Insulin Resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 3545–3550. [Google Scholar] [CrossRef]
- Petrochilos, D.; Shojaie, A.; Gennari, J.; Abernethy, N. Using Random Walks to Identify Cancer-Associated Modules in Expression Data. BioData Min. 2013, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Luo, X.; Liu, X.; Fang, Z.; Xu, J.; Li, L. DUSP9 Suppresses Proliferation and Migration of Clear Cell Renal Cell Carcinoma via the MTOR Pathway. OncoTargets Ther. 2020, 13, 1321–1330. [Google Scholar] [CrossRef]
- Wu, F.; Lv, T.; Chen, G.; Ye, H.; Wu, W.; Li, G.; Zhi, F.-C. Epigenetic Silencing of DUSP9 Induces the Proliferation of Human Gastric Cancer by Activating JNK Signaling. Oncol. Rep. 2015, 34, 121–128. [Google Scholar] [CrossRef]
- Liu, Y.; Lagowski, J.; Sundholm, A.; Sundberg, A.; Kulesz-Martin, M. Microtubule Disruption and Tumor Suppression by Mitogen-Activated Protein Kinase Phosphatase 4. Cancer Res. 2007, 67, 10711–10719. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wang, H.; Xiao, H.; Lan, B.; Liu, J.; Yang, Z. EEF1A2 and ERN2 Could Potentially Discriminate Metastatic Status of Mediastinal Lymph Node in Lung Adenocarcinomas Harboring EGFR 19Del/L858R Mutations. Thorac. Cancer 2020, 11, 2755–2766. [Google Scholar] [CrossRef]
- Cairo, S.; Armengol, C.; De Reyniès, A.; Wei, Y.; Thomas, E.; Renard, C.-A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic Stem-like Phenotype and Interplay of Wnt/β-Catenin and Myc Signaling in Aggressive Childhood Liver Cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef]
- Hooks, K.B.; Audoux, J.; Fazli, H.; Lesjean, S.; Ernault, T.; Dugot-Senant, N.; Leste-Lasserre, T.; Hagedorn, M.; Rousseau, B.; Danet, C.; et al. New Insights into Diagnosis and Therapeutic Options for Proliferative Hepatoblastoma. Hepatology 2018, 68, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M.; Liu, P.; Hayashi, N.; Lluch, A.; Ferrer-Lozano, J.; Hortobágyi, G.N. High ERK Protein Expression Levels Correlate with Shorter Survival in Triple-Negative Breast Cancer Patients. Oncologist 2012, 17, 766–774. [Google Scholar] [CrossRef]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of Apc Allows Phenotypic Manifestation of the Transforming Properties of an Endogenous K-Ras Oncogene in Vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127. [Google Scholar] [CrossRef] [PubMed]
- Jenner, S.; Wiedorn, K.H.; Techel, D. Development of a DUSP9 Methylation Screening Assay. Pathol. Oncol. Res. 2015, 21, 123–130. [Google Scholar] [CrossRef]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in Targeted Therapies for Hepatocellular Carcinoma in the Genomic Era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ni, W.; Xiao, M.; Jiang, F.; Ni, R. Decreased Expression and Prognostic Role of Mitogen-Activated Protein Kinase Phosphatase 4 in Hepatocellular Carcinoma. J. Gastrointest. Surg. 2013, 17, 756–765. [Google Scholar] [CrossRef] [PubMed]
- López-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 Is a Hepatoblastoma-Derived Cell Line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef]
- Sanders, J.A.; Brilliant, K.E.; Clift, D.; Patel, A.; Cerretti, B.; Claro, P.; Mills, D.R.; Hixson, D.C.; Gruppuso, P.A. The Inhibitory Effect of Rapamycin on the Oval Cell Response and Development of Preneoplastic Foci in the Rat. Exp. Mol. Pathol. 2012, 93, 40–49. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.; Zang, R.K.; Du, Y.N. HSA_CIRC_0004050 on Proliferation and Apoptosis of A549 Cells through ERK/JNK Signaling Pathway. J. Biol. Regul. Homeost. Agents 2020, 34, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Laczmanska, I.; Laczmanski, L.; Sasiadek, M.M. Expression Analysis of Tyrosine Phosphatase Genes at Different Stages of Renal Cell Carcinoma. Anticancer Res. 2020, 40, 5667–5671. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated Profiling of MicroRNAs and MRNAs: MicroRNAs Located on Xq27.3 Associate with Clear Cell Renal Cell Carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T. Drug Discovery and Protein Tyrosine Phosphatases. Curr. Med. Chem. 2009, 16, 2095–2176. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Z.-Y. PTP1B as a Drug Target: Recent Developments in PTP1B Inhibitor Discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Lee, J.H.; Kwon, S.J.; Kang, H.J.; Chung, S.J. Ginkgolic Acid as a Dual-Targeting Inhibitor for Protein Tyrosine Phosphatases Relevant to Insulin Resistance. Bioorg. Chem. 2018, 81, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Na, N.; Sheng, H.; Feng, B.; Wang, H.; Zhu, P.; Zhang, W.; Zhang, M.; Deng, Z. Ginkgolic Acid Inhibits the Growth of Renal Cell Carcinoma Cells via Inactivation of the EGFR Signaling Pathway. Exp. Ther. Med. 2020, 19, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.-R.; Yang, H. Ginkgolic Acid (GA) Suppresses Gastric Cancer Growth by Inducing Apoptosis and Suppressing STAT3/JAK2 Signaling Regulated by ROS. Biomed. Pharmacother. 2020, 125, 109585. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Classification | Gene Symbol | Synonyms | Chromosomal Localization | Cell Localization | MAPK Substrates (Others) | Inducible by MAPKs | Main Functions in Physiological and Pathophysiological States |
|---|---|---|---|---|---|---|---|
| Typical MKPs | DUSP1 | MKP1 | 5 | Nuclear | JNK, p38 > ERK | ERK, p38 | Involved in infectious diseases, pulmonary diseases, inflammatory disorders, atherosclerosis, tumorigenesis and tumor progression [22]. |
| DUSP2 | PAC1 | 2 | Nuclear | ERK, JNK, p38 | ERK, JNK | Involved in immune and inflammatory responses, cancer, CLN3 disease and endometriosis [23]. | |
| DUSP4 | MKP2 | 8 | Nuclear | ERK, JNK > p38 | ERK | Involved in inflammatory cytokine secretion, susceptibility to sepsis shock, and resistance to Leishmania mexicana infection [24,25]. | |
| DUSP5 | hVH3 | 10 | Nuclear | ERK | ERK | Plays an anti-inflammatory role and has tumor suppressive functions in several types of cancer [26]. | |
| DUSP6 | MKP3 | 12 | Cytoplasmic | ERK | ERK | Plays a role in carcinogenesis in several cancers as an oncogene or a tumor suppressor [27]. | |
| DUSP7 | MKPX | 3 | Cytoplasmic | ERK, JNK, p38 | N/D | Involved in some cancers [28]. | |
| DUSP8 | hVH5 | 11 | Dually-located | ERK, JNK, p38 | N/D | Plays a role in the central nervous system, circulatory system, urinary system, immune system, genetic diseases and cancers [29]. | |
| DUSP9 | MKP4 | X | Cytoplasmic | ERK >> p38, JNK | N/D | Involved in development of cardiac dystrophy, metabolic diseases and cancers [30,31,32,33,34]. | |
| (MAP3K5/ASK1) | |||||||
| DUSP10 | MKP5 | 1 | Dually-located | JNK, p38 >> ERK | N/D | Involved in immune response, anti-inflammatory response and some cancers [35]. | |
| DUSP16 | MKP7 | 12 | Dually-located | JNK | N/D | Involved in non-alcoholic steatohepatitis and some cancers [36]. | |
| Atypical MKPs | DUSP14 | MKP6 | 17 | Dually-located | ERK, JNK, p38 | N/D | Involved in immune response, bone diseases and cancers [37]. |
| DUSP26 | MKP8 | 8 | Nuclear | p38 | N/D | Regulates neuronal cell proliferation and acts as an oncogene or a tumor suppressor depending on the cellular context [38]. |
| Organ | Cancer | Expression | Role | Main Results |
|---|---|---|---|---|
| Kidneys | Clear cell renal carcinoma | Low | Tumor suppressor | DUSP9 blocks the growth and migration of clear cell renal cell carcinoma cells in vitro and tumor development in mice through mTOR pathway inhibition [69]. |
| Stomach | Gastric carcinoma | Low | Tumor suppressor | DUSP9 inhibits growth of MKN-1 cells through cell cycle arrest in S-G2/M phases and JNK pathway inactivation [70]. |
| Skin | Skin carcinoma | Low | Tumor suppressor | DUSP9 triggers tumoral cell death by arresting cells in G2/M phase and by inducing microtubule disruption and mitotic catastrophe [11,71]. |
| Colon | Colorectal carcinoma | Low | Tumor suppressor | In vitro and in vivo, DUSP9 affects proliferation, migration, invasion and epithelial-mesenchymal transition of colorectal cancer cells [56]. |
| Lungs | Lung cancer | Low | Tumor suppressor | DUSP9 down expression is associated with tumor progression, invasion and metastasis [72]. |
| Breast | Breast cancer | Low | Involved in chemoresistance | DUSP9 promotes cancer stem cell enrichment and chemotherapy resistance of triple negative breast cancer through HIF1α [17,35]. |
| Liver | Hepatocellular carcinoma | Low or High | Oncogene or Tumor suppressor | Conflicting results report a tumor suppressive or an oncogenic role of DUSP9 in adult liver cancer [15,21]. |
| Hepatoblastoma | High | Associated with poor prognosis | DUSP9 is increased in poor prognosis C2 or C2A group [73,74]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoubai, F.Z.; Grosset, C.F. DUSP9, a Dual-Specificity Phosphatase with a Key Role in Cell Biology and Human Diseases. Int. J. Mol. Sci. 2021, 22, 11538. https://doi.org/10.3390/ijms222111538
Khoubai FZ, Grosset CF. DUSP9, a Dual-Specificity Phosphatase with a Key Role in Cell Biology and Human Diseases. International Journal of Molecular Sciences. 2021; 22(21):11538. https://doi.org/10.3390/ijms222111538
Chicago/Turabian StyleKhoubai, Fatma Zohra, and Christophe F. Grosset. 2021. "DUSP9, a Dual-Specificity Phosphatase with a Key Role in Cell Biology and Human Diseases" International Journal of Molecular Sciences 22, no. 21: 11538. https://doi.org/10.3390/ijms222111538
APA StyleKhoubai, F. Z., & Grosset, C. F. (2021). DUSP9, a Dual-Specificity Phosphatase with a Key Role in Cell Biology and Human Diseases. International Journal of Molecular Sciences, 22(21), 11538. https://doi.org/10.3390/ijms222111538