
Resilience in Long-Term Viral Infection: Genetic Determinants and Interactions

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Expression of Gm41561, a Long Non-Coding RNA Gene, Was Significantly Affected by TMEV Infection Regardless of Mouse Strain
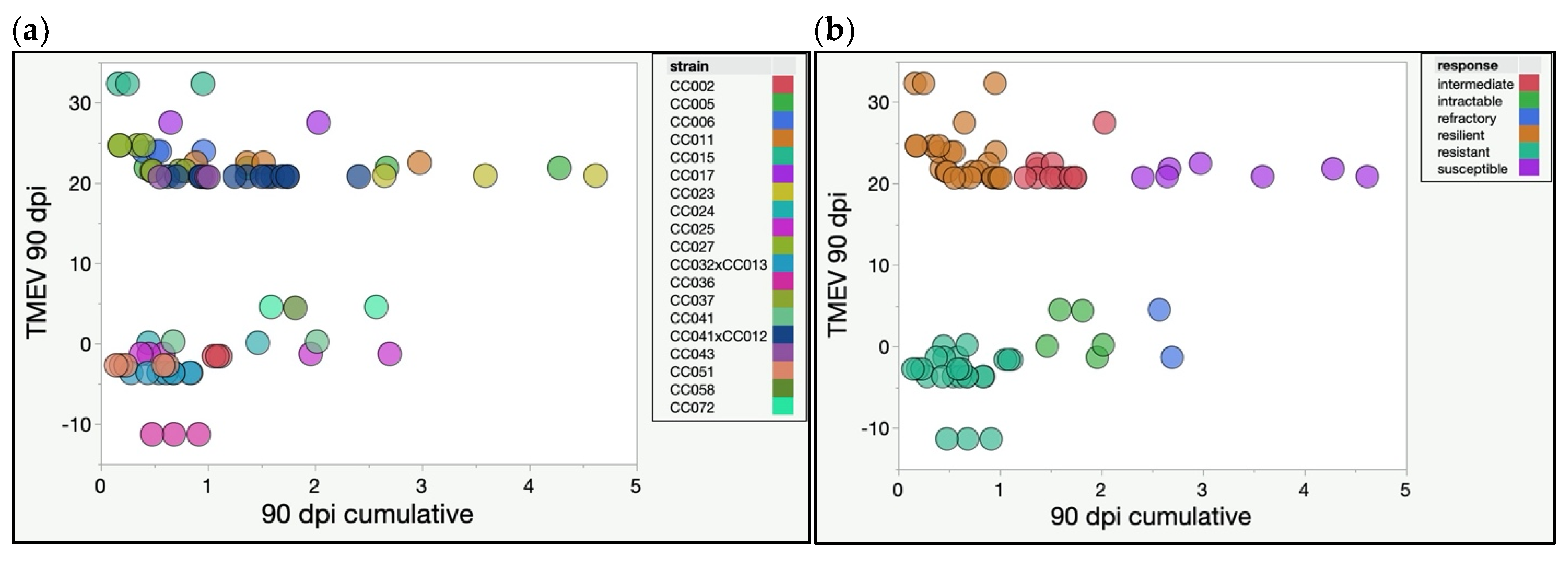
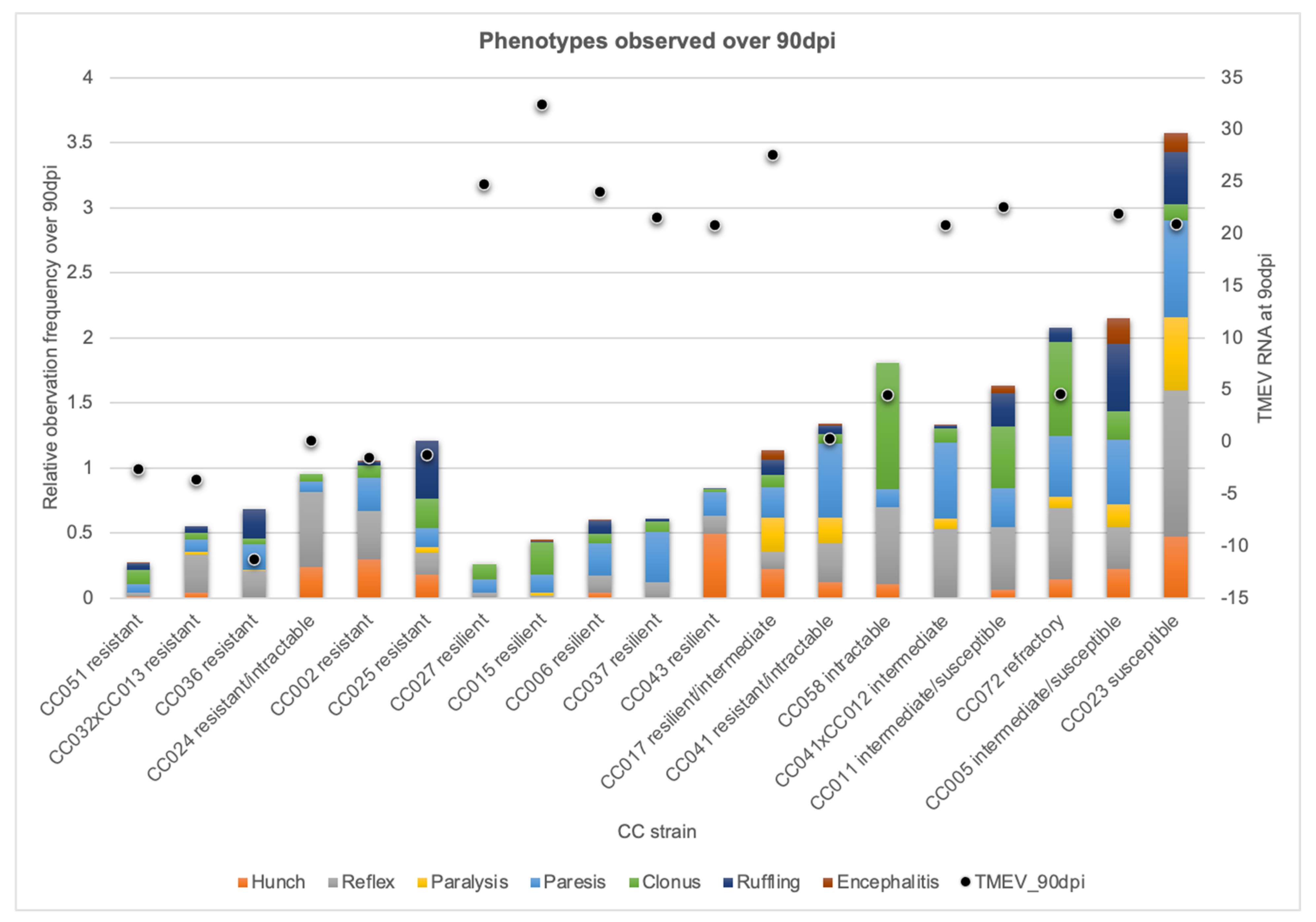
2.2. CC Strains Demonstrated Novel Responses to TMEV Based on TMEV Persistence and Phenotypic Severity
2.3. Genetic Diversity Contributed to Protection, Compensation, or Capitulation in the Face of TMEV Infection
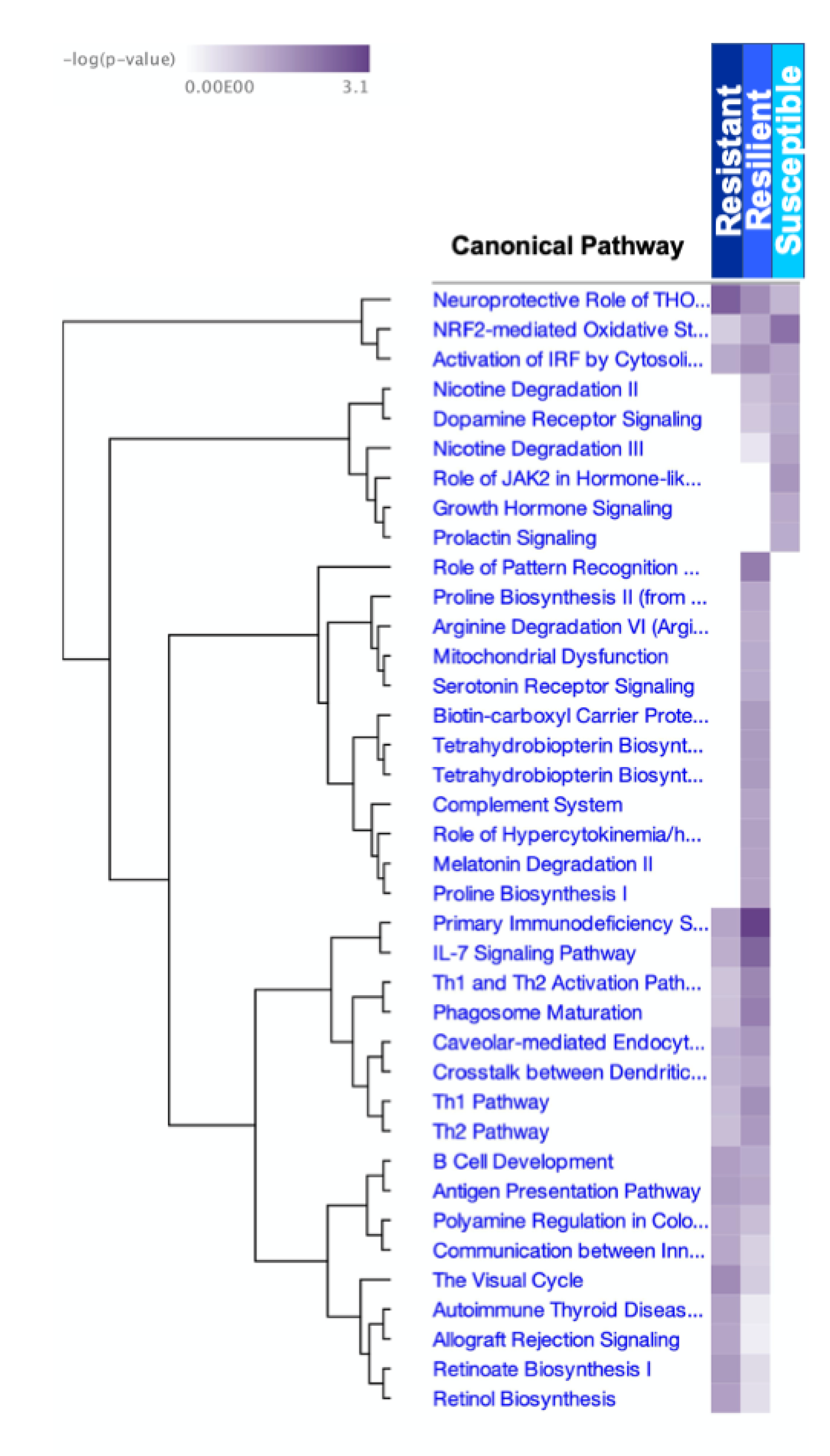
2.4. Upstream Regulators of Biological Functions and Their Molecular Targets, Varied by TMEV Response Group
2.5. Unique Biomarkers Distinguished TMEV Response Categories
2.6. Haplotypes Provided Context for Pleiotropy and Predictive Alleles
3. Discussion
4. Materials and Methods
4.1. Mice and Phenotyping
4.2. RNA Isolation and Sequencing
4.3. Identification of Key Pathways, Networks, and Regulatory Molecules
4.4. Haplotypes and Sequence Variation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Threadgill, D.W.; Hunter, K.W.; Williams, R.W. Genetic dissection of complex and quantitative traits: From fantasy to reality via a community effort. Mamm. Genome 2002, 13, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Threadgill, D.W.; Miller, D.R.; Churchill, G.A.; de Villena, F.P. The collaborative cross: A recombinant inbred mouse population for the systems genetic era. ILAR J. 2011, 52, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; Gelfond, J.A.; Airey, D.C.; Lu, L.; Manly, K.F.; Williams, R.W.; Threadgill, D.W. Quantitative trait locus analysis using recombinant inbred intercrosses: Theoretical and empirical considerations. Genetics 2005, 170, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Brahic, M.; Bureau, J.F.; Michiels, T. The genetics of the persistent infection and demyelinating disease caused by Theiler’s virus. Annu. Rev. Microbiol. 2005, 59, 279–298. [Google Scholar] [CrossRef]
- Butterfield, R.J.; Roper, R.J.; Rhein, D.M.; Melvold, R.W.; Haynes, L.; Ma, R.Z.; Doerge, R.W.; Teuscher, C. Sex-specific quantitative trait loci govern susceptibility to Theiler’s murine encephalomyelitis virus-induced demyelination. Genetics 2003, 163, 1041–1046. [Google Scholar] [CrossRef]
- Clatch, R.J.; Melvold, R.W.; Miller, S.D.; Lipton, H.L. Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease in mice is influenced by the H-2D region: Correlation with TEMV-specific delayed-type hypersensitivity. J. Immunol. 1985, 135, 1408–1414. [Google Scholar]
- Fiette, L.; Aubert, C.; Muller, U.; Huang, S.; Aguet, M.; Brahic, M.; Bureau, J.F. Theiler’s virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J. Exp. Med. 1995, 181, 2069–2076. [Google Scholar] [CrossRef]
- Rodriguez, M.; David, C.S. Demyelination induced by Theiler’s virus: Influence of the H-2 haplotype. J. Immunol. 1985, 135, 2145–2148. [Google Scholar]
- Rodriguez, M.; Leibowitz, J.; David, C.S. Susceptibility to Theiler’s virus-induced demyelination. Mapping of the gene within the H-2D region. J. Exp. Med. 1986, 163, 620–631. [Google Scholar]
- Levillayer, F.; Mas, M.; Levi-Acobas, F.; Brahic, M.; Bureau, J.F. Interleukin 22 is a candidate gene for Tmevp3, a locus controlling Theiler’s virus-induced neurological diseases. Genetics 2007, 176, 1835–1844. [Google Scholar] [CrossRef]
- Aubagnac, S.; Brahic, M.; Bureau, J.F. Viral load and a locus on chromosome 11 affect the late clinical disease caused by Theiler’s virus. J. Virol. 1999, 73, 7965–7971. [Google Scholar] [CrossRef]
- Bieber, A.J.; Suwansrinon, K.; Kerkvliet, J.; Zhang, W.; Pease, L.R.; Rodriguez, M. Allelic variation in the Tyk2 and EGF genes as potential genetic determinants of CNS repair. Proc. Natl. Acad. Sci. USA 2010, 107, 792–797. [Google Scholar] [CrossRef]
- Bihl, F.; Brahic, M.; Bureau, J.F. Two loci, Tmevp2 and Tmevp3, located on the telomeric region of chromosome 10, control the persistence of Theiler’s virus in the central nervous system of mice. Genetics 1999, 152, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Brahic, M.; Bureau, J.F. Genetics of susceptibility to Theiler’s virus infection. Bioessays 1998, 20, 627–633. [Google Scholar] [CrossRef]
- Bureau, J.F.; Drescher, K.M.; Pease, L.R.; Vikoren, T.; Delcroix, M.; Zoecklein, L.; Brahic, M.; Rodriguez, M. Chromosome 14 contains determinants that regulate susceptibility to Theiler’s virus-induced demyelination in the mouse. Genetics 1998, 148, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Melvold, R.W.; Jokinen, D.M.; Knobler, R.L.; Lipton, H.L. Variations in genetic control of susceptibility to Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease. I. Differences between susceptible SJL/J and resistant BALB/c strains map near the T cell beta-chain constant gene on chromosome 6. J. Immunol. 1987, 138, 1429–1433. [Google Scholar]
- Melvold, R.W.; Jokinen, D.M.; Miller, S.D.; Dal Canto, M.C.; Lipton, H.L. Identification of a locus on mouse chromosome 3 involved in differential susceptibility to Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Virol. 1990, 64, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Teuscher, C.; Rhein, D.M.; Livingstone, K.D.; Paynter, R.A.; Doerge, R.W.; Nicholson, S.M.; Melvold, R.W. Evidence that Tmevd2 and eae3 may represent either a common locus or members of a gene complex controlling susceptibility to immunologically mediated demyelination in mice. J. Immunol. 1997, 159, 4930–4934. [Google Scholar]
- Bureau, J.F.; Montagutelli, X.; Bihl, F.; Lefebvre, S.; Guenet, J.L.; Brahic, M. Mapping loci influencing the persistence of Theiler’s virus in the murine central nervous system. Nat. Genet. 1993, 5, 87–91. [Google Scholar] [CrossRef]
- Lipton, H.L.; Melvold, R. Genetic analysis of susceptibility to Theiler’s virus-induced demyelinating disease in mice. J. Immunol. 1984, 132, 1821–1825. [Google Scholar]
- Khan, M.R.; Wellinger, R.J.; Laurent, B. Exploring the Alternative Splicing of Long Noncoding RNAs. Trends Genet. 2021. [Google Scholar] [CrossRef]
- Sun, L.; Goff, L.A.; Trapnell, C.; Alexander, R.; Lo, K.A.; Hacisuleyman, E.; Sauvageau, M.; Tazon-Vega, B.; Kelley, D.R.; Hendrickson, D.G.; et al. Long noncoding RNAs regulate adipogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3387–3392. [Google Scholar] [CrossRef]
- Eldridge, R.; Osorio, D.; Amstalden, K.; Edwards, C.; Young, C.R.; Cai, J.J.; Konganti, K.; Hillhouse, A.; Threadgill, D.W.; Welsh, C.J.; et al. Antecedent presentation of neurological phenotypes in the Collaborative Cross reveals four classes with complex sex-dependencies. Sci. Rep. 2020, 10, 7918. [Google Scholar] [CrossRef]
- Lipton, H.L.; Dal Canto, M.C. Susceptibility of inbred mice to chronic central nervous system infection by Theiler’s murine encephalomyelitis virus. Infect. Immun. 1979, 26, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Clatch, R.J.; Lipton, H.L.; Miller, S.D. Class II-restricted T cell responses in Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease. II. Survey of host immune responses and central nervous system virus titers in inbred mouse strains. Microb. Pathog. 1987, 3, 327–337. [Google Scholar] [CrossRef]
- Klein, J.P.; Sun, Z.; Staff, N.P. Association between ALS and retroviruses: Evidence from bioinformatics analysis. BMC Bioinform. 2019, 20, 680. [Google Scholar] [CrossRef] [PubMed]
- Juranic Lisnic, V.; Babic Cac, M.; Lisnic, B.; Trsan, T.; Mefferd, A.; Das Mukhopadhyay, C.; Cook, C.H.; Jonjic, S.; Trgovcich, J. Dual analysis of the murine cytomegalovirus and host cell transcriptomes reveal new aspects of the virus-host cell interface. PLoS Pathog. 2013, 9, e1003611. [Google Scholar] [CrossRef]
- Calculating and Interpreting the p-values for Functions, Pathways, and Lists in IPA; Qiagen: Germantown, MD, USA. 2010. Available online: https://qiagen.secure.force.com/KnowledgeBase/KnowledgeIPAPage?id=kA41i000000L5nQCAS (accessed on 14 October 2021).
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Li Puma, D.D.; Piacentini, R.; Leone, L.; Gironi, K.; Marcocci, M.E.; De Chiara, G.; Palamara, A.T.; Grassi, C. Herpes Simplex Virus Type-1 Infection Impairs Adult Hippocampal Neurogenesis via Amyloid-beta Protein Accumulation. Stem Cells 2019, 37, 1467–1480. [Google Scholar] [CrossRef]
- Hagg, E.; Astrom, L.; Steen, L. Persistent hypothalamic-pituitary insufficiency following acute meningoencephalitis. A report of two cases. Acta Med. Scand. 1978, 203, 231–235. [Google Scholar] [CrossRef]
- Kupari, M.; Pelkonen, R.; Valtonen, V. Post-encephalitic hypothalamic-pituitary insufficiency. Acta Endocrinol. (Copenh.) 1980, 94, 433–438. [Google Scholar] [CrossRef]
- Beatrice, A.M.; Selvan, C.; Mukhopadhyay, S. Pituitary dysfunction in infective brain diseases. Indian J. Endocrinol. Metab. 2013, 17, S608–S611. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; De Bellis, A.; Teksahin, H.; Alp, E.; Bizzarro, A.; Sinisi, A.A.; Bellastella, G.; Paglionico, V.A.; Bellastella, A.; Unluhizarci, K.; et al. Prospective investigation of pituitary functions in patients with acute infectious meningitis: Is acute meningitis induced pituitary dysfunction associated with autoimmunity? Pituitary 2012, 15, 579–588. [Google Scholar] [CrossRef]
- Sarasin-Filipowicz, M.; Krol, J.; Markiewicz, I.; Heim, M.H.; Filipowicz, W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat. Med. 2009, 15, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Israelow, B.; Narbus, C.M.; Sourisseau, M.; Evans, M.J. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 2014, 60, 1170–1179. [Google Scholar] [CrossRef]
- Accessing and Using Upstream Regulators; QIAGEN Digital Insights: Germantown, MD, USA. 2021. Available online: https://qiagen.secure.force.com/KnowledgeBase/KnowledgeIPAPage?id=kA41i000000L5sECAS (accessed on 14 October 2021).
- Vazirinejad, R.; Ahmadi, Z.; Kazemi Arababadi, M.; Hassanshahi, G.; Kennedy, D. The biological functions, structure and sources of CXCL10 and its outstanding part in the pathophysiology of multiple sclerosis. Neuroimmunomodulation 2014, 21, 322–330. [Google Scholar] [CrossRef]
- Djelloul, M.; Popa, N.; Pelletier, F.; Raguenez, G.; Boucraut, J. RAE-1 expression is induced during experimental autoimmune encephalomyelitis and is correlated with microglia cell proliferation. Brain Behav. Immun. 2016, 58, 209–217. [Google Scholar] [CrossRef]
- Arapovic, J.; Lenac, T.; Antulov, R.; Polic, B.; Ruzsics, Z.; Carayannopoulos, L.N.; Koszinowski, U.H.; Krmpotic, A.; Jonjic, S. Differential susceptibility of RAE-1 isoforms to mouse cytomegalovirus. J. Virol. 2009, 83, 8198–8207. [Google Scholar] [CrossRef]
- Spreafico, A.; Delord, J.P.; De Mattos-Arruda, L.; Berge, Y.; Rodon, J.; Cottura, E.; Bedard, P.L.; Akimov, M.; Lu, H.; Pain, S.; et al. A first-in-human phase I, dose-escalation, multicentre study of HSP990 administered orally in adult patients with advanced solid malignancies. Br. J. Cancer 2015, 112, 650–659. [Google Scholar] [CrossRef]
- Mutsvunguma, L.Z.; Moetlhoa, B.; Edkins, A.L.; Luke, G.A.; Blatch, G.L.; Knox, C. Theiler’s murine encephalomyelitis virus infection induces a redistribution of heat shock proteins 70 and 90 in BHK-21 cells, and is inhibited by novobiocin and geldanamycin. Cell Stress Chaperones 2011, 16, 505–515. [Google Scholar] [CrossRef]
- Ross, C.; Upfold, N.; Luke, G.A.; Bishop, O.T.; Knox, C. Subcellular localisation of Theiler’s murine encephalomyelitis virus (TMEV) capsid subunit VP1 vis-a-vis host protein Hsp90. Virus Res. 2016, 222, 53–63. [Google Scholar] [CrossRef]
- Eaton, A.; Bernier, F.P.; Goedhart, C.; Caluseriu, O.; Lamont, R.E.; Boycott, K.M.; Parboosingh, J.S.; Innes, A.M.; Care4Rare Canada, C. Is PNPT1-related hearing loss ever non-syndromic? Whole exome sequencing of adult siblings expands the natural history of PNPT1-related disorders. Am. J. Med. Genet. A 2018, 176, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Arai-Ichinoi, N.; Kikuchi, A.; Matsuhashi, T.; Numata-Uematsu, Y.; Uematsu, M.; Fujii, Y.; Murayama, K.; Ohtake, A.; Abe, T.; et al. Novel biallelic mutations in the PNPT1 gene encoding a mitochondrial-RNA-import protein PNPase cause delayed myelination. Clin. Genet. 2018, 93, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hunn, J.P.; Howard, J.C. The mouse resistance protein Irgm1 (LRG-47): A regulator or an effector of pathogen defense? PLoS Pathog. 2010, 6, e1001008. [Google Scholar] [CrossRef] [PubMed]
- Maric-Biresev, J.; Hunn, J.P.; Krut, O.; Helms, J.B.; Martens, S.; Howard, J.C. Loss of the interferon-gamma-inducible regulatory immunity-related GTPase (IRG), Irgm1, causes activation of effector IRG proteins on lysosomes, damaging lysosomal function and predicting the dramatic susceptibility of Irgm1-deficient mice to infection. BMC Biol. 2016, 14, 33. [Google Scholar] [CrossRef]
- Rai, P.; Janardhan, K.S.; Meacham, J.; Madenspacher, J.H.; Lin, W.C.; Karmaus, P.W.F.; Martinez, J.; Li, Q.Z.; Yan, M.; Zeng, J.; et al. IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol. 2021, 22, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.M.; Zhu, Y.F.; Wang, W.; Song, Z.Y.; Shao, X.Q.; Li, L.; Song, R.H.; An, X.F.; Qin, Q.; Li, Q.; et al. Polymorphisms in Autophagy-Related Gene IRGM Are Associated with Susceptibility to Autoimmune Thyroid Diseases. Biomed. Res. Int. 2018, 2018, 7959707. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res. Ther. 2010, 12 (Suppl. 1), S5. [Google Scholar] [CrossRef]
- Li, S.F.; Gong, M.J.; Zhao, F.R.; Shao, J.J.; Xie, Y.L.; Zhang, Y.G.; Chang, H.Y. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef]
- Christodoulou-Vafeiadou, E.; Ioakeimidis, F.; Andreadou, M.; Giagkas, G.; Stamatakis, G.; Reczko, M.; Samiotaki, M.; Papanastasiou, A.D.; Karakasiliotis, I.; Kontoyiannis, D.L. Divergent Innate and Epithelial Functions of the RNA-Binding Protein HuR in Intestinal Inflammation. Front. Immunol. 2018, 9, 2732. [Google Scholar] [CrossRef]
- Kafasla, P.; Skliris, A.; Kontoyiannis, D.L. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat. Immunol. 2014, 15, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Molineros, J.E.; Maiti, A.K.; Sun, C.; Looger, L.L.; Han, S.; Kim-Howard, X.; Glenn, S.; Adler, A.; Kelly, J.A.; Niewold, T.B.; et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet. 2013, 9, e1003222. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Wilkins, C.; Thomas, S.; Sekine, A.; Hendrick, D.M.; Voss, K.; Ireton, R.C.; Mooney, M.; Go, J.T.; Choonoo, G.; et al. Oas1b-dependent Immune Transcriptional Profiles of West Nile Virus Infection in the Collaborative Cross. G3 (Bethesda) 2017, 7, 1665–1682. [Google Scholar] [CrossRef] [PubMed]
- Kreit, M.; Vertommen, D.; Gillet, L.; Michiels, T. The Interferon-Inducible Mouse Apolipoprotein L9 and Prohibitins Cooperate to Restrict Theiler’s Virus Replication. PLoS ONE 2015, 10, e0133190. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, A.; Wixler, L.; Boergeling, Y.; Wixler, V.; Ludwig, S. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 2012, 26, 1290–1300. [Google Scholar] [CrossRef]
- Florio, T.; Barbieri, F.; Spaziante, R.; Zona, G.; Hofland, L.J.; van Koetsveld, P.M.; Feelders, R.A.; Stalla, G.K.; Theodoropoulou, M.; Culler, M.D.; et al. Efficacy of a dopamine-somatostatin chimeric molecule, BIM-23A760, in the control of cell growth from primary cultures of human non-functioning pituitary adenomas: A multi-center study. Endocr. Relat. Cancer 2008, 15, 583–596. [Google Scholar] [CrossRef]
- Ibanez-Costa, A.; Lopez-Sanchez, L.M.; Gahete, M.D.; Rivero-Cortes, E.; Vazquez-Borrego, M.C.; Galvez, M.A.; de la Riva, A.; Venegas-Moreno, E.; Jimenez-Reina, L.; Moreno-Carazo, A.; et al. BIM-23A760 influences key functional endpoints in pituitary adenomas and normal pituitaries: Molecular mechanisms underlying the differential response in adenomas. Sci. Rep. 2017, 7, 42002. [Google Scholar] [CrossRef]
- Jaquet, P.; Gunz, G.; Saveanu, A.; Barlier, A.; Dufour, H.; Taylor, J.; Dong, J.; Kim, S.; Moreau, J.P.; Culler, M.D. BIM-23A760, a chimeric molecule directed towards somatostatin and dopamine receptors, vs universal somatostatin receptors ligands in GH-secreting pituitary adenomas partial responders to octreotide. J. Endocrinol. Investig. 2005, 28, 21–27. [Google Scholar]
- Li, K.; Ma, Y.B.; Zhang, Z.; Tian, Y.H.; Xu, X.L.; He, Y.Q.; Xu, L.; Gao, Y.; Pan, W.T.; Song, W.J.; et al. Upregulated IQUB promotes cell proliferation and migration via activating Akt/GSK3beta/beta-catenin signaling pathway in breast cancer. Cancer Med. 2018, 7, 3875–3888. [Google Scholar] [CrossRef]
- Tanabe, K.; Tachibana, T.; Yamashita, T.; Che, Y.H.; Yoneda, Y.; Ochi, T.; Tohyama, M.; Yoshikawa, H.; Kiyama, H. The small GTP-binding protein TC10 promotes nerve elongation in neuronal cells, and its expression is induced during nerve regeneration in rats. J. Neurosci. 2000, 20, 4138–4144. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kato, M.; Miki, H.; Takenawa, T.; Endo, T. Small GTPase Tc10 and its homologue RhoT induce N-WASP-mediated long process formation and neurite outgrowth. J. Cell Sci. 2003, 116, 155–168. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burbage, M.; Keppler, S.J.; Montaner, B.; Mattila, P.K.; Batista, F.D. The Small Rho GTPase TC10 Modulates B Cell Immune Responses. J. Immunol. 2017, 199, 1682–1695. [Google Scholar] [CrossRef]
- Sheng, Y.; Hong, J.H.; Doherty, R.; Srikumar, T.; Shloush, J.; Avvakumov, G.V.; Walker, J.R.; Xue, S.; Neculai, D.; Wan, J.W.; et al. A human ubiquitin conjugating enzyme (E2)-HECT E3 ligase structure-function screen. Mol. Cell Proteom. 2012, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Xie, X.C.; Liu, L.L.; Lai, W.D. Aberrant methylation of UBE2Q1 promoter is associated with poor prognosis of acute-on-chronic hepatitis B pre-liver failure. Medicine 2021, 100, e26066. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Fan, X.P.; Fan, Y.C.; Chen, L.Y.; Qiao, C.Y.; Han, L.Y.; Wang, K. Hypomethylated Ubiquitin-Conjugating Enzyme2 Q1 (UBE2Q1) Gene Promoter in the Serum Is a Promising Biomarker for Hepatitis B Virus-Associated Hepatocellular Carcinoma. Tohoku J. Exp. Med. 2017, 242, 93–100. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, B.; Deng, C.; Wang, L.; Zhou, F.; Zhang, S.; Kang, W.; Zhan, P.; Chen, J.; Shen, S.; Guo, H.; et al. Upregulation of UBE2Q1 via gene copy number gain in hepatocellular carcinoma promotes cancer progression through beta-catenin-EGFR-PI3K-Akt-mTOR signaling pathway. Mol. Carcinog. 2018, 57, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Topno, R.; Singh, I.; Kumar, M.; Agarwal, P. Integrated bioinformatic analysis identifies UBE2Q1 as a potential prognostic marker for high grade serous ovarian cancer. BMC Cancer 2021, 21, 220. [Google Scholar] [CrossRef] [PubMed]
- Grzmil, P.; Altmann, M.E.; Adham, I.M.; Engel, U.; Jarry, H.; Schweyer, S.; Wolf, S.; Manz, J.; Engel, W. Embryo implantation failure and other reproductive defects in Ube2q1-deficient female mice. Reproduction 2013, 145, 45–56. [Google Scholar] [CrossRef]
- Belli, M.; Shimasaki, S. Molecular Aspects and Clinical Relevance of GDF9 and BMP15 in Ovarian Function. Vitam. Horm. 2018, 107, 317–348. [Google Scholar] [CrossRef]
- Sanfins, A.; Rodrigues, P.; Albertini, D.F. GDF-9 and BMP-15 direct the follicle symphony. J. Assist. Reprod. Genet. 2018, 35, 1741–1750. [Google Scholar] [CrossRef]
- Leung, Y.T.; Maurer, K.; Song, L.; Convissar, J.; Sullivan, K.E. Prolactin activates IRF1 and leads to altered balance of histone acetylation: Implications for systemic lupus erythematosus. Mod. Rheumatol. 2020, 30, 532–543. [Google Scholar] [CrossRef]
- Tufa, D.M.; Shank, T.; Yingst, A.M.; Trahan, G.D.; Shim, S.; Lake, J.; Woods, R.; Jones, K.; Verneris, M.R. Prolactin Acts on Myeloid Progenitors to Modulate SMAD7 Expression and Enhance Hematopoietic Stem Cell Differentiation into the NK Cell Lineage. Sci. Rep. 2020, 10, 6335. [Google Scholar] [CrossRef]
- Falcon, A.M.; Fortes, P.; Marion, R.M.; Beloso, A.; Ortin, J. Interaction of influenza virus NS1 protein and the human homologue of Staufen in vivo and in vitro. Nucl. Acids Res. 1999, 27, 2241–2247. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Boulay, K.; Mouland, A.J.; Desgroseillers, L. The host protein Staufen1 interacts with the Pr55Gag zinc fingers and regulates HIV-1 assembly via its N-terminus. Retrovirology 2008, 5, 41. [Google Scholar] [CrossRef]
- Hanke, K.; Hohn, O.; Liedgens, L.; Fiddeke, K.; Wamara, J.; Kurth, R.; Bannert, N. Staufen-1 interacts with the human endogenous retrovirus family HERV-K(HML-2) rec and gag proteins and increases virion production. J. Virol. 2013, 87, 11019–11030. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Pietzsch, C.; Ramanathan, P.; Santos, R.I.; Ilinykh, P.A.; Garcia-Blanco, M.A.; Bukreyev, A.; Bradrick, S.S. Staufen1 Interacts with Multiple Components of the Ebola Virus Ribonucleoprotein and Enhances Viral RNA Synthesis. mBio 2018, 9, e01771-18. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Clark, A.G. Impact of microRNA regulation on variation in human gene expression. Genome Res. 2012, 22, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Cammaerts, S.; Strazisar, M.; De Rijk, P.; Del Favero, J. Genetic variants in microRNA genes: Impact on microRNA expression, function, and disease. Front. Genet. 2015, 6, 186. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucl. Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Pollio, G.; Hoozemans, J.J.; Andersen, C.A.; Roncarati, R.; Rosi, M.C.; van Haastert, E.S.; Seredenina, T.; Diamanti, D.; Gotta, S.; Fiorentini, A.; et al. Increased expression of the oligopeptidase THOP1 is a neuroprotective response to Abeta toxicity. Neurobiol. Dis. 2008, 31, 145–158. [Google Scholar] [CrossRef]
- Sundstrom, J.M.; Hernandez, C.; Weber, S.R.; Zhao, Y.; Dunklebarger, M.; Tiberti, N.; Laremore, T.; Simo-Servat, O.; Garcia-Ramirez, M.; Barber, A.J.; et al. Proteomic Analysis of Early Diabetic Retinopathy Reveals Mediators of Neurodegenerative Brain Diseases. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeyer-Langford, C.L.; Rech, R.; Amstalden, K.; Kochan, K.J.; Hillhouse, A.E.; Young, C.; Welsh, C.J.; Threadgill, D.W. Host genetic background influences diverse neurological responses to viral infection in mice. Sci. Rep. 2017, 7, 12194. [Google Scholar] [CrossRef]
- Carneiro-Sampaio, M.; Coutinho, A. Tolerance and autoimmunity: Lessons at the bedside of primary immunodeficiencies. Adv. Immunol. 2007, 95, 51–82. [Google Scholar] [CrossRef]
- Azizi, G.; Ghanavatinejad, A.; Abolhassani, H.; Yazdani, R.; Rezaei, N.; Mirshafiey, A.; Aghamohammadi, A. Autoimmunity in primary T-cell immunodeficiencies. Expert Rev. Clin. Immunol. 2016, 12, 989–1006. [Google Scholar] [CrossRef]
- Comrie, W.A.; Lenardo, M.J. Molecular Classification of Primary Immunodeficiencies of T Lymphocytes. Adv. Immunol. 2018, 138, 99–193. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.R.; Leskovsek, N.V.; Rose, N.R. The odd couple: A fresh look at autoimmunity and immunodeficiency. J. Autoimmun 2010, 35, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Todoric, K.; Koontz, J.B.; Mattox, D.; Tarrant, T.K. Autoimmunity in immunodeficiency. Curr. Allergy Asthma Rep. 2013, 13, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Gerhauser, I.; Hansmann, F.; Ciurkiewicz, M.; Loscher, W.; Beineke, A. Facets of Theiler’s Murine Encephalomyelitis Virus-Induced Diseases: An Update. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Turrin, N.P. Central nervous system Toll-like receptor expression in response to Theiler’s murine encephalomyelitis virus-induced demyelination disease in resistant and susceptible mouse strains. Virol. J. 2008, 5, 154. [Google Scholar] [CrossRef]
- Nicholson, S.M.; Peterson, J.D.; Miller, S.D.; Wang, K.; Dal Canto, M.C.; Melvold, R.W. BALB/c substrain differences in susceptibility to Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neuroimmunol. 1994, 52, 19–24. [Google Scholar] [CrossRef]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef]
- Yasuda, K.; Torigoe, T.; Mariya, T.; Asano, T.; Kuroda, T.; Matsuzaki, J.; Ikeda, K.; Yamauchi, M.; Emori, M.; Asanuma, H.; et al. Fibroblasts induce expression of FGF4 in ovarian cancer stem-like cells/cancer-initiating cells and upregulate their tumor initiation capacity. Lab. Investig. 2014, 94, 1355–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Liu, S.; Zhang, X.; Li, W.B.; Chen, Y.; Huang, X.; Sun, L.; Luo, W.; Netzer, W.J.; Threadgill, R.; et al. A functional mouse retroposed gene Rps23r1 reduces Alzheimer's beta-amyloid levels and tau phosphorylation. Neuron 2009, 64, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, Y.; Li, W.B.; Cohen, S.N.; Liao, F.F.; Li, L.; Xu, H.; Zhang, Y.W. The Rps23rg gene family originated through retroposition of the ribosomal protein s23 mRNA and encodes proteins that decrease Alzheimer's beta-amyloid level and tau phosphorylation. Hum. Mol. Genet. 2010, 19, 3835–3843. [Google Scholar] [CrossRef]
- Zhao, D.; Meng, J.; Zhao, Y.; Huo, Y.; Liu, Y.; Zheng, N.; Zhang, M.; Gao, Y.; Chen, Z.; Sun, H.; et al. RPS23RG1 Is Required for Synaptic Integrity and Rescues Alzheimer's Disease-Associated Cognitive Deficits. Biol. Psychiatry 2019, 86, 171–184. [Google Scholar] [CrossRef]
- Boding, L.; Hansen, A.K.; Meroni, G.; Johansen, B.B.; Braunstein, T.H.; Bonefeld, C.M.; Kongsbak, M.; Jensen, B.A.; Woetmann, A.; Thomsen, A.R.; et al. Midline 1 directs lytic granule exocytosis and cytotoxicity of mouse killer T cells. Eur J. Immunol. 2014, 44, 3109–3118. [Google Scholar] [CrossRef] [PubMed]
- Collison, A.; Hatchwell, L.; Verrills, N.; Wark, P.A.; de Siqueira, A.P.; Tooze, M.; Carpenter, H.; Don, A.S.; Morris, J.C.; Zimmermann, N.; et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat. Med. 2013, 19, 232–237. [Google Scholar] [CrossRef]
- Dorshkind, K.; Horseman, N.D. The roles of prolactin, growth hormone, insulin-like growth factor-I, and thyroid hormones in lymphocyte development and function: Insights from genetic models of hormone and hormone receptor deficiency. Endocr. Rev. 2000, 21, 292–312. [Google Scholar] [CrossRef] [PubMed]
- Torner, L.; Karg, S.; Blume, A.; Kandasamy, M.; Kuhn, H.G.; Winkler, J.; Aigner, L.; Neumann, I.D. Prolactin prevents chronic stress-induced decrease of adult hippocampal neurogenesis and promotes neuronal fate. J. Neurosci. 2009, 29, 1826–1833. [Google Scholar] [CrossRef]
- Walker, T.L.; Vukovic, J.; Koudijs, M.M.; Blackmore, D.G.; Mackay, E.W.; Sykes, A.M.; Overall, R.W.; Hamlin, A.S.; Bartlett, P.F. Prolactin stimulates precursor cells in the adult mouse hippocampus. PLoS ONE 2012, 7, e44371. [Google Scholar] [CrossRef] [PubMed]
- Zhornitsky, S.; Johnson, T.A.; Metz, L.M.; Weiss, S.; Yong, V.W. Prolactin in combination with interferon-beta reduces disease severity in an animal model of multiple sclerosis. J. Neuroinflamm. 2015, 12, 55. [Google Scholar] [CrossRef]
- Tang, M.W.; Garcia, S.; Gerlag, D.M.; Tak, P.P.; Reedquist, K.A. Insight into the Endocrine System and the Immune System: A Review of the Inflammatory Role of Prolactin in Rheumatoid Arthritis and Psoriatic Arthritis. Front. Immunol. 2017, 8, 720. [Google Scholar] [CrossRef]
- Buskila, D.; Sukenik, S.; Shoenfeld, Y. The possible role of prolactin in autoimmunity. Am. J. Reprod. Immunol. 1991, 26, 118–123. [Google Scholar] [CrossRef]
- Borba, V.V.; Zandman-Goddard, G.; Shoenfeld, Y. Prolactin and Autoimmunity. Front. Immunol. 2018, 9, 73. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F.; Ysrraelit, M.C. Role of prolactin in B cell regulation in multiple sclerosis. J. Neuroimmunol. 2014, 269, 76–86. [Google Scholar] [CrossRef]
- Costanza, M.; Binart, N.; Steinman, L.; Pedotti, R. Prolactin: A versatile regulator of inflammation and autoimmune pathology. Autoimmun. Rev. 2015, 14, 223–230. [Google Scholar] [CrossRef]
- Costanza, M.; Pedotti, R. Prolactin: Friend or Foe in Central Nervous System Autoimmune Inflammation? Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.B.; Smith, M.L.; Williams, A.C.; Waring, R.H.; Ramsden, D.B. Expression of nicotinamide N-methyltransferase (E.C. 2.1.1.1) in the Parkinsonian brain. J. Neuropathol. Exp. Neurol. 2002, 61, 111–124. [Google Scholar] [CrossRef]
- Parsons, R.B.; Smith, S.W.; Waring, R.H.; Williams, A.C.; Ramsden, D.B. High expression of nicotinamide N-methyltransferase in patients with idiopathic Parkinson's disease. Neurosci. Lett. 2003, 342, 13–16. [Google Scholar] [CrossRef]
- Schmeisser, K.; Parker, J.A. Nicotinamide-N-methyltransferase controls behavior, neurodegeneration and lifespan by regulating neuronal autophagy. PLoS Genet. 2018, 14, e1007561. [Google Scholar] [CrossRef] [PubMed]
- Kocinaj, A.; Chaudhury, T.; Uddin, M.S.; Junaid, R.R.; Ramsden, D.B.; Hondhamuni, G.; Klamt, F.; Parsons, L.; Parsons, R.B. High Expression of Nicotinamide N-Methyltransferase in Patients with Sporadic Alzheimer's Disease. Mol. Neurobiol. 2021, 58, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.B.; Waring, R.H.; Parsons, R.B.; Barlow, D.J.; Williams, A.C. Nicotinamide N-Methyltransferase: Genomic Connection to Disease. Int. J. Tryptophan Res. 2020, 13, 1178646920919770. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, A.; Lerer, E.; Udawela, M.; Scarr, E.; Dean, B.; Belmaker, R.H.; Ebstein, R.; Agam, G. Nicotinamide-N-methyltransferase (NNMT) in schizophrenia: Genetic association and decreased frontal cortex mRNA levels. Int. J. Neuropsychopharmacol. 2012, 15, 727–737. [Google Scholar] [CrossRef]
- Hu, Q.; Liu, F.; Yang, L.; Fang, Z.; He, J.; Wang, W.; You, P. Lower serum nicotinamide N-methyltransferase levels in patients with bipolar disorder during acute episodes compared to healthy controls: A cross-sectional study. BMC Psychiatry 2020, 20, 33. [Google Scholar] [CrossRef]
- Milani, Z.H.; Ramsden, D.B.; Parsons, R.B. Neuroprotective effects of nicotinamide N-methyltransferase and its metabolite 1-methylnicotinamide. J. Biochem. Mol. Toxicol. 2013, 27, 451–456. [Google Scholar] [CrossRef]
- Holwerda, S.J.; de Laat, W. CTCF: The protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120369. [Google Scholar] [CrossRef]
- Klenova, E.M.; Nicolas, R.H.; Paterson, H.F.; Carne, A.F.; Heath, C.M.; Goodwin, G.H.; Neiman, P.E.; Lobanenkov, V.V. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell. Biol. 1993, 13, 7612–7624. [Google Scholar] [CrossRef]
- Ohlsson, R.; Renkawitz, R.; Lobanenkov, V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001, 17, 520–527. [Google Scholar] [CrossRef]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef]
- Sun, G.; Yan, J.; Noltner, K.; Feng, J.; Li, H.; Sarkis, D.A.; Sommer, S.S.; Rossi, J.J. SNPs in human miRNA genes affect biogenesis and function. RNA 2009, 15, 1640–1651. [Google Scholar] [CrossRef]
- Shafi, G.; Aliya, N.; Munshi, A. MicroRNA signatures in neurological disorders. Can. J. Neurol. Sci. 2010, 37, 177–185. [Google Scholar] [CrossRef]
- Christensen, M.; Schratt, G.M. microRNA involvement in developmental and functional aspects of the nervous system and in neurological diseases. Neurosci. Lett. 2009, 466, 55–62. [Google Scholar] [CrossRef]
- Junn, E.; Mouradian, M.M. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 2012, 133, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Vohra, M.; Sharma, A.R.; Prabhu, B.N.; Rai, P.S. SNPs in Sites for DNA Methylation, Transcription Factor Binding, and miRNA Targets Leading to Allele-Specific Gene Expression and Contributing to Complex Disease Risk: A Systematic Review. Public Health Genom. 2020, 23, 155–170. [Google Scholar] [CrossRef]
- Afrasiabi, A.; Fewings, N.L.; Schibeci, S.D.; Keane, J.T.; Booth, D.R.; Parnell, G.P.; Swaminathan, S. The Interaction of Human and Epstein-Barr Virus miRNAs with Multiple Sclerosis Risk Loci. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Hassani, A.; Khan, G. Epstein-Barr Virus and miRNAs: Partners in Crime in the Pathogenesis of Multiple Sclerosis? Front. Immunol. 2019, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Kim, H.; Kartika, A.V.; Kanehiro, Y.; Yoshiyama, H. Role of Viral and Host microRNAs in Immune Regulation of Epstein-Barr Virus-Associated Diseases. Front. Immunol. 2020, 11, 367. [Google Scholar] [CrossRef]
- Moyano, A.L.; Steplowski, J.; Wang, H.; Son, K.N.; Rapolti, D.I.; Marshall, J.; Elackattu, V.; Marshall, M.S.; Hebert, A.K.; Reiter, C.R.; et al. microRNA-219 Reduces Viral Load and Pathologic Changes in Theiler’s Virus-Induced Demyelinating Disease. Mol. Ther. 2018, 26, 730–743. [Google Scholar] [CrossRef]
- De Cock, A.; Michiels, T. Cellular microRNAs Repress Vesicular Stomatitis Virus but Not Theiler’s Virus Replication. Viruses 2016, 8, 75. [Google Scholar] [CrossRef]
- Campbell, T.; Meagher, M.W.; Sieve, A.; Scott, B.; Storts, R.; Welsh, T.H.; Welsh, C.J. The effects of restraint stress on the neuropathogenesis of Theiler’s virus infection: I. Acute disease. Brain Behav. Immun. 2001, 15, 235–254. [Google Scholar] [CrossRef]
- Welsh, C.J.; Tonks, P.; Nash, A.A.; Blakemore, W.F. The effect of L3T4 T cell depletion on the pathogenesis of Theiler’s murine encephalomyelitis virus infection in CBA mice. J. Gen. Virol. 1987, 68 Pt 6, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Consortium. ; C.C. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 2012, 190, 389–401. [CrossRef]
- Srivastava, A.; Morgan, A.P.; Najarian, M.L.; Sarsani, V.K.; Sigmon, J.S.; Shorter, J.R.; Kashfeen, A.; McMullan, R.C.; Williams, L.H.; Giusti-Rodriguez, P.; et al. Genomes of the Mouse Collaborative Cross. Genetics 2017, 206, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.; Kang, H.M.; Wade, C.M.; Cotsapas, C.; Kostem, E.; Han, B.; Furlotte, N.; Kang, E.Y.; Rivas, M.; Bogue, M.A.; et al. Fine mapping in 94 inbred mouse strains using a high-density haplotype resource. Genetics 2010, 185, 1081–1095. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.P.; Fu, C.P.; Kao, C.Y.; Welsh, C.E.; Didion, J.P.; Yadgary, L.; Hyacinth, L.; Ferris, M.T.; Bell, T.A.; Miller, D.R.; et al. The Mouse Universal Genotyping Array: From Substrains to Subspecies. G3 (Bethesda) 2015, 6, 263–279. [Google Scholar] [CrossRef]
- Blake, J.A.; Baldarelli, R.; Kadin, J.A.; Richardson, J.E.; Smith, C.L.; Bult, C.J.; Mouse Genome Database, G. Mouse Genome Database (MGD): Knowledgebase for mouse-human comparative biology. Nucl. Acids Res. 2021, 49, D981–D987. [Google Scholar] [CrossRef]
- Keane, T.M.; Goodstadt, L.; Danecek, P.; White, M.A.; Wong, K.; Yalcin, B.; Heger, A.; Agam, A.; Slater, G.; Goodson, M.; et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011, 477, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, B.; Wong, K.; Agam, A.; Goodson, M.; Keane, T.M.; Gan, X.; Nellaker, C.; Goodstadt, L.; Nicod, J.; Bhomra, A.; et al. Sequence-based characterization of structural variation in the mouse genome. Nature 2011, 477, 326–329. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Top 5 Canonical Pathways | p-Value | Molecules |
|---|---|---|
| Overall | ||
| Neuroinflammation Signaling Pathway | 1.32 × 10−2 | GABRA6 ↓ |
| GABA Receptor Signaling | 2.94 × 10−2 | GABRA6 ↓ |
| Resistant | ||
| Neuroprotective Role of THOP1 in Alzheimer’s Disease | 2.59 × 10−3 | HLA-A ↑, PRSS41 ↑ |
| The Visual Cycle | 1.29 × 10−2 | RDH13 ↑ |
| Retinoate Biosynthesis I | 2.31 × 10−2 | RDH13 ↑ |
| Antigen Presentation Pathway | 2.50 × 10−2 | HLA-A ↑ |
| B Cell Development | 2.75 × 10−2 | HLA-A ↑ |
| Resilient | ||
| Primary Immunodeficiency Signaling | 8.23 × 10−4 | CD4 ↑, Igha ↑, IGHG1 ↑, Ighg2b ↑ |
| IL-7 Signaling Pathway | 3.21 × 10−3 | Igha ↑, IGHG1 ↑, Ighg2b ↑, Ighg2c ↑ |
| Role of Pattern Recognition Receptors in Recognition of Bacteria and Viruses | 7.53 × 10−3 | IFIH1 ↑, IL25 ↑, LTA ↑, Oas1b ↑, Oas1d ↓ (includes others) |
| Phagosome Maturation | 8.14 × 10−3 | DYNLT1 ↑, HLA-A ↑, HLA-E ↑, PRDX1 ↑, TUBD1 ↑ |
| Th1 and Th2 Activation Pathway | 1.12 × 10−2 | Aph1c ↓, CD4 ↑, HLA-A ↑, IL25 ↑, LTA ↑ |
| Susceptible | ||
| NRF2-mediated Oxidative Stress Response | 5.01 × 10−3 | AOX1 ↑, PPIB ↑ |
| Role of JAK2 in Hormone-like Cytokine Signaling | 1.92 × 10−2 | PRL ↓ |
| Nicotine Degradation III | 3.20 × 10−2 | AOX1 ↑ |
| Activation of IRF by Cytosolic Pattern Recognition Receptors | 3.53 × 10−2 | PPIB ↑ |
| Nicotine Degradation II | 3.64 × 10−2 | AOX1 ↑ |
| Top 5 Upstream Regulators | p-Value of Overlap | Target Molecules |
|---|---|---|
| Overall (Infected vs. Sham) | ||
| NFIA ↑ | 1.37 × 10−3 | GABRA6 ↓ |
| miR-122-5p (miRNAs w/seed GGAGUGU) | 4.42 × 10−3 | TBX19 ↓ |
| TAF7L ↓ | 5.16 × 10−3 | TBX19 ↓ |
| EP300 ↓ | 2.39 × 10−2 | TBX19 ↓ |
| GATA2 ↑ | 2.69 × 10−2 | TBX19 ↓ |
| Resistant | ||
| MSH2 ↑ | 1.30 × 10−5 | IGHG1 ↑, IGKC ↑ |
| IL21R ↑ | 7.54 × 10−5 | IGHG1 ↑, IGKC ↑ |
| CXCL10 ↑ | 1.77 × 10−4 | Ccl6 ↓, IGKC ↑ |
| HSP-990 | 6.24 × 10−4 | HLA-A ↑ |
| Raet1d ↑/Raet1e ↑ | 6.24 × 10−4 | HLA-A ↑ |
| Resilient | ||
| MSH2 ↑ | 4.33 × 10−5 | IGHG1 ↑, Ighg2b ↑, IGKC ↑ |
| PNPT1 ↑ | 5.38 × 10−5 | Apol9a ↑/Apol9b ↑, GBP6 ↑, IFI16 ↑, IFIH1 ↑, Oas1b ↑ |
| Irgm1 ↑ | 1.54 × 10−4 | Apol9a ↑/Apol9b ↑, GBP6 ↑, IFI16 ↑, IFIH1 ↑, Oas1b ↑, Oas1d↓ (includes others) |
| IFNB1 ↑ | 1.63 × 10−4 | GBP3 ↑, GBP6 ↑, GLP2R ↑, HLA-A ↑, IFI16 ↑, IFIH1 ↑, MCM10 ↑, Oas1b ↑, Oas1d ↓ (includes others), TRIM6-TRIM34 ↑ |
| ELAVL1 ↓ | 3.09 × 10−4 | CASP9 ↓, GBP6 ↑, HLA-A ↑, IFI16 ↑, IFIH1 ↑, Igha ↑, Igkv8-30 ↑, Oas1b ↑ |
| Susceptible | ||
| GNAS ↑ | 5.07 × 10−4 | GDF9 ↓, PRL ↓ |
| BIM 23A760 | 5.82 × 10−4 | PRL ↓ |
| IQUB ↑ | 5.82 × 10−4 | PRL ↓ |
| RHOQ ↓ | 5.82 × 10−4 | PRL ↓ |
| UBE2Q1 ↑ | 5.82 × 10−4 | PRL ↓ |
| Symbol | Entrez Gene Name | Expr Log Ratio | Expr p-Value |
|---|---|---|---|
| Overall | |||
| STAU1 | staufen double-stranded RNA binding protein 1 | −0.002 | 0.999 |
| Resistant | |||
| HLA-A | major histocompatibility complex, class I, A | 10.265 | 1.92 × 10−2 |
| Resilient | |||
| CDPF1 | cysteine rich DPF motif domain-containing 1 | −1.384 | 1.62 × 10−3 |
| FGF4 | fibroblast growth factor 4 | −16.685 | 3.07 × 10−4 |
| Susceptible | |||
| EIF3J | eukaryotic translation initiation factor 3 subunit J | −6.53 | 8.03 × 10−3 |
| GDF9 | growth differentiation factor 9 | −3.6 | 3.57 × 10−2 |
| Gm5148/Rps23rg1 | ribosomal protein S23, retrogene 1 | −8.529 | 4.00 × 10−2 |
| MID1 | midline 1 | −5.28 | 8.03 × 10−3 |
| PRL | Prolactin | −17.432 | 6.52 × 10−3 |
| Strain | Infected F | Infected M | Sham F | Sham M | Total n | TMEV 90 dpi | 90 dpi Cumulative Score |
|---|---|---|---|---|---|---|---|
| CC002 | 1 | 2 | 1 | 1 | 5 | −1.52 | 1.08 |
| CC005 | 2 | 4 | 3 | 3 | 12 | 21.90 | 2.18 |
| CC006 | 3 | 2 | 4 | 2 | 11 | 23.98 | 0.60 |
| CC011 | 3 | 4 | 3 | 3 | 13 | 22.55 | 1.68 |
| CC015 | 1 | 2 | 1 | 2 | 6 | 32.38 | 0.45 |
| CC017 | 1 | 3 | 2 | 3 | 9 | 27.56 | 1.34 |
| CC023 | 1 | 1 | 3 | 2 | 7 | 20.95 | 3.61 |
| CC024 | 1 | 1 | 1 | 0 | 3 | 0.15 | 0.95 |
| CC025 | 1 | 1 | 1 | 0 | 3 | −1.24 | 1.21 |
| CC027 | 3 | 1 | 2 | 3 | 9 | 24.72 | 0.27 |
| CC032×CC013 | 2 | 4 | 1 | 1 | 8 | −3.60 | 0.61 |
| CC036 | 1 | 6 | 1 | 2 | 10 | −11.25 | 0.69 |
| CC037 | 3 | 4 | 2 | 3 | 12 | 21.53 | 0.61 |
| CC041 | 2 | 2 | 2 | 0 | 6 | 0.29 | 1.34 |
| CC041×CC012 | 5 | 4 | 2 | 1 | 12 | 20.85 | 1.35 |
| CC043 | 0 | 2 | 1 | 1 | 4 | 20.79 | 0.84 |
| CC051 | 5 | 1 | 2 | 1 | 9 | −2.67 | 0.28 |
| CC058 | 1 | 0 | 1 | 1 | 3 | 4.48 | 1.81 |
| CC072 | 0 | 2 | 1 | 0 | 3 | 4.60 | 2.08 |
| Total | 36 | 46 | 34 | 29 | 145 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brinkmeyer-Langford, C.; Amstalden, K.; Konganti, K.; Hillhouse, A.; Lawley, K.; Perez-Gomez, A.; Young, C.R.; Welsh, C.J.; Threadgill, D.W. Resilience in Long-Term Viral Infection: Genetic Determinants and Interactions. Int. J. Mol. Sci. 2021, 22, 11379. https://doi.org/10.3390/ijms222111379
Brinkmeyer-Langford C, Amstalden K, Konganti K, Hillhouse A, Lawley K, Perez-Gomez A, Young CR, Welsh CJ, Threadgill DW. Resilience in Long-Term Viral Infection: Genetic Determinants and Interactions. International Journal of Molecular Sciences. 2021; 22(21):11379. https://doi.org/10.3390/ijms222111379
Chicago/Turabian StyleBrinkmeyer-Langford, Candice, Katia Amstalden, Kranti Konganti, Andrew Hillhouse, Koedi Lawley, Aracely Perez-Gomez, Colin R. Young, C. Jane Welsh, and David W. Threadgill. 2021. "Resilience in Long-Term Viral Infection: Genetic Determinants and Interactions" International Journal of Molecular Sciences 22, no. 21: 11379. https://doi.org/10.3390/ijms222111379
APA StyleBrinkmeyer-Langford, C., Amstalden, K., Konganti, K., Hillhouse, A., Lawley, K., Perez-Gomez, A., Young, C. R., Welsh, C. J., & Threadgill, D. W. (2021). Resilience in Long-Term Viral Infection: Genetic Determinants and Interactions. International Journal of Molecular Sciences, 22(21), 11379. https://doi.org/10.3390/ijms222111379