
The Genetics of Diabetes: What We Can Learn from Drosophila




Abstract
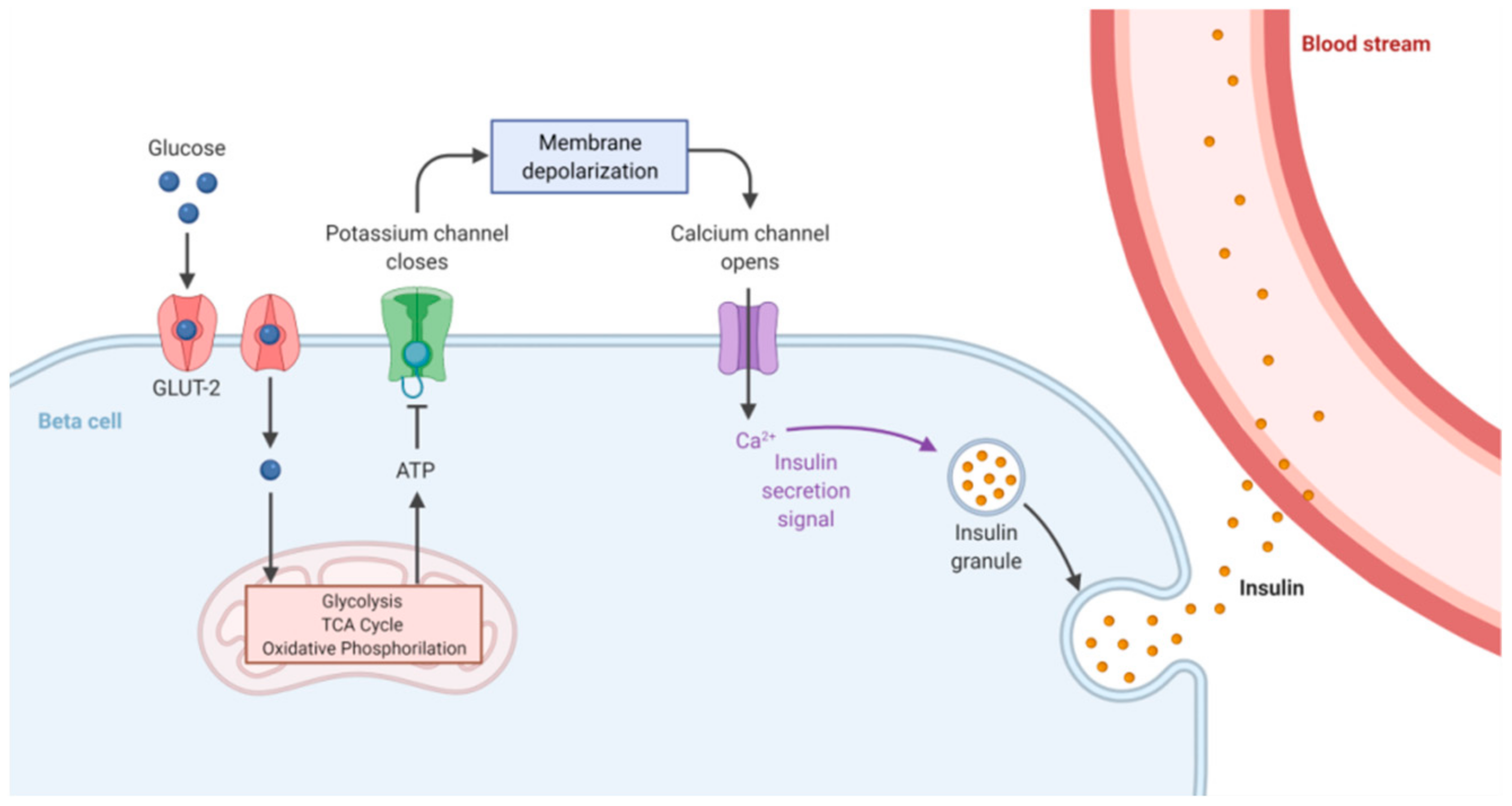
1. Glucose Homeostasis Maintenance
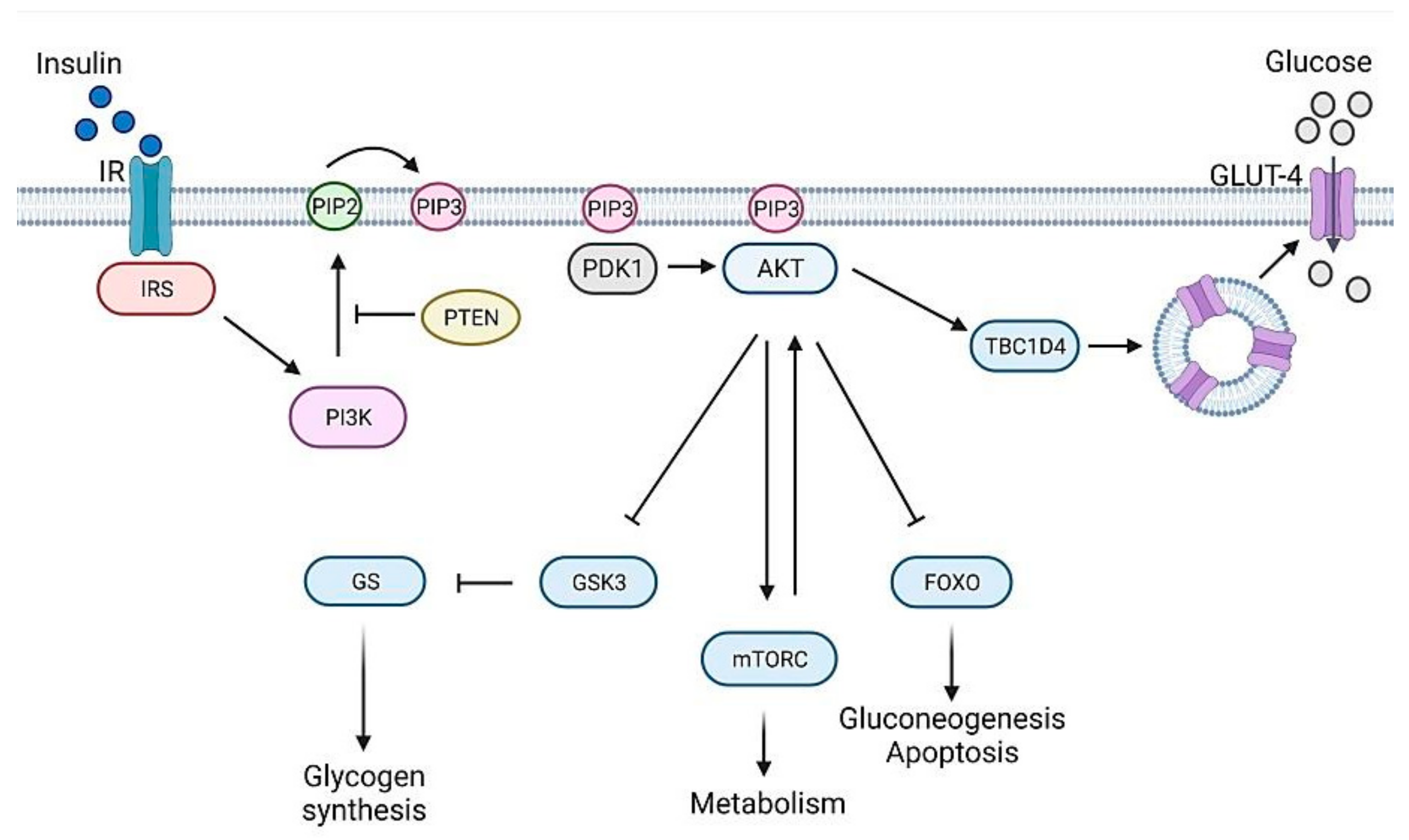
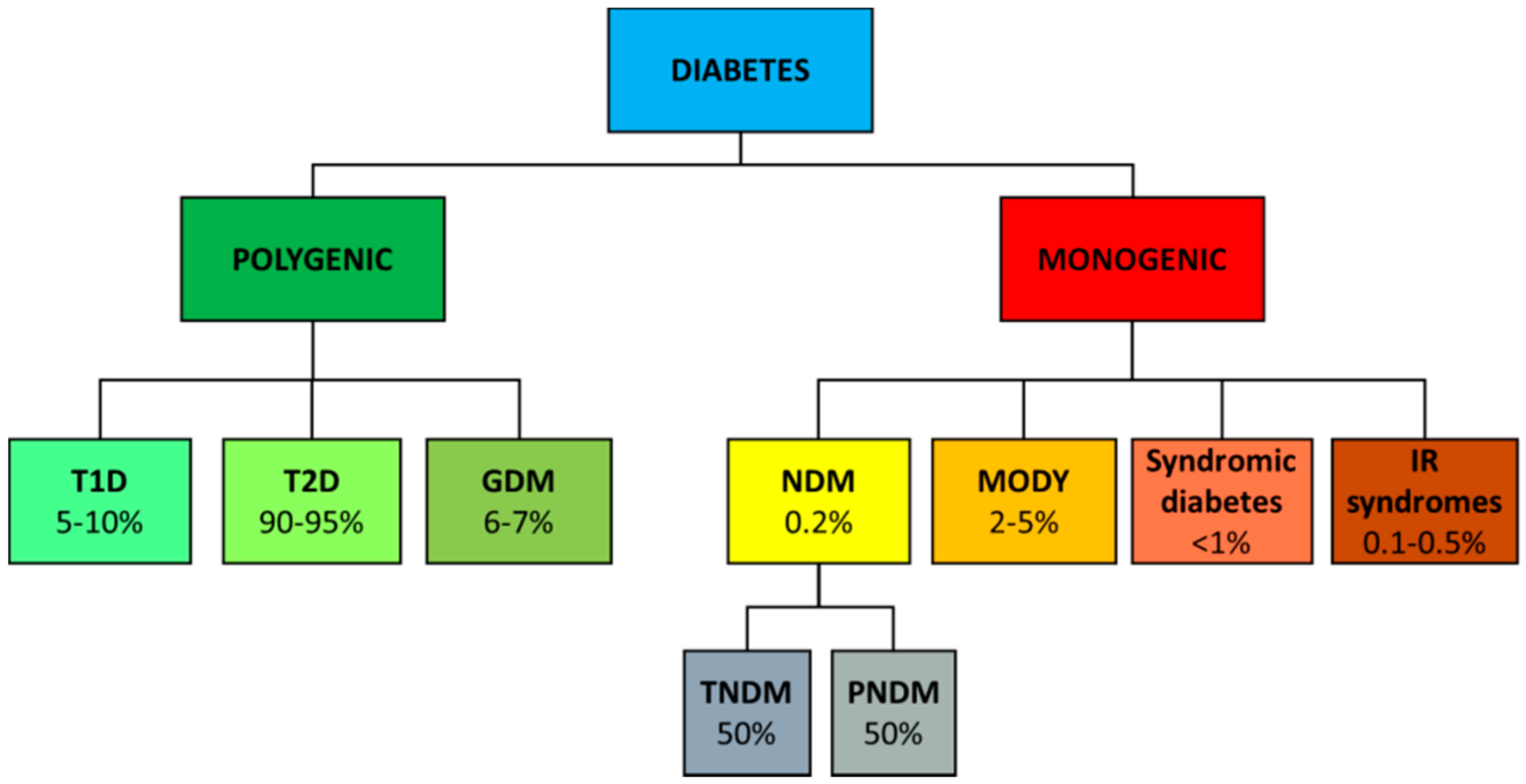
2. Genetics of Diabetes
2.1. Type 1 Diabetes
2.2. Type 2 Diabetes
2.3. Gestational Diabetes
2.4. Monogenic Diabetes
2.4.1. NDM
2.4.2. MODY
2.4.3. Monogenic Forms of Insulin Resistance
2.5. Other Diabetes
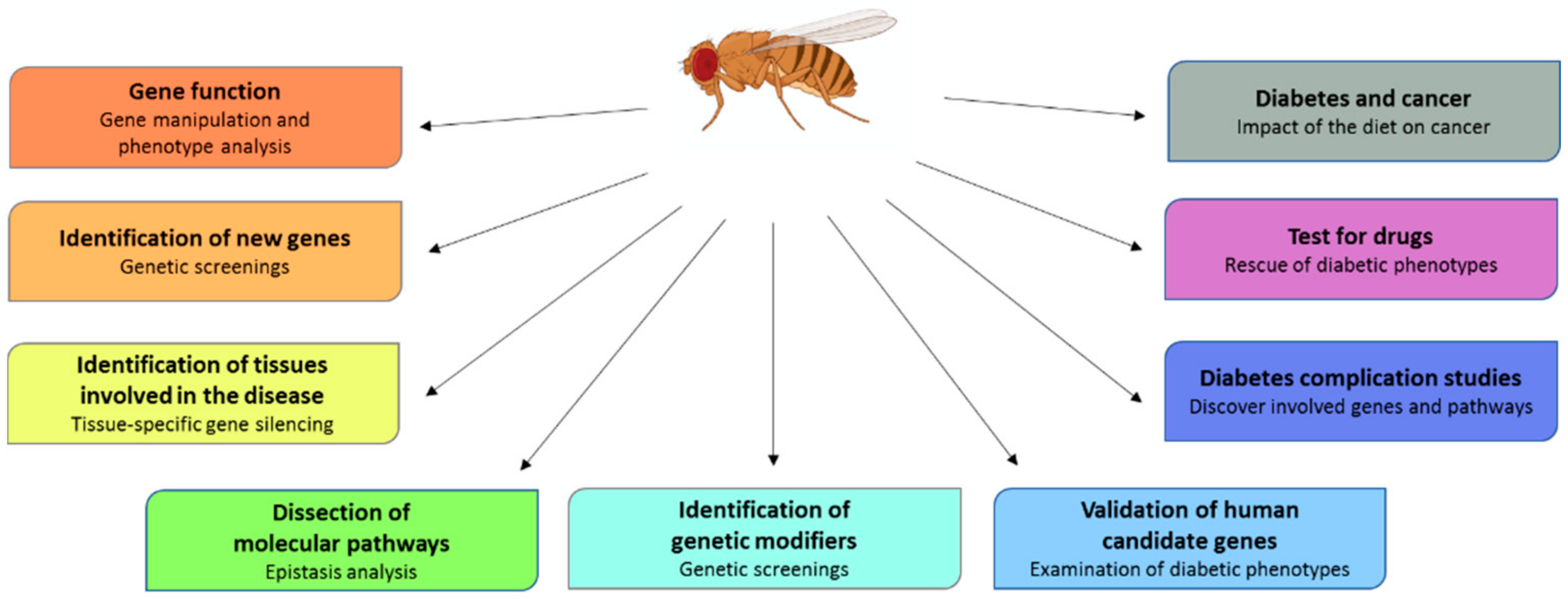
3. Drosophila as a Diabetes Model
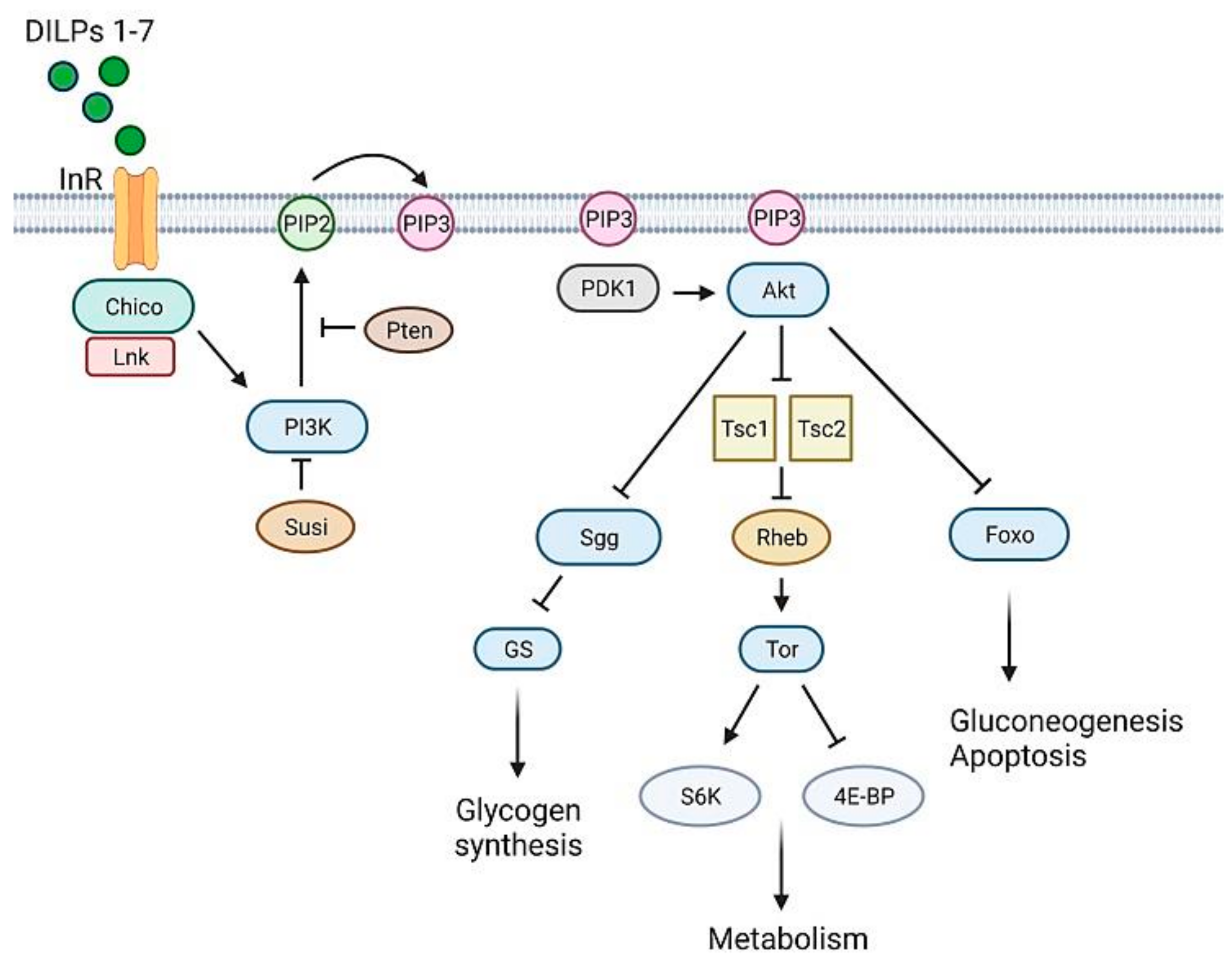
3.1. Glucose Homeostasis in Drosophila
3.2. T1D Fly Models
3.3. T2D Fly Models
3.4. Monogenic Diabetes Fly Models
4. Drosophila as a Mean to Validate Human Candidate Genes
5. Screening to Isolate New Genes Involved in Diabetes
6. How to Study Diabetes Complications in Drosophila

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drosophila Diabetes Models | ||
| Type | Generation Method | Ref |
| T1D | IPC ablation | [112,113,114] |
| T1D | Dilp 1-5 gene deletion | [116] |
| T1D | Dominant negative Hsc70-3 expression | [121] |
| T2D | HSD feeding | [98,122] |
| T2D | IIS gene silencing | [105,124,125,126] |
| T2D | Pdxk mutations | [130] |
| T2D | Sgll gene silencing | [131] |
| MODY1 | dHNF4 gene silencing | [140] |
| MODY2 | HexA or HexC gene silencing | [144] |
| Diabetes Candidate Genes Validated in Drosophila | ||
| Candidate Gene | Diabetes Type | Ref |
| HHEX | T2D | [145] |
| BCL11A | T2D | [146] |
| Screenings Performed in Drosophila to Isolate New Diabetes-Related Genes | ||
| Gene | Pathway | Ref |
| CSNK1α1 | Glucose metabolism | [99] |
| Unc-104 | Insulin trafficking | [150] |
| Rab1 | Insulin trafficking | |
| Pointed | Glucose level maintenance | [153] |
| sulfateless | Beta cell destruction | [154] |
| Diabetes Complications Drosophila Models | ||
| Complication | Diabetes Type | Ref |
| Heart dysfunction | HSD-induced T2D | [158] |
| Retinal degeneration | HSD-induced T2D | [161] |
| Diabetic nephropathy | HSD-induced T2D | [162] |
7. Diabetes and Cancer Risk in Drosophila
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Association, A.D. 2. Classification and Diagnosis of Diabetes. Diabetes Care 2021, 44, S15–S33. [Google Scholar] [CrossRef]
- Aronoff, S.L.; Berkowitz, K.; Shreiner, B.; Want, L. Glucose Metabolism and Regulation: Beyond Insulin and Glucagon. Diabetes Spectrum 2004, 17, 183–190. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef]
- Gupta, A.; Dey, C.S. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol. Biol. Cell 2012, 23, 3882–3898. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef] [PubMed]
- Julien, L.A.; Carriere, A.; Moreau, J.; Roux, P.P. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef]
- Kearney, A.L.; Norris, D.M.; Ghomlaghi, M.; Kin Lok Wong, M.; Humphrey, S.J.; Carroll, L.; Yang, G.; Cooke, K.C.; Yang, P.; Geddes, T.A.; et al. Akt phosphorylates insulin receptor substrate to limit PI3K-mediated PIP3 synthesis. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef]
- Pociot, F.; Lernmark, Å. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Ram, R.; Mehta, M.; Nguyen, Q.T.; Larma, I.; Boehm, B.O.; Pociot, F.; Concannon, P.; Morahan, G. Systematic Evaluation of Genes and Genetic Variants Associated with Type 1 Diabetes Susceptibility. J. Immunol. 2016, 196, 3043–3053. [Google Scholar] [CrossRef]
- Ounissi-Benkalha, H.; Polychronakos, C. The molecular genetics of type 1 diabetes: New genes and emerging mechanisms. Trends Mol. Med. 2008, 14, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wong, F.S.; Wen, L. Type 1 diabetes and gut microbiota: Friend or foe? Pharmacol. Res. 2015, 98, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F. Type 1 diabetes genome-wide association studies: Not to be lost in translation. Clin. Transl. Immunol. 2017, 6, e162. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Várnai, C.; Thiecke, M.J.; et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef]
- Ram, R.; Morahan, G. Using Systems Genetics to Understanding the Etiology of Complex Disease. Methods Mol. Biol. 2017, 1488, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Fortune, M.D.; Burren, O.S.; Schofield, E.; Todd, J.A.; Wallace, C. Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum. Mol. Genet. 2015, 24, 3305–3313. [Google Scholar] [CrossRef] [PubMed]
- Mambiya, M.; Shang, M.; Wang, Y.; Li, Q.; Liu, S.; Yang, L.; Zhang, Q.; Zhang, K.; Liu, M.; Nie, F.; et al. The Play of Genes and Non-genetic Factors on Type 2 Diabetes. Front. Public Health 2019, 7, 349. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B.; Cupples, L.A.; Wilson, P.W. Parental transmission of type 2 diabetes: The Framingham Offspring Study. Diabetes 2000, 49, 2201–2207. [Google Scholar] [CrossRef]
- Poulsen, P.; Kyvik, K.O.; Vaag, A.; Beck-Nielsen, H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance--a population-based twin study. Diabetologia 1999, 42, 139–145. [Google Scholar] [CrossRef]
- Silander, K.; Mohlke, K.L.; Scott, L.J.; Peck, E.C.; Hollstein, P.; Skol, A.D.; Jackson, A.U.; Deloukas, P.; Hunt, S.; Stavrides, G.; et al. Genetic variation near the hepatocyte nuclear factor-4 alpha gene predicts susceptibility to type 2 diabetes. Diabetes 2004, 53, 1141–1149. [Google Scholar] [CrossRef]
- Grant, S.F.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Scott, L.J.; Saxena, R.; Voight, B.F.; Marchini, J.L.; Hu, T.; de Bakker, P.I.; Abecasis, G.R.; Almgren, P.; Andersen, G.; et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008, 40, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Spracklen, C.N.; Horikoshi, M.; Kim, Y.J.; Lin, K.; Bragg, F.; Moon, S.; Suzuki, K.; Tam, C.H.T.; Tabara, Y.; Kwak, S.H.; et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 2020, 582, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Consortium, W.T.C.C. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Ndiaye, F.K.; Ortalli, A.; Canouil, M.; Huyvaert, M.; Salazar-Cardozo, C.; Lecoeur, C.; Verbanck, M.; Pawlowski, V.; Boutry, R.; Durand, E.; et al. Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol. Metab. 2017, 6, 459–470. [Google Scholar] [CrossRef]
- Adeyemo, A.A.; Zaghloul, N.A.; Chen, G.; Doumatey, A.P.; Leitch, C.C.; Hostelley, T.L.; Nesmith, J.E.; Zhou, J.; Bentley, A.R.; Shriner, D.; et al. ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Nat. Commun. 2019, 10, 3195. [Google Scholar] [CrossRef]
- Pasquali, L.; Gaulton, K.J.; Rodríguez-Seguí, S.A.; Mularoni, L.; Miguel-Escalada, I.; Akerman, İ.; Tena, J.J.; Morán, I.; Gómez-Marín, C.; van de Bunt, M.; et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 2014, 46, 136–143. [Google Scholar] [CrossRef]
- Dimas, A.S.; Lagou, V.; Barker, A.; Knowles, J.W.; Mägi, R.; Hivert, M.F.; Benazzo, A.; Rybin, D.; Jackson, A.U.; Stringham, H.M.; et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 2014, 63, 2158–2171. [Google Scholar] [CrossRef]
- Vassy, J.L.; Hivert, M.F.; Porneala, B.; Dauriz, M.; Florez, J.C.; Dupuis, J.; Siscovick, D.S.; Fornage, M.; Rasmussen-Torvik, L.J.; Bouchard, C.; et al. Polygenic type 2 diabetes prediction at the limit of common variant detection. Diabetes 2014, 63, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Froguel, P. Rare and common genetic events in type 2 diabetes: What should biologists know? Cell Metab. 2015, 21, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Mercader, J.M.; Fuchsberger, C.; Udler, M.S.; Mahajan, A.; Wessel, J.; Teslovich, T.M.; Caulkins, L.; Koesterer, R.; Barajas-Olmos, F.; et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 2019, 570, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Alonso, L.C. T2D Risk Genes: Exome Sequencing Goes Straight to the Source. Cell Metab. 2019, 30, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Clément, N.; Fawcett, K.; Yengo, L.; Vaillant, E.; Guillaume, J.L.; Dechaume, A.; Payne, F.; Roussel, R.; Czernichow, S.; et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet. 2012, 44, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef]
- Majithia, A.R.; Flannick, J.; Shahinian, P.; Guo, M.; Bray, M.A.; Fontanillas, P.; Gabriel, S.B.; Rosen, E.D.; Altshuler, D.; Consortium, G.D.; et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 13127–13132. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Usman, K.; Banerjee, M. Pharmacogenetic studies update in type 2 diabetes mellitus. World J. Diabetes 2016, 7, 302–315. [Google Scholar] [CrossRef]
- Mannino, G.C.; Sesti, G. Individualized therapy for type 2 diabetes: Clinical implications of pharmacogenetic data. Mol. Diagn Ther. 2012, 16, 285–302. [Google Scholar] [CrossRef]
- Sorenson, R.L.; Brelje, T.C. Adaptation of islets of Langerhans to pregnancy: Beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm. Metab. Res. 1997, 29, 301–307. [Google Scholar] [CrossRef]
- Burlina, S.; Dalfrà, M.G.; Lapolla, A. Short- and long-term consequences for offspring exposed to maternal diabetes: A review. J. Matern. Fetal Neonatal Med. 2019, 32, 687–694. [Google Scholar] [CrossRef]
- Dalfrà, M.G.; Burlina, S.; Del Vescovo, G.G.; Lapolla, A. Genetics and Epigenetics: New Insight on Gestational Diabetes Mellitus. Front. Endocrinol. (Lausanne) 2020, 11, 602477. [Google Scholar] [CrossRef]
- Kwak, S.H.; Kim, S.H.; Cho, Y.M.; Go, M.J.; Cho, Y.S.; Choi, S.H.; Moon, M.K.; Jung, H.S.; Shin, H.D.; Kang, H.M.; et al. A genome-wide association study of gestational diabetes mellitus in Korean women. Diabetes 2012, 61, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Huopio, H.; Cederberg, H.; Vangipurapu, J.; Hakkarainen, H.; Pääkkönen, M.; Kuulasmaa, T.; Heinonen, S.; Laakso, M. Association of risk variants for type 2 diabetes and hyperglycemia with gestational diabetes. Eur. J. Endocrinol. 2013, 169, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Freathy, R.M.; Hayes, M.G.; Urbanek, M.; Lowe, L.P.; Lee, H.; Ackerman, C.; Frayling, T.M.; Cox, N.J.; Dunger, D.B.; Dyer, A.R.; et al. Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study: Common genetic variants in GCK and TCF7L2 are associated with fasting and postchallenge glucose levels in pregnancy and with the new consensus definition of gestational diabetes mellitus from the International Association of Diabetes and Pregnancy Study Groups. Diabetes 2010, 59, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Bennink, H.J.; Schreurs, W.H. Improvement of oral glucose tolerance in gestational diabetes by pyridoxine. Br. Med. J. 1975, 3, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Spellacy, W.N.; Buhi, W.C.; Birk, S.A. Vitamin B6 treatment of gestational diabetes mellitus: Studies of blood glucose and plasma insulin. Am. J. Obstet. Gynecol. 1977, 127, 599–602. [Google Scholar] [CrossRef]
- Fields, A.M.; Welle, K.; Ho, E.S.; Mesaros, C.; Susiarjo, M. Vitamin B6 deficiency disrupts serotonin signaling in pancreatic islets and induces gestational diabetes in mice. Commun. Biol. 2021, 4, 421. [Google Scholar] [CrossRef]
- Zhang, H.; Colclough, K.; Gloyn, A.L.; Pollin, T.I. Monogenic diabetes: A gateway to precision medicine in diabetes. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Weiss, M.; Tura, A.; Kautzky-Willer, A.; Pacini, G.; D’Argenio, D.Z. Human insulin dynamics in women: A physiologically based model. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R268–R274. [Google Scholar] [CrossRef]
- Fajans, S.S.; Bell, G.I. MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011, 34, 1878–1884. [Google Scholar] [CrossRef]
- Iafusco, D.; Massa, O.; Pasquino, B.; Colombo, C.; Iughetti, L.; Bizzarri, C.; Mammì, C.; Lo Presti, D.; Suprani, T.; Schiaffini, R.; et al. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol. 2012, 49, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, A.S.; Shields, B.M.; Flanagan, S.E.; Bruining, G.J.; Noordam, K.; Gach, A.; Mlynarski, W.; Malecki, M.T.; Hattersley, A.T.; Ellard, S. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia 2009, 52, 1683–1685. [Google Scholar] [CrossRef] [PubMed]
- Polak, M.; Cavé, H. Neonatal diabetes mellitus: A disease linked to multiple mechanisms. Orphanet J. Rare Dis. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Pearson, E.R.; Antcliff, J.F.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.; Molnes, J.; et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 2004, 350, 1838–1849. [Google Scholar] [CrossRef]
- Temple, I.K.; Shield, J.P. 6q24 transient neonatal diabetes. Rev. Endocr. Metab. Disord. 2010, 11, 199–204. [Google Scholar] [CrossRef]
- Babenko, A.P.; Polak, M.; Cavé, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006, 355, 456–466. [Google Scholar] [CrossRef]
- Naylor, R.N.; Greeley, S.A.; Bell, G.I.; Philipson, L.H. Genetics and pathophysiology of neonatal diabetes mellitus. J. Diabetes Investig. 2011, 2, 158–169. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Clauin, S.; Bellanné-Chantelot, C.; de Lonlay, P.; Harries, L.W.; Gloyn, A.L.; Ellard, S. Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum. Mutat. 2009, 30, 170–180. [Google Scholar] [CrossRef]
- Støy, J.; Edghill, E.L.; Flanagan, S.E.; Ye, H.; Paz, V.P.; Pluzhnikov, A.; Below, J.E.; Hayes, M.G.; Cox, N.J.; Lipkind, G.M.; et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 15040–15044. [Google Scholar] [CrossRef]
- Garin, I.; Edghill, E.L.; Akerman, I.; Rubio-Cabezas, O.; Rica, I.; Locke, J.M.; Maestro, M.A.; Alshaikh, A.; Bundak, R.; del Castillo, G.; et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3105–3110. [Google Scholar] [CrossRef]
- Njølstad, P.R.; Sagen, J.V.; Bjørkhaug, L.; Odili, S.; Shehadeh, N.; Bakry, D.; Sarici, S.U.; Alpay, F.; Molnes, J.; Molven, A.; et al. Permanent neonatal diabetes caused by glucokinase deficiency: Inborn error of the glucose-insulin signaling pathway. Diabetes 2003, 52, 2854–2860. [Google Scholar] [CrossRef]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef]
- Laver, T. Redefining the pathogenicity of Maturity Onset Diabetes of the Young. (MODY) genes: BLK, PAX4 and KLF11 do not cause MODY. In Proceedings of the Abstracts of the Diabetes UK Professional Conference 2018, London ExCeL, London, UK, 14–16 March 2018; Volume 35, pp. 5–205. [Google Scholar] [CrossRef]
- Patel, K.A.; Kettunen, J.; Laakso, M.; Stančáková, A.; Laver, T.W.; Colclough, K.; Johnson, M.B.; Abramowicz, M.; Groop, L.; Miettinen, P.J.; et al. Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nat. Commun. 2017, 8, 888. [Google Scholar] [CrossRef]
- Angelidi, A.M.; Filippaios, A.; Mantzoros, C.S. Severe insulin resistance syndromes. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Kahn, C.R.; Flier, J.S.; Bar, R.S.; Archer, J.A.; Gorden, P.; Martin, M.M.; Roth, J. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N. Engl. J. Med. 1976, 294, 739–745. [Google Scholar] [CrossRef]
- Taylor, S.I.; Cama, A.; Accili, D.; Barbetti, F.; Quon, M.J.; de la Luz Sierra, M.; Suzuki, Y.; Koller, E.; Levy-Toledano, R.; Wertheimer, E. Mutations in the insulin receptor gene. Endocr. Rev. 1992, 13, 566–595. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, K.K.; Winnay, J.; Johansson, S.; Claudi, T.; König, R.; Haldorsen, I.; Johansson, B.; Woo, J.R.; Aarskog, D.; Sagen, J.V.; et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am. J. Hum. Genet. 2013, 93, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Kushi, R.; Hirota, Y.; Ogawa, W. Insulin resistance and exaggerated insulin sensitivity triggered by single-gene mutations in the insulin signaling pathway. Diabetol. Int. 2021, 12, 62–67. [Google Scholar] [CrossRef]
- Feingold, K.R.; Anawalt, B.; Boyce, A. Atypical Forms of Diabetes. [Updated 2019 March 19]. In Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279128/ (accessed on 15 September 2021).
- Al-Awar, A.; Kupai, K.; Veszelka, M.; Szűcs, G.; Attieh, Z.; Murlasits, Z.; Török, S.; Pósa, A.; Varga, C. Experimental Diabetes Mellitus in Different Animal Models. J. Diabetes Res. 2016, 2016, 9051426. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef]
- Corradini, N.; Rossi, F.; Giordano, E.; Caizzi, R.; Verní, F.; Dimitri, P. Drosophila melanogaster as a model for studying protein-encoding genes that are resident in constitutive heterochromatin. Heredity (Edinb) 2007, 98, 3–12. [Google Scholar] [CrossRef][Green Version]
- Mohr, S.E.; Hu, Y.; Kim, K.; Housden, B.E.; Perrimon, N. Resources for functional genomics studies in Drosophila melanogaster. Genetics 2014, 197, 1–18. [Google Scholar] [CrossRef]
- De Nobrega, A.K.; Lyons, L.C. Aging and the clock: Perspective from flies to humans. Eur. J. Neurosci. 2020, 51, 454–481. [Google Scholar] [CrossRef]
- Liguori, F.; Amadio, S.; Volonté, C. Fly for ALS: Drosophila modeling on the route to amyotrophic lateral sclerosis modifiers. Cell Mol. Life Sci. 2021, 78, 6143–6160. [Google Scholar] [CrossRef]
- Specchia, V.; Puricella, A.; D’Attis, S.; Massari, S.; Giangrande, A.; Bozzetti, M.P. Drosophila melanogaster as a Model to Study the Multiple Phenotypes, Related to Genome Stability of the Fragile-X Syndrome. Front. Genet. 2019, 10, 10. [Google Scholar] [CrossRef]
- Tsuda, L.; Lim, Y.M. Alzheimer’s Disease Model System Using Drosophila. Adv. Exp. Med. Biol. 2018, 1076, 25–40. [Google Scholar] [CrossRef]
- Zhu, S.; Han, Z.; Luo, Y.; Chen, Y.; Zeng, Q.; Wu, X.; Yuan, W. Molecular mechanisms of heart failure: Insights from Drosophila. Heart Fail. Rev. 2017, 22, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Millet-Boureima, C.; Porras Marroquin, J.; Gamberi, C. Modeling Renal Disease “On the Fly”. Biomed. Res. Int. 2018, 2018, 5697436. [Google Scholar] [CrossRef] [PubMed]
- Graham, P.; Pick, L. Drosophila as a Model for Diabetes and Diseases of Insulin Resistance. Curr. Top. Dev. Biol. 2017, 121, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, K.N. The epicurean fly: Using Drosophila melanogaster to study metabolism. Pediatr. Res. 2009, 65, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Grönke, S.; Clarke, D.F.; Broughton, S.; Andrews, T.D.; Partridge, L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010, 6, e1000857. [Google Scholar] [CrossRef]
- Nässel, D.R.; Kubrak, O.I.; Liu, Y.; Luo, J.; Lushchak, O.V. Factors that regulate insulin producing cells and their output in Drosophila. Front. Physiol. 2013, 4, 252. [Google Scholar] [CrossRef]
- Brogiolo, W.; Stocker, H.; Ikeya, T.; Rintelen, F.; Fernandez, R.; Hafen, E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 2001, 11, 213–221. [Google Scholar] [CrossRef]
- Kim, S.K.; Rulifson, E.J. Conserved mechanisms of glucose sensing and regulation by Drosophila corpora cardiaca cells. Nature 2004, 431, 316–320. [Google Scholar] [CrossRef]
- Lee, G.; Park, J.H. Hemolymph sugar homeostasis and starvation-induced hyperactivity affected by genetic manipulations of the adipokinetic hormone-encoding gene in Drosophila melanogaster. Genetics 2004, 167, 311–323. [Google Scholar] [CrossRef]
- Pasco, M.Y.; Léopold, P. High sugar-induced insulin resistance in Drosophila relies on the lipocalin Neural Lazarillo. PLoS ONE 2012, 7, e36583. [Google Scholar] [CrossRef] [PubMed]
- Ugrankar, R.; Berglund, E.; Akdemir, F.; Tran, C.; Kim, M.S.; Noh, J.; Schneider, R.; Ebert, B.; Graff, J.M. Drosophila glucome screening identifies Ck1alpha as a regulator of mammalian glucose metabolism. Nat. Commun. 2015, 6, 7102. [Google Scholar] [CrossRef]
- Fridell, Y.W.; Hoh, M.; Kréneisz, O.; Hosier, S.; Chang, C.; Scantling, D.; Mulkey, D.K.; Helfand, S.L. Increased uncoupling protein (UCP) activity in Drosophila insulin-producing neurons attenuates insulin signaling and extends lifespan. Aging (Albany NY) 2009, 1, 699–713. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kréneisz, O.; Chen, X.; Fridell, Y.W.; Mulkey, D.K. Glucose increases activity and Ca2+ in insulin-producing cells of adult Drosophila. Neuroreport 2010, 21, 1116–1120. [Google Scholar] [CrossRef]
- Inoue, Y.H.; Katsube, H.; Hinami, Y. Drosophila Models to Investigate Insulin Action and Mechanisms Underlying Human Diabetes Mellitus. Adv. Exp. Med. Biol. 2018, 1076, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.; Tabarini, D.; Azpiazu, N.; Frasch, M.; Schlessinger, J. The Drosophila insulin receptor homolog: A gene essential for embryonic development encodes two receptor isoforms with different signaling potential. EMBO J. 1995, 14, 3373–3384. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fernandez, R.; Roth, R.A. Comparison of the signaling abilities of the Drosophila and human insulin receptors in mammalian cells. Biochemistry 1995, 34, 4962–4968. [Google Scholar] [CrossRef]
- Böhni, R.; Riesgo-Escovar, J.; Oldham, S.; Brogiolo, W.; Stocker, H.; Andruss, B.F.; Beckingham, K.; Hafen, E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 1999, 97, 865–875. [Google Scholar] [CrossRef]
- Almudi, I.; Poernbacher, I.; Hafen, E.; Stocker, H. The Lnk/SH2B adaptor provides a fail-safe mechanism to establish the Insulin receptor-Chico interaction. Cell Commun. Signal. 2013, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Werz, C.; Köhler, K.; Hafen, E.; Stocker, H. The Drosophila SH2B family adaptor Lnk acts in parallel to chico in the insulin signaling pathway. PLoS Genet. 2009, 5, e1000596. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R.; Wullschleger, S.; Sakamoto, K.; García-Martínez, J.M.; Clacher, C.; Komander, D.; van Aalten, D.M.; Boini, K.M.; Lang, F.; Lipina, C.; et al. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol. Cell Biol. 2008, 28, 3258–3272. [Google Scholar] [CrossRef] [PubMed]
- Goberdhan, D.C.; Paricio, N.; Goodman, E.C.; Mlodzik, M.; Wilson, C. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 1999, 13, 3244–3258. [Google Scholar] [CrossRef]
- Wittwer, F.; Jaquenoud, M.; Brogiolo, W.; Zarske, M.; Wüstemann, P.; Fernandez, R.; Stocker, H.; Wymann, M.P.; Hafen, E. Susi, a negative regulator of Drosophila PI3-kinase. Dev. Cell 2005, 8, 817–827. [Google Scholar] [CrossRef]
- Crivat, G.; Lizunov, V.A.; Li, C.R.; Stenkula, K.G.; Zimmerberg, J.; Cushman, S.W.; Pick, L. Insulin stimulates translocation of human GLUT4 to the membrane in fat bodies of transgenic Drosophila melanogaster. PLoS ONE 2013, 8, e77953. [Google Scholar] [CrossRef]
- Rulifson, E.J.; Kim, S.K.; Nusse, R. Ablation of insulin-producing neurons in flies: Growth and diabetic phenotypes. Science 2002, 296, 1118–1120. [Google Scholar] [CrossRef]
- Broughton, S.J.; Piper, M.D.; Ikeya, T.; Bass, T.M.; Jacobson, J.; Driege, Y.; Martinez, P.; Hafen, E.; Withers, D.J.; Leevers, S.J.; et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. USA 2005, 102, 3105–3110. [Google Scholar] [CrossRef]
- Ikeya, T.; Galic, M.; Belawat, P.; Nairz, K.; Hafen, E. Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr. Biol. 2002, 12, 1293–1300. [Google Scholar] [CrossRef]
- Haselton, A.; Sharmin, E.; Schrader, J.; Sah, M.; Poon, P.; Fridell, Y.W. Partial ablation of adult Drosophila insulin-producing neurons modulates glucose homeostasis and extends life span without insulin resistance. Cell Cycle 2010, 9, 3063–3071. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Li, C.R.; Momen, B.; Kohanski, R.A.; Pick, L. Deletion of Drosophila insulin-like peptides causes growth defects and metabolic abnormalities. Proc. Natl. Acad. Sci. USA 2009, 106, 19617–19622. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan-Murphy, B.; Urano, F. ER stress as a trigger for β-cell dysfunction and autoimmunity in type 1 diabetes. Diabetes 2012, 61, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Tomer, Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J. Autoimmun. 2017, 80, 48–55. [Google Scholar] [CrossRef]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Ngyuen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef]
- Katsube, H.; Hinami, Y.; Yamazoe, T.; Inoue, Y.H. Endoplasmic reticulum stress-induced cellular dysfunction and cell death in insulin-producing cells results in diabetes-like phenotypes in. Biol. Open 2019, 8. [Google Scholar] [CrossRef]
- Musselman, L.P.; Fink, J.L.; Narzinski, K.; Ramachandran, P.V.; Hathiramani, S.S.; Cagan, R.L.; Baranski, T.J. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis. Model. Mech. 2011, 4, 842–849. [Google Scholar] [CrossRef]
- van Dam, E.; van Leeuwen, L.A.G.; Dos Santos, E.; James, J.; Best, L.; Lennicke, C.; Vincent, A.J.; Marinos, G.; Foley, A.; Buricova, M.; et al. Sugar-Induced Obesity and Insulin Resistance Are Uncoupled from Shortened Survival in Drosophila. Cell Metab. 2020, 31, 710–725.e7. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Maldonado, J.M.; Sánchez-Chávez, G.; Salgado, L.M.; Salceda, R.; Riesgo-Escovar, J.R. Drosophila insulin pathway mutants affect visual physiology and brain function besides growth, lipid, and carbohydrate metabolism. Diabetes 2011, 60, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- Merigliano, C.; Mascolo, E.; La Torre, M.; Saggio, I.; Vernì, F. Protective role of vitamin B6 (PLP) against DNA damage in Drosophila models of type 2 diabetes. Sci. Rep. 2018, 8, 11432. [Google Scholar] [CrossRef] [PubMed]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef]
- Park, S.; Alfa, R.W.; Topper, S.M.; Kim, G.E.; Kockel, L.; Kim, S.K. A genetic strategy to measure circulating Drosophila insulin reveals genes regulating insulin production and secretion. PLoS Genet. 2014, 10, e1004555. [Google Scholar] [CrossRef]
- Percudani, R.; Peracchi, A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef]
- Mascolo, E.; Vernì, F. Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 3669. [Google Scholar] [CrossRef]
- Marzio, A.; Merigliano, C.; Gatti, M.; Vernì, F. Sugar and chromosome stability: Clastogenic effects of sugars in vitamin B6-deficient cells. PLoS Genet. 2014, 10, e1004199. [Google Scholar] [CrossRef]
- Mascolo, E.; Amoroso, N.; Saggio, I.; Merigliano, C.; Vernì, F. Pyridoxine/pyridoxamine 5′-phosphate oxidase (Sgll/PNPO) is important for DNA integrity and glucose homeostasis maintenance in Drosophila. J. Cell Physiol. 2020, 235, 504–512. [Google Scholar] [CrossRef]
- Mascolo, E.; Barile, A.; Mecarelli, L.S.; Amoroso, N.; Merigliano, C.; Massimi, A.; Saggio, I.; Hansen, T.; Tramonti, A.; Di Salvo, M.L.; et al. The expression of four pyridoxal kinase (PDXK) human variants in Drosophila impacts on genome integrity. Sci. Rep. 2019, 9, 14188. [Google Scholar] [CrossRef]
- Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; Thomas, H.E.; Gough, D.J. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016, 283, 3002–3015. [Google Scholar] [CrossRef]
- Lourido, F.; Quenti, D.; Salgado-Canales, D.; Tobar, N. Domeless receptor loss in fat body tissue reverts insulin resistance induced by a high-sugar diet in Drosophila melanogaster. Sci. Rep. 2021, 11, 3263. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, S.; Fournier, D.; Mounier, C.; Rassart, E. Human apolipoprotein D overexpression in transgenic mice induces insulin resistance and alters lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E802–E811. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Increased Plasma Levels of Xanthurenic and Kynurenic Acids in Type 2 Diabetes. Mol. Neurobiol. 2015, 52, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Navrotskaya, V.; Oxenkrug, G.; Vorobyova, L.; Summergrad, P. Attenuation of high sucrose diet-induced insulin resistance in tryptophan 2,3-dioxygenase deficient Drosophila melanogaster vermilion mutants. Integr. Obes. Diabetes 2015, 1, 93–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barry, W.E.; Thummel, C.S. The Drosophila HNF4 nuclear receptor promotes glucose-stimulated insulin secretion and mitochondrial function in adults. Elife 2016, 5. [Google Scholar] [CrossRef]
- Palu, R.A.; Thummel, C.S. Sir2 Acts through Hepatocyte Nuclear Factor 4 to maintain insulin Signaling and Metabolic Homeostasis in Drosophila. PLoS Genet. 2016, 12, e1005978. [Google Scholar] [CrossRef] [PubMed]
- Reis, T.; Van Gilst, M.R.; Hariharan, I.K. A buoyancy-based screen of Drosophila larvae for fat-storage mutants reveals a role for Sir2 in coupling fat storage to nutrient availability. PLoS Genet. 2010, 6, e1001206. [Google Scholar] [CrossRef]
- Storelli, G.; Nam, H.J.; Simcox, J.; Villanueva, C.J.; Thummel, C.S. Drosophila HNF4 Directs a Switch in Lipid Metabolism that Supports the Transition to Adulthood. Dev. Cell 2019, 48, 200–214.e6. [Google Scholar] [CrossRef]
- Mascolo, E.; Liguori, F.; Stufera Mecarelli, L.; Amoroso, N.; Merigliano, C.; Amadio, S.; Volonté, C.; Contestabile, R.; Tramonti, A.; Vernì, F. Functional Inactivation of Drosophila GCK orthologs causes genomic instability and oxidative stress in a fly model of MODY-2. Int. J. Mol. Sci. 2021, 22, 918. [Google Scholar] [CrossRef] [PubMed]
- Pendse, J.; Ramachandran, P.V.; Na, J.; Narisu, N.; Fink, J.L.; Cagan, R.L.; Collins, F.S.; Baranski, T.J. A Drosophila functional evaluation of candidates from human genome-wide association studies of type 2 diabetes and related metabolic traits identifies tissue-specific roles for dHHEX. BMC Genom. 2013, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Peiris, H.; Park, S.; Louis, S.; Gu, X.; Lam, J.Y.; Asplund, O.; Ippolito, G.C.; Bottino, R.; Groop, L.; Tucker, H.; et al. Discovering human diabetes-risk gene function with genetics and physiological assays. Nat. Commun. 2018, 9, 3855. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Yamazaki, H.; Sekine-Aizawa, Y.; Hirokawa, N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 1995, 81, 769–780. [Google Scholar] [CrossRef]
- Pack-Chung, E.; Kurshan, P.T.; Dickman, D.K.; Schwarz, T.L. A Drosophila kinesin required for synaptic bouton formation and synaptic vesicle transport. Nat. Neurosci. 2007, 10, 980–989. [Google Scholar] [CrossRef]
- Barkus, R.V.; Klyachko, O.; Horiuchi, D.; Dickson, B.J.; Saxton, W.M. Identification of an axonal kinesin-3 motor for fast anterograde vesicle transport that facilitates retrograde transport of neuropeptides. Mol. Biol. Cell 2008, 19, 274–283. [Google Scholar] [CrossRef]
- Cao, J.; Ni, J.; Ma, W.; Shiu, V.; Milla, L.A.; Park, S.; Spletter, M.L.; Tang, S.; Zhang, J.; Wei, X.; et al. Insight into insulin secretion from transcriptome and genetic analysis of insulin-producing cells of Drosophila. Genetics 2014, 197, 175–192. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Jünger, M.A.; Rintelen, F.; Stocker, H.; Wasserman, J.D.; Végh, M.; Radimerski, T.; Greenberg, M.E.; Hafen, E. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J. Biol. 2003, 2, 20. [Google Scholar] [CrossRef]
- Zhang, W.; Thompson, B.J.; Hietakangas, V.; Cohen, S.M. MAPK/ERK signaling regulates insulin sensitivity to control glucose metabolism in Drosophila. PLoS Genet. 2011, 7, e1002429. [Google Scholar] [CrossRef]
- He, B.Z.; Ludwig, M.Z.; Dickerson, D.A.; Barse, L.; Arun, B.; Vilhjálmsson, B.J.; Jiang, P.; Park, S.Y.; Tamarina, N.A.; Selleck, S.B.; et al. Effect of genetic variation in a Drosophila model of diabetes-associated misfolded human proinsulin. Genetics 2014, 196, 557–567. [Google Scholar] [CrossRef]
- Park, S.Y.; Ludwig, M.Z.; Tamarina, N.A.; He, B.Z.; Carl, S.H.; Dickerson, D.A.; Barse, L.; Arun, B.; Williams, C.L.; Miles, C.M.; et al. Genetic complexity in a Drosophila model of diabetes-associated misfolded human proinsulin. Genetics 2014, 196, 539–555. [Google Scholar] [CrossRef]
- Mackay, T.F.; Richards, S.; Stone, E.A.; Barbadilla, A.; Ayroles, J.F.; Zhu, D.; Casillas, S.; Han, Y.; Magwire, M.M.; Cridland, J.M.; et al. The Drosophila melanogaster Genetic Reference Panel. Nature 2012, 482, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Musselman, L.P.; Pendse, J.; Baranski, T.J.; Bodmer, R.; Ocorr, K.; Cagan, R. A Drosophila model of high sugar diet-induced cardiomyopathy. PLoS Genet. 2013, 9, e1003175. [Google Scholar] [CrossRef]
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 2002, 109, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kahn, C.R. Altered insulin signaling in retinal tissue in diabetic states. J. Biol. Chem. 2004, 279, 37997–38006. [Google Scholar] [CrossRef] [PubMed]
- Catalani, E.; Silvestri, F.; Bongiorni, S.; Taddei, A.R.; Fanelli, G.; Rinalducci, S.; De Palma, C.; Perrotta, C.; Prantera, G.; Cervia, D. Retinal damage in a new model of hyperglycemia induced by high-sucrose diets. Pharmacol. Res. 2021, 166, 105488. [Google Scholar] [CrossRef]
- Rani, L.; Saini, S.; Shukla, N.; Chowdhuri, D.K.; Gautam, N.K. High sucrose diet induces morphological, structural and functional impairments in the renal tubules of Drosophila melanogaster: A model for studying type-2 diabetes mediated renal tubular dysfunction. Insect Biochem. Mol. Biol. 2020, 125, 103441. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Scully, T.; Ettela, A.; LeRoith, D.; Gallagher, E.J. Obesity, Type 2 Diabetes, and Cancer Risk. Front. Oncol. 2020, 10, 615375. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, S.; Baranski, T.J.; Cagan, R.L. Transformed Drosophila cells evade diet-mediated insulin resistance through wingless signaling. Cell 2013, 154, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, M.; Montisano, D.F.; Toledo, S.; Barrieux, A. High glucose induces DNA damage in cultured human endothelial cells. J. Clin. Investig. 1986, 77, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Arabski, M.; Krupa, R.; Wozniak, K.; Zadrozny, M.; Kasznicki, J.; Zurawska, M.; Drzewoski, J. DNA damage and repair in type 2 diabetes mellitus. Mutat. Res. 2004, 554, 297–304. [Google Scholar] [CrossRef]
- Tatsch, E.; Bochi, G.V.; Piva, S.J.; De Carvalho, J.A.; Kober, H.; Torbitz, V.D.; Duarte, T.; Signor, C.; Coelho, A.C.; Duarte, M.M.; et al. Association between DNA strand breakage and oxidative, inflammatory and endothelial biomarkers in type 2 diabetes. Mutat. Res. 2012, 732, 16–20. [Google Scholar] [CrossRef]
- Lee, S.C.; Chan, J.C. Evidence for DNA damage as a biological link between diabetes and cancer. Chin. Med. J. 2015, 128, 1543–1548. [Google Scholar] [CrossRef]
- Ames, B.N. Micronutrients prevent cancer and delay aging. Toxicol. Lett. 1998, 102–103, 5–18. [Google Scholar] [CrossRef]
- Ames, B.N. Micronutrient deficiencies. A major cause of DNA damage. Ann. N. Y. Acad. Sci. 1999, 889, 87–106. [Google Scholar] [CrossRef]





| MODY Subtype | Gene |
|---|---|
| 1 | HNF4A |
| 2 | GCK |
| 3 | HNF1A |
| 4 | PDX1 |
| 5 | HNF1B |
| 6 | NEUROD1 |
| 7 | KLF11 |
| 8 | CEL |
| 9 | PAX4 |
| 10 | INS |
| 11 | BLK |
| 12 | KCNJ11 |
| 13 | ABCC8 |
| 14 | APPL1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liguori, F.; Mascolo, E.; Vernì, F. The Genetics of Diabetes: What We Can Learn from Drosophila. Int. J. Mol. Sci. 2021, 22, 11295. https://doi.org/10.3390/ijms222011295
Liguori F, Mascolo E, Vernì F. The Genetics of Diabetes: What We Can Learn from Drosophila. International Journal of Molecular Sciences. 2021; 22(20):11295. https://doi.org/10.3390/ijms222011295
Chicago/Turabian StyleLiguori, Francesco, Elisa Mascolo, and Fiammetta Vernì. 2021. "The Genetics of Diabetes: What We Can Learn from Drosophila" International Journal of Molecular Sciences 22, no. 20: 11295. https://doi.org/10.3390/ijms222011295
APA StyleLiguori, F., Mascolo, E., & Vernì, F. (2021). The Genetics of Diabetes: What We Can Learn from Drosophila. International Journal of Molecular Sciences, 22(20), 11295. https://doi.org/10.3390/ijms222011295