
In Silico and In Vivo Analysis of Amino Acid Substitutions That Cause Laminopathies

Abstract
:1. Introduction
2. Results
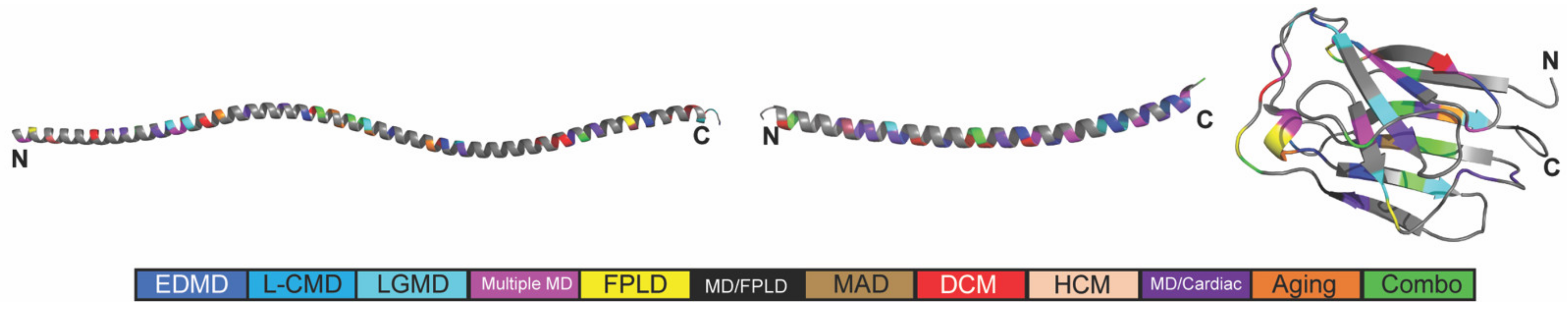
2.1. The Position and Predicted Solvent Accessibility of an Amino Acid Substitution Do Not Correspond to Disease Phenotypes
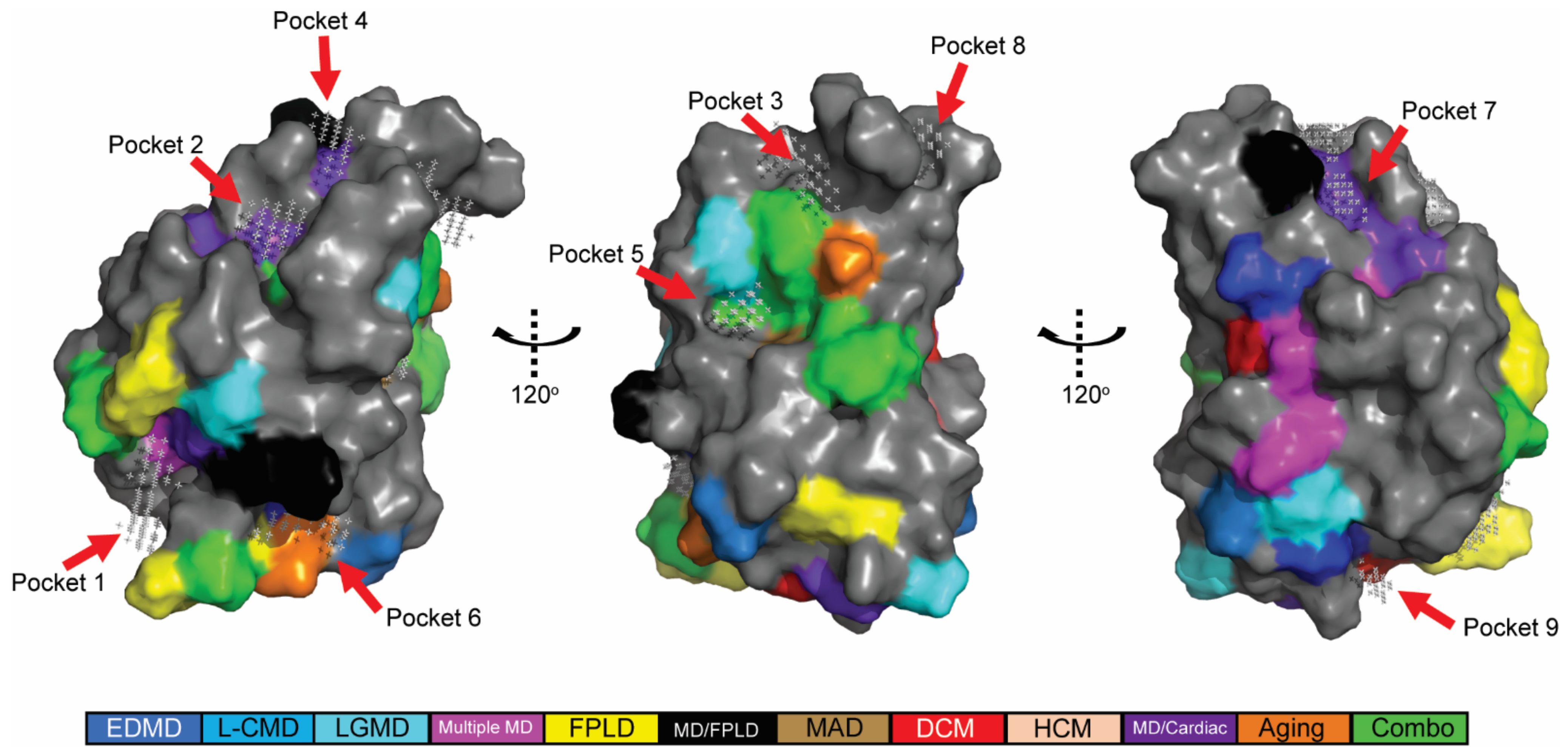
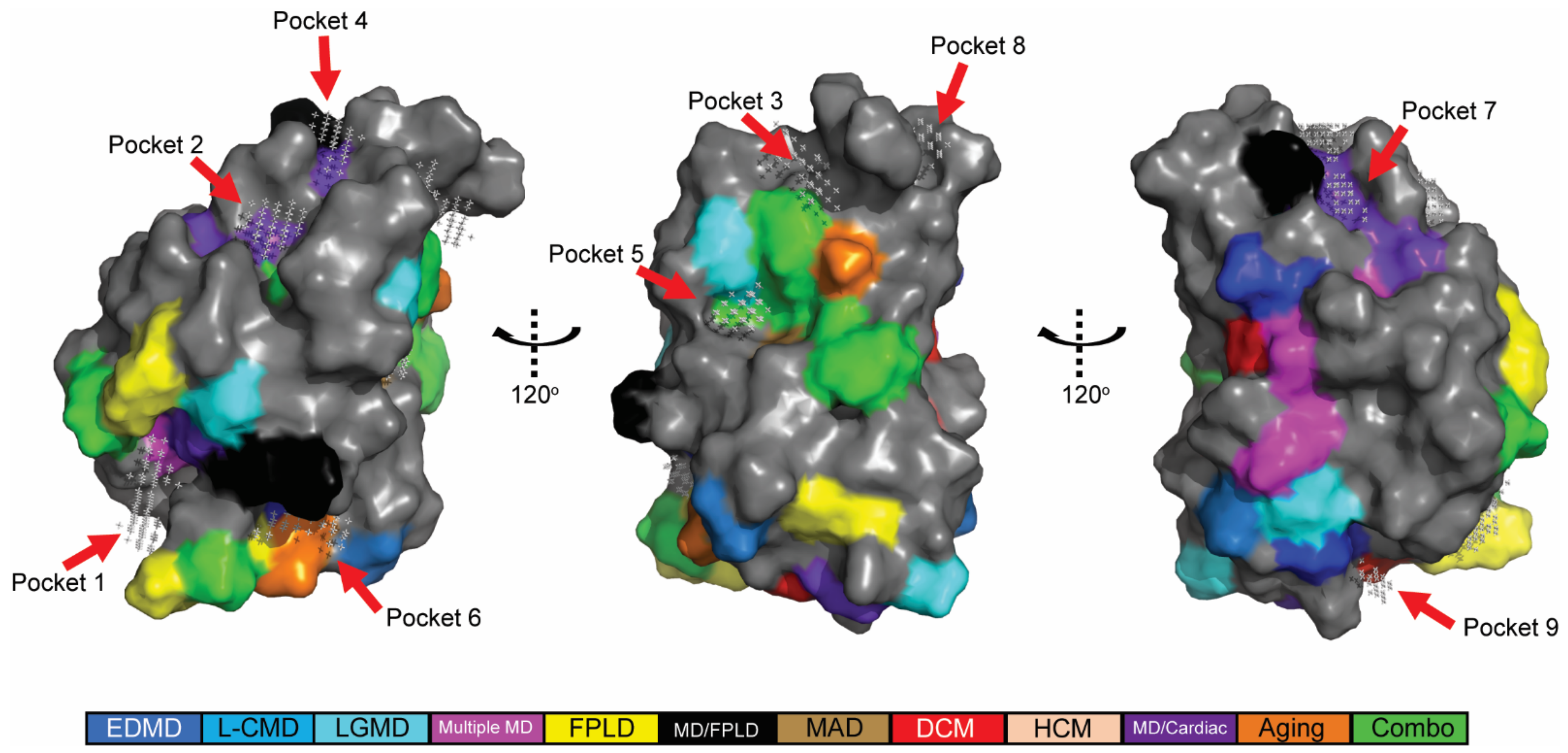
2.2. In Silico Predicted Binding Pockets of the Ig-like Fold Map to Disease-Causing Amino Acid Substitutions
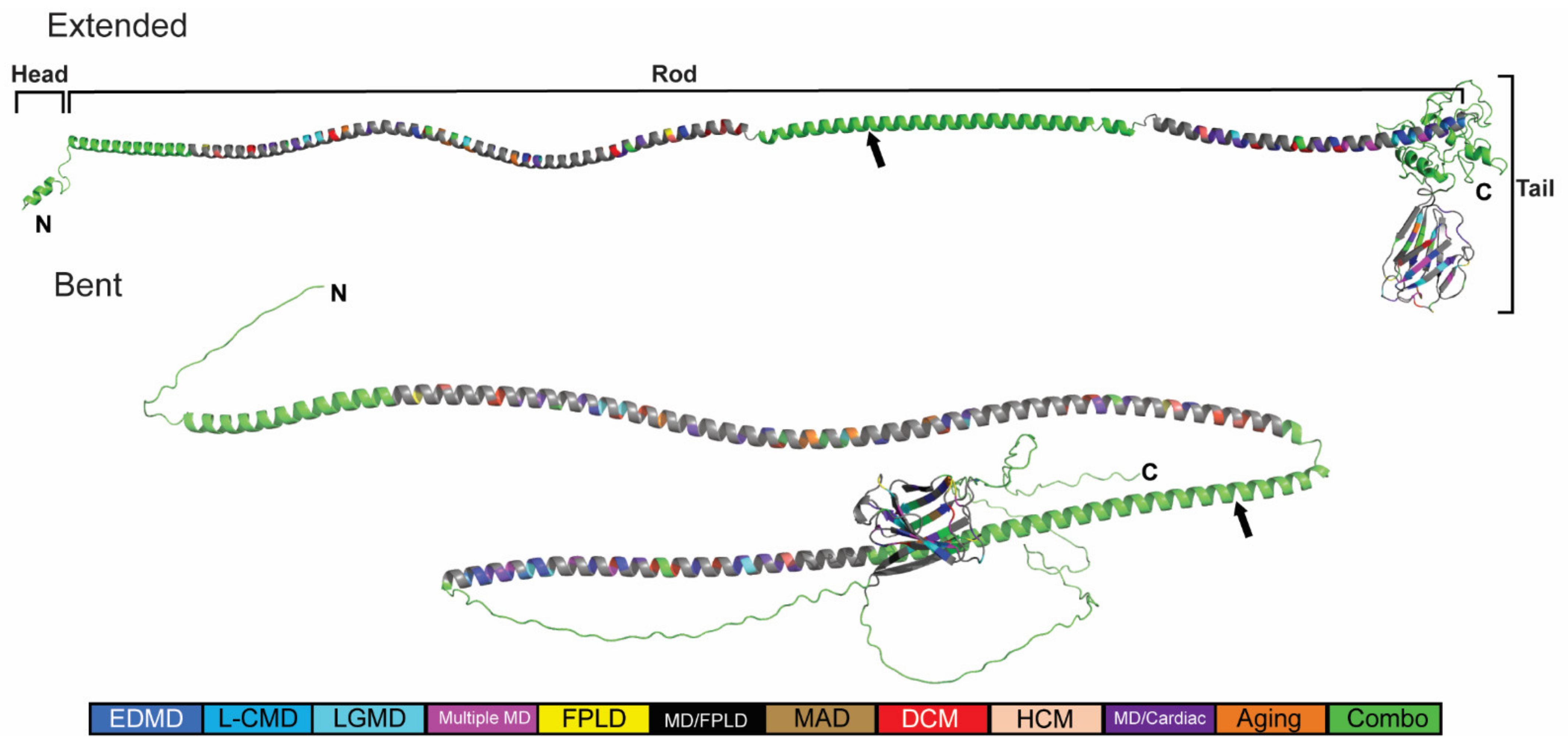
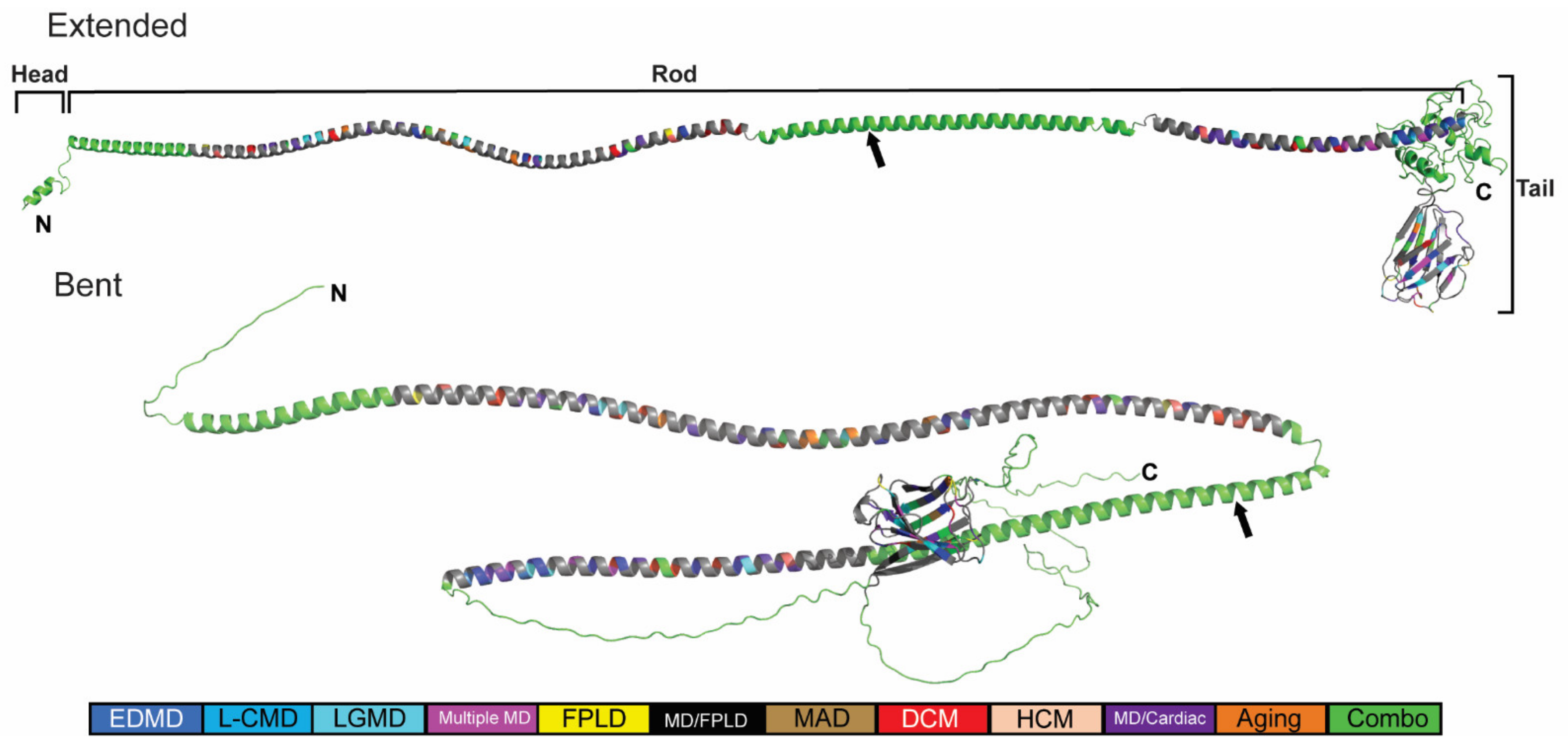
2.3. Generation of an In Silico Full-Length Human Lamin A/C
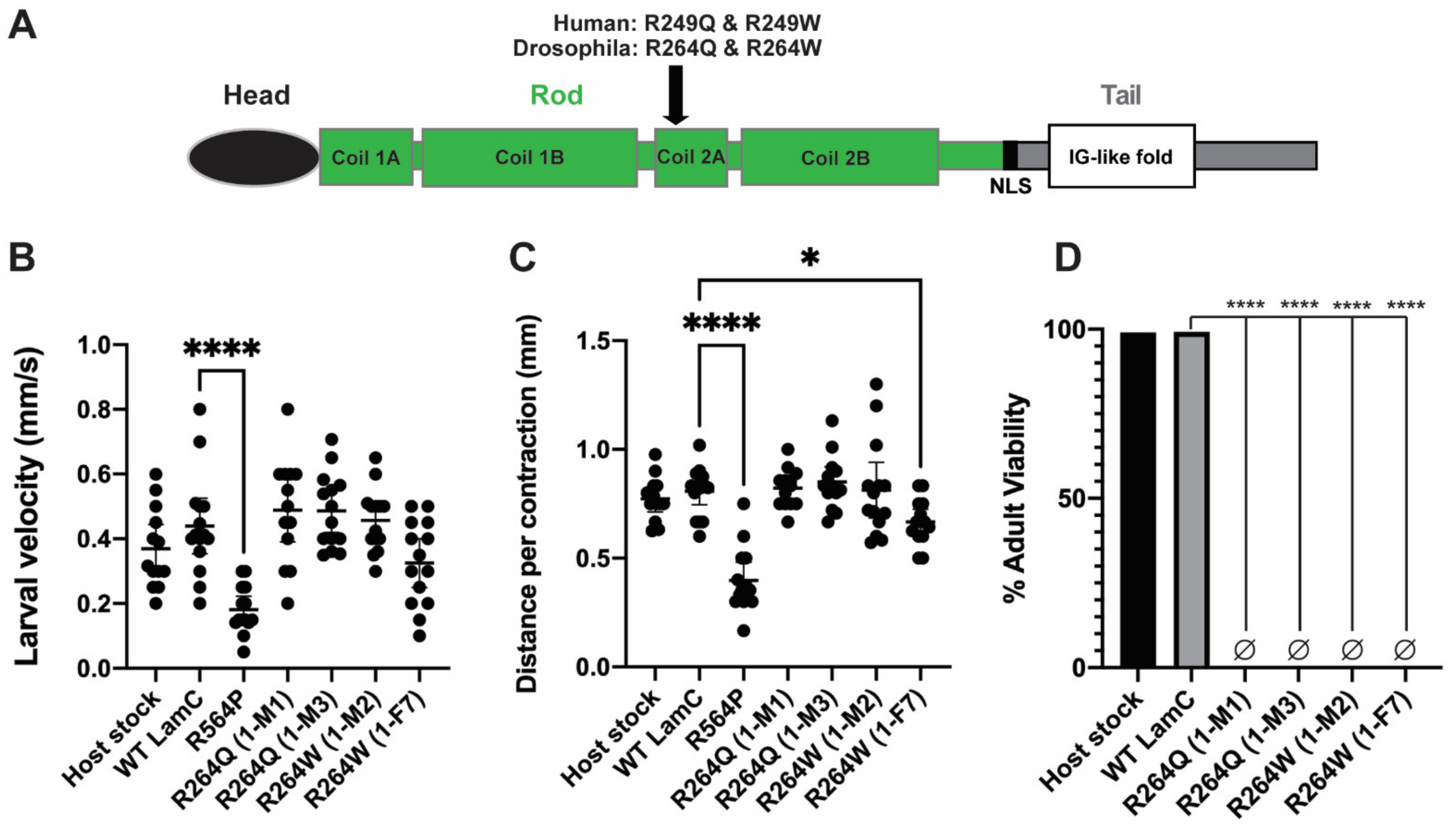
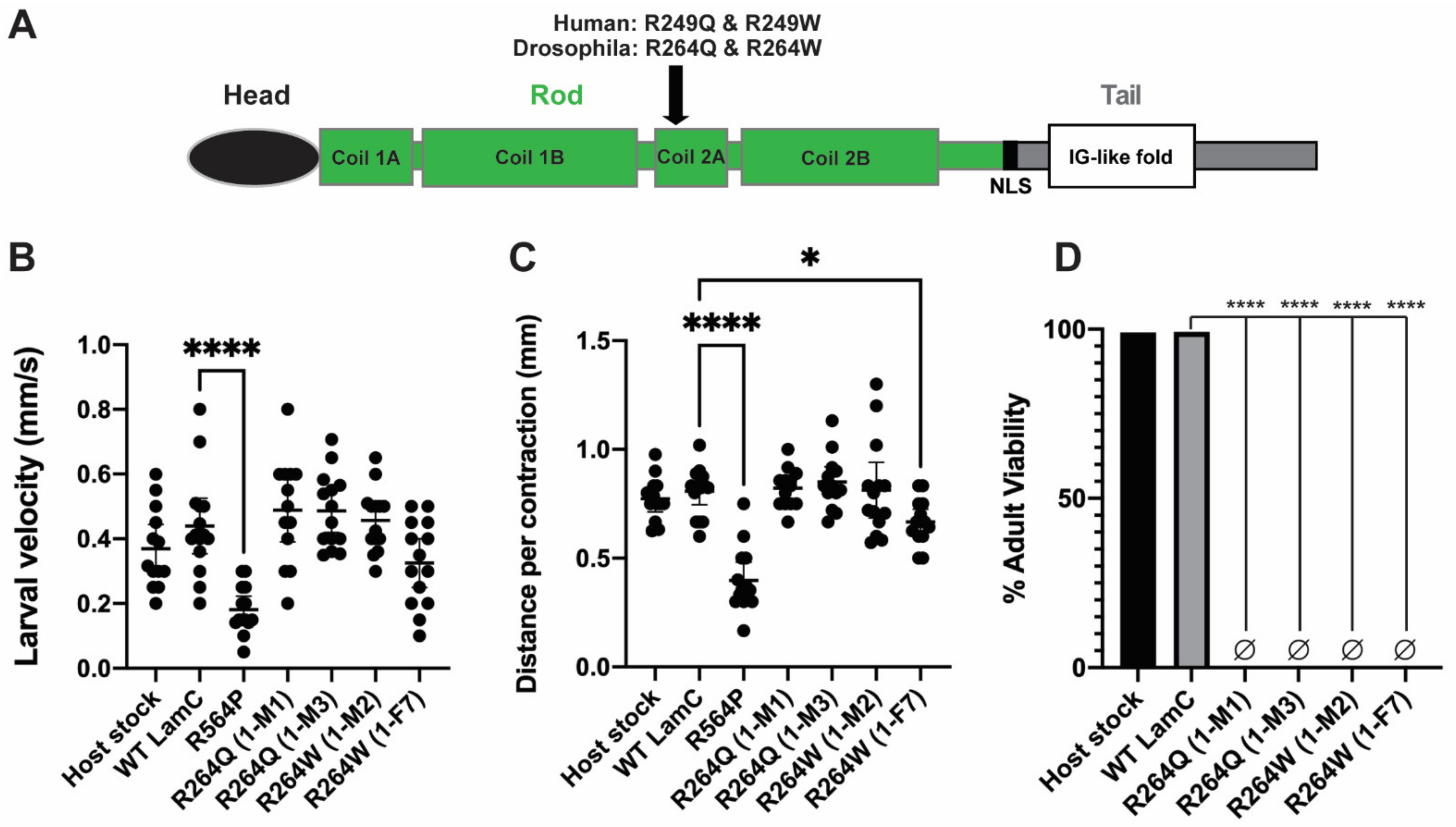
2.4. Generation of In Silico and In Vivo Models of Different Amino Acid Substitutions at the Same Position of LamC
2.5. Larval Body Wall Muscle-Specific Expression of R264Q and R264W Did Not Affect Motility, but Caused Pupal Lethality
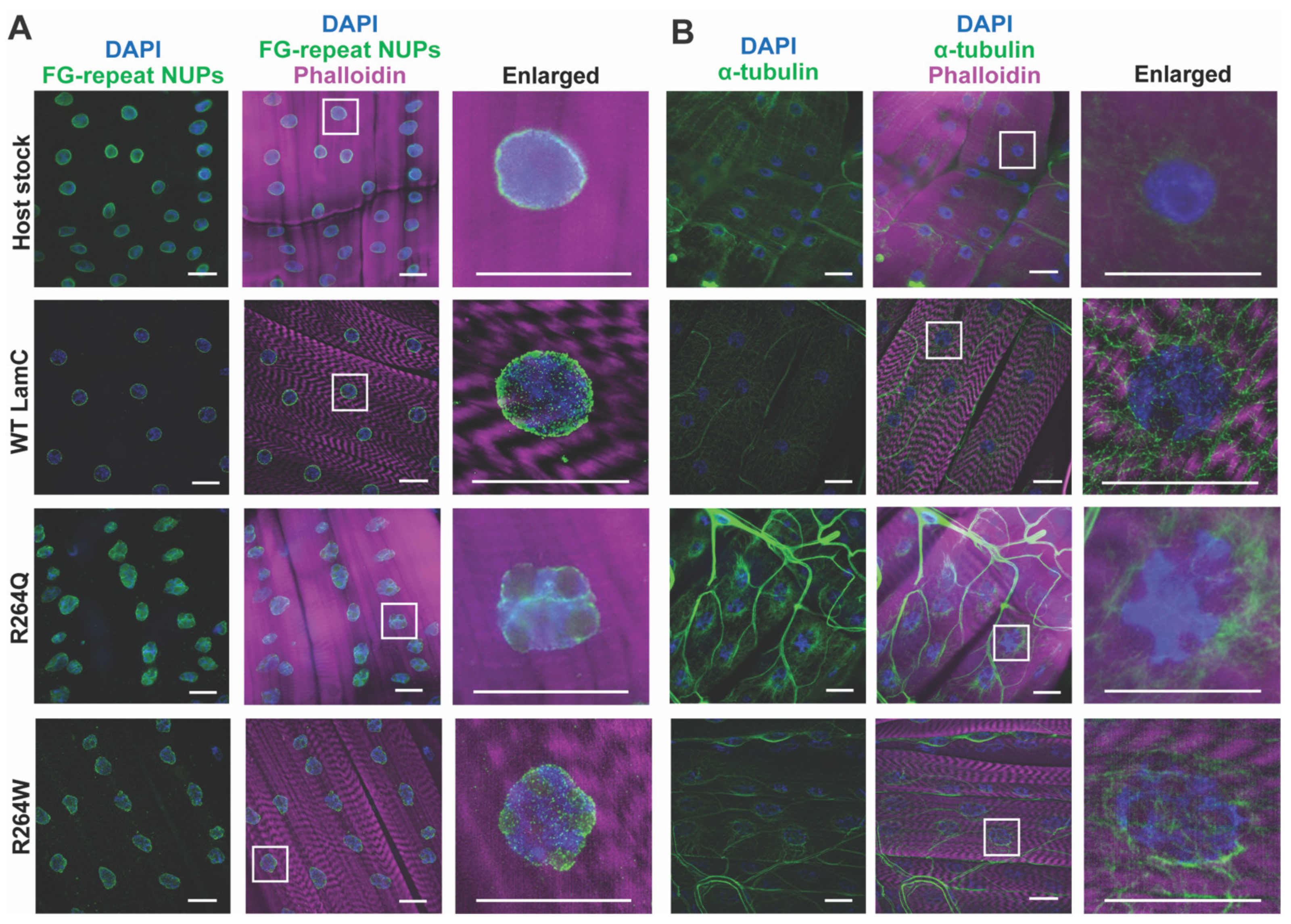
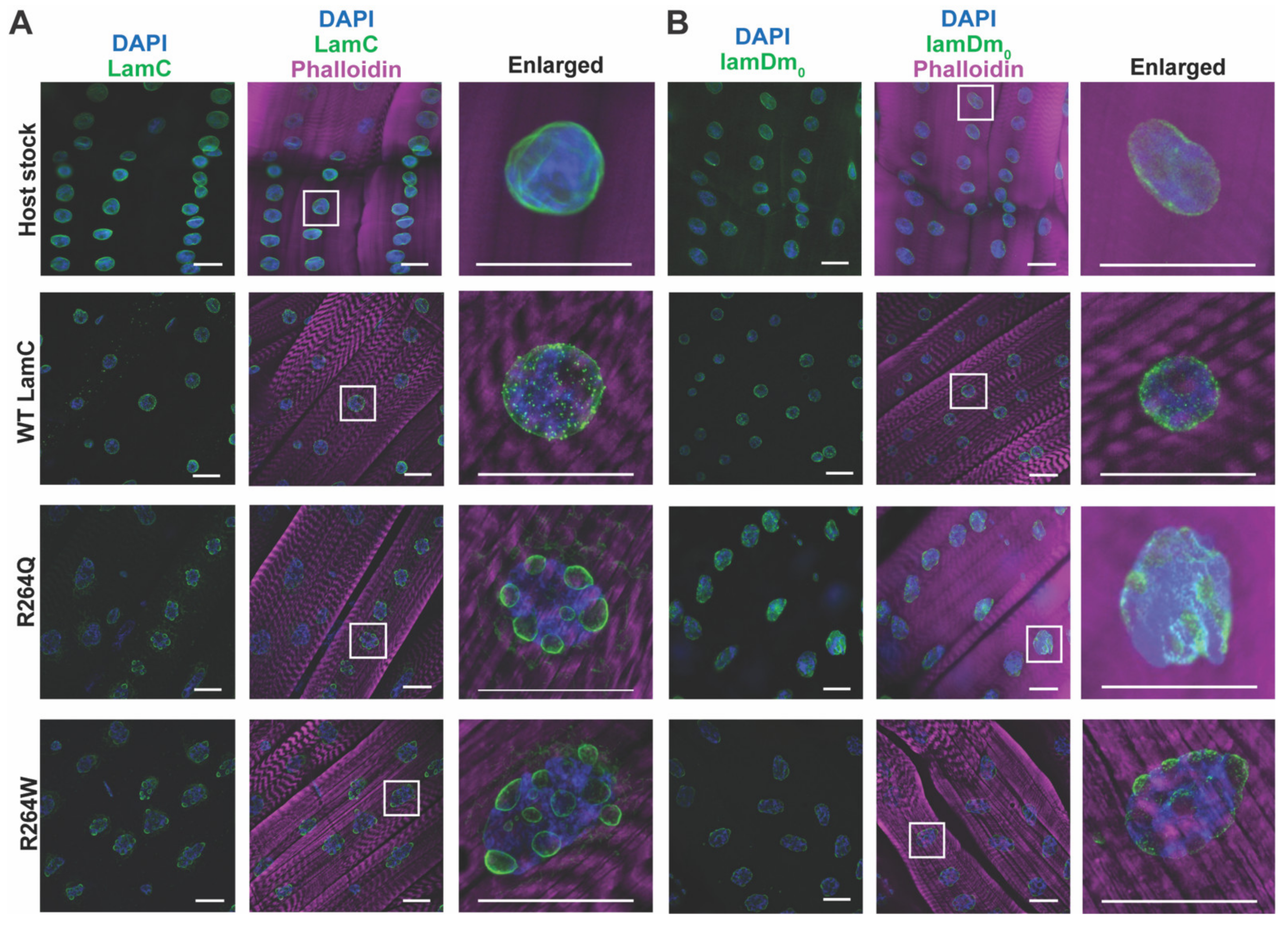
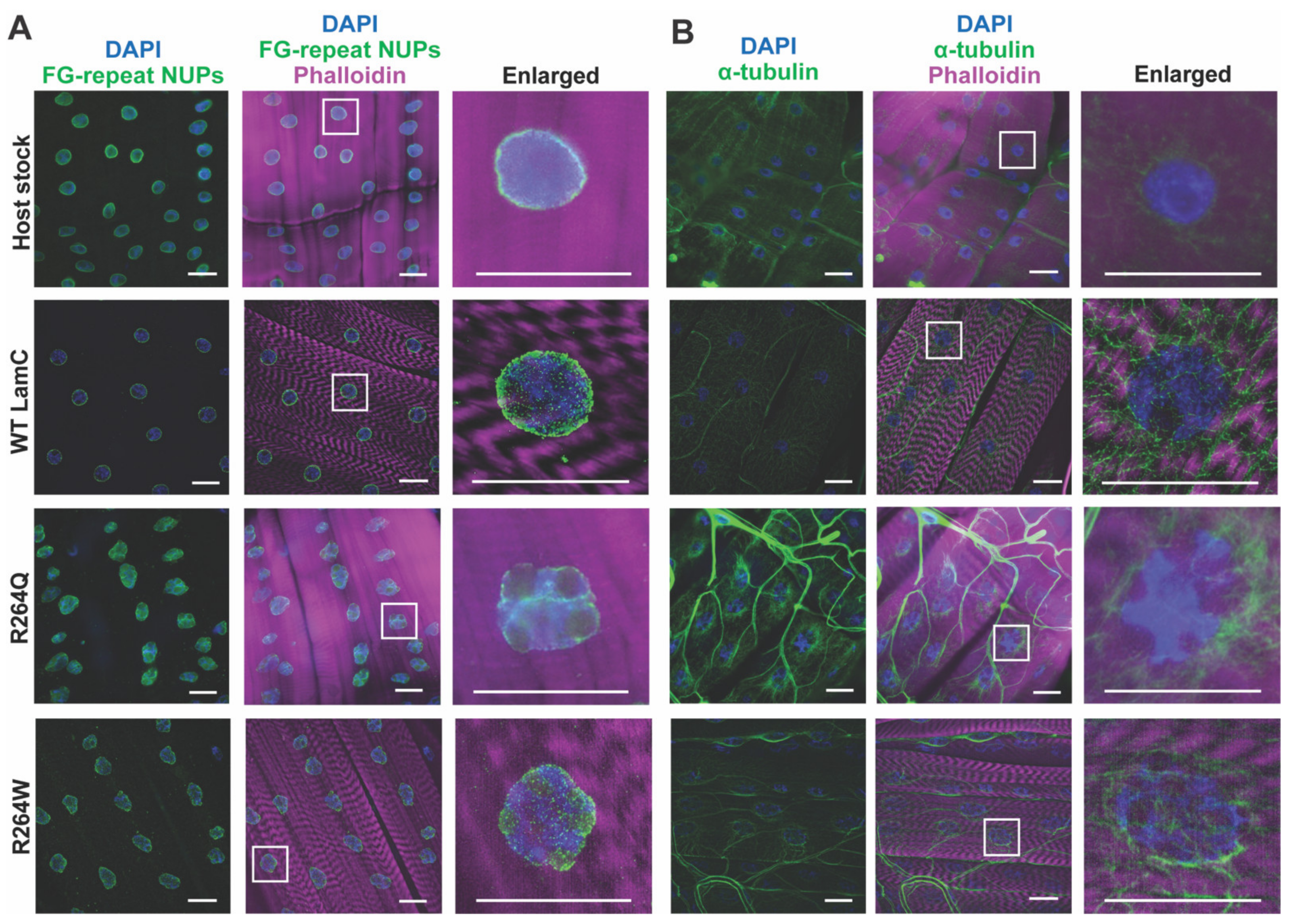
2.6. LamC R264Q and R264W Cause Nuclear Lobulation and Nuclear Envelope Protein Mislocalization
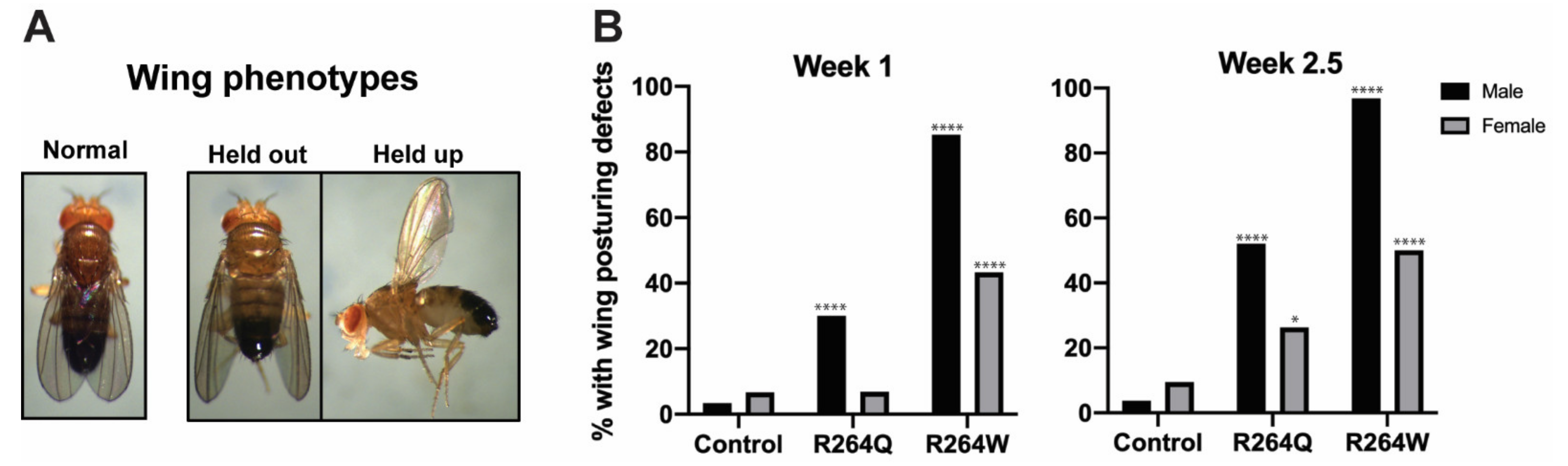
2.7. Expression of R264Q and R264W in Indirect Flight Muscles Caused Wing Posturing Defects
3. Discussion
4. Materials and Methods
4.1. In Silico Analyses
4.1.1. Mapping Disease Related Amino Acid onto Lamin A/C
4.1.2. Deep Learning Prediction of Full-Length Lamin
4.1.3. Determination of Potential Partner Protein Binding Pockets within the Ig-Like Fold Domain
4.1.4. Determination of Surface and Buried Resides within the Ig-Like Fold Domain
4.2. In Vivo Analyses
4.2.1. Drosophila Culturing
4.2.2. Western Analysis
4.2.3. Larval Motility Assays
4.2.4. Larval Body Wall Muscle Cytology
4.2.5. Colocalization of Fluorescent Signals
4.2.6. Adult Viability Assays
4.2.7. Analysis of Indirect Flight Muscle Function
4.3. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD-EDMD | Autosomal dominant Emery–Dreifuss muscular dystrophy |
| AMOEBA | Atomic Multipoles Optimized Energetics for Biomolecular Applications |
| BSA | Bovine serum albumin |
| DDX43 | Dead-box helicase 43 |
| DTT | Dithiothreitol |
| FSG | Fish skin gelatin |
| IFM | Indirect flight muscle |
| Ig | Immunoglobulin |
| L-CMD | LMNA-related congenital muscular dystrophy |
| LAD | Lamin-associated domain |
| LamC | Lamin C |
| lamDm0 | lamin Dm0 |
| LINC | Linker of nucleoskeleton and cytoskeleton |
| NUP | Nuclear pore protein |
| PBS | Phosphate buffer solution |
| POCASA | POcket-CAvity Search Application |
| R249Q | LMNA p.Arg249Gln |
| R249W | LMNA p.Arg249Trp |
| R264Q | Lamin C p.Arg264Gln |
| R264W | Lamin C p.Arg264Trp |
| RMS | Root mean square |
| ROI | Region of interest |
| SASA | Surface-accessible solvent area |
| SDS | Sodium dodecylsulfate |
| TBS | Tris-buffered saline |
| TBS-T | Tris-buffered saline-tween |
| TM | Template modeling |
| UAS | Upstream activation sequence |
| WT | Wild-type |
References
- Wong, X.; Stewart, C.L. The Laminopathies and the Insights They Provide into the Structural and Functional Organization of the Nucleus. Annu. Rev. Genom. Hum. Genet. 2020, 21, 263–288. [Google Scholar] [CrossRef]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef]
- Al-Saaidi, R.; Bross, P. Do lamin A and lamin C have unique roles? Chromosoma 2015, 124, 1–12. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Takamori, Y.; Hirahara, Y.; Wakabayashi, T.; Mori, T.; Koike, T.; Kataoka, Y.; Tamura, Y.; Kurebayashi, S.; Kurokawa, K.; Yamada, H. Differential expression of nuclear lamin subtypes in the neural cells of the adult rat cerebral cortex. IBRO Rep. 2018, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Lee, J.M.; Yang, S.H.; Young, S.G.; Fong, L.G. Nuclear lamins in the brain—New insights into function and regulation. Mol. Neurobiol. 2013, 47, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [Green Version]
- Dhe-Paganon, S.; Werner, E.D.; Chi, Y.I.; Shoelson, S.E. Structure of the globular tail of nuclear lamin. J. Biol. Chem. 2002, 277, 17381–17384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Jo, I.; Kang, S.M.; Hong, S.; Kim, S.; Jeong, S.; Kim, Y.H.; Park, B.J.; Ha, N.C. Structural basis for lamin assembly at the molecular level. Nat. Commun. 2019, 10, 3757. [Google Scholar] [CrossRef]
- Stalmans, G.; Lilina, A.V.; Vermeire, P.J.; Fiala, J.; Novák, P.; Strelkov, S.V. Addressing the Molecular Mechanism of Longitudinal Lamin Assembly Using Chimeric Fusions. Cells 2020, 9, 1633. [Google Scholar] [CrossRef] [PubMed]
- Kapinos, L.E.; Schumacher, J.; Mücke, N.; Machaidze, G.; Burkhard, P.; Aebi, U.; Strelkov, S.V.; Herrmann, H. Characterization of the head-to-tail overlap complexes formed by human lamin A, B1 and B2 “half-minilamin” dimers. J. Mol. Biol. 2010, 396, 719–731. [Google Scholar] [CrossRef]
- Kittisopikul, M.; Virtanen, L.; Taimen, P.; Goldman, R.D. Quantitative Analysis of Nuclear Lamins Imaged by Super-Resolution Light Microscopy. Cells 2019, 8, 361. [Google Scholar] [CrossRef] [Green Version]
- Turgay, Y.; Medalia, O. The structure of lamin filaments in somatic cells as revealed by cryo-electron tomography. Nucleus 2017, 8, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef] [PubMed]
- Moir, R.D.; Yoon, M.; Khuon, S.; Goldman, R.D. Nuclear lamins A and B1: Different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 2000, 151, 1155–1168. [Google Scholar] [CrossRef] [Green Version]
- Worman, H.J.; Ostlund, C.; Wang, Y. Diseases of the nuclear envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000760. [Google Scholar] [CrossRef] [PubMed]
- Schulze, S.R.; Curio-Penny, B.; Li, Y.; Imani, R.A.; Rydberg, L.; Geyer, P.K.; Wallrath, L.L. Molecular genetic analysis of the nested Drosophila melanogaster lamin C gene. Genetics 2005, 171, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osouda, S.; Nakamura, Y.; de Saint Phalle, B.; McConnell, M.; Horigome, T.; Sugiyama, S.; Fisher, P.A.; Furukawa, K. Null mutants of Drosophila B-type lamin Dm(0) show aberrant tissue differentiation rather than obvious nuclear shape distortion or specific defects during cell proliferation. Dev. Biol. 2005, 284, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Wong, X.; Melendez-Perez, A.J.; Reddy, K.L. The Nuclear Lamina. Cold Spring Harb. Perspect. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 2019, 49, 920–935. [Google Scholar] [CrossRef] [PubMed]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2020, 19, 464–473. [Google Scholar] [CrossRef]
- Wong, X.; Cutler, J.A.; Hoskins, V.E.; Gordon, M.; Madugundu, A.K.; Pandey, A.; Reddy, K.L. Mapping the micro-proteome of the nuclear lamina and lamina-associated domains. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Briand, N.; Collas, P. Lamina-associated domains: Peripheral matters and internal affairs. Genome Biol. 2020, 21, 85. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.L.; Foisner, R. Lamin-binding Proteins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000554. [Google Scholar] [CrossRef] [Green Version]
- Kittisopikul, M.; Shimi, T.; Tatli, M.; Tran, J.R.; Zheng, Y.; Medalia, O.; Jaqaman, K.; Adam, S.A.; Goldman, R.D. Computational analyses reveal spatial relationships between nuclear pore complexes and specific lamins. J. Cell Biol. 2021, 220, e202007082. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Pennypacker, J.K. RB and lamins in cell cycle regulation and aging. Adv. Exp. Med. Biol. 2014, 773, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.M.; Lammerding, J. Broken nuclei—Lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puckelwartz, M.J.; Depreux, F.F.; McNally, E.M. Gene expression, chromosome position and lamin A/C mutations. Nucleus 2011, 2, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worman, H.J.; Schirmer, E.C. Nuclear membrane diversity: Underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Ostlund, C.; Worman, H.J. Nuclear envelope proteins and neuromuscular diseases. Muscle Nerve 2003, 27, 393–406. [Google Scholar] [CrossRef]
- Vidak, S.; Foisner, R. Molecular insights into the premature aging disease progeria. Histochem. Cell Biol. 2016, 145, 401–417. [Google Scholar] [CrossRef] [Green Version]
- Genschel, J.; Schmidt, H.H. Mutations in the LMNA gene encoding lamin A/C. Hum. Mutat. 2000, 16, 451–459. [Google Scholar] [CrossRef]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Bécane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Sewry, C.A.; Brown, S.C.; Mercuri, E.; Bonne, G.; Feng, L.; Camici, G.; Morris, G.E.; Muntoni, F. Skeletal muscle pathology in autosomal dominant Emery-Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol. Appl. Neurobiol. 2001, 27, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Felice, K.J.; Schwartz, R.C.; Brown, C.A.; Leicher, C.R.; Grunnet, M.L. Autosomal dominant Emery-Dreifuss dystrophy due to mutations in rod domain of the lamin A/C gene. Neurology 2000, 55, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, R.M., Jr.; Costa-Riquetto, A.D.; Fernandes, V.O.; Montenegro, A.; de Santana, L.S.; Jorge, A.A.L.; Karbage, L.; Aguiar, L.B.; Carvalho, F.H.C.; Teles, M.G.; et al. Homozygous and Heterozygous Nuclear Lamin A p.R582C Mutation: Different Lipodystrophic Phenotypes in the Same Kindred. Front. Endocrinol. 2018, 9, 458. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Poppe, M.; Quinlivan, R.; Messina, S.; Kinali, M.; Demay, L.; Bourke, J.; Richard, P.; Sewry, C.; Pike, M.; et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: From congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch. Neurol. 2004, 61, 690–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharner, J.; Lu, H.C.; Fraternali, F.; Ellis, J.A.; Zammit, P.S. Mapping disease-related missense mutations in the immunoglobulin-like fold domain of lamin A/C reveals novel genotype-phenotype associations for laminopathies. Proteins 2014, 82, 904–915. [Google Scholar] [CrossRef]
- Fraczkiewicz, R.; Braun, W. Exact and Efficient Analytical Calculation of the Accessible Surface Areas and Their Gradients for Macromolecules. J. Comp. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, T.A.; Sahni, N.; Kubben, N.; Hill, D.E.; Vidal, M.; Burgess, R.C.; Roukos, V.; Misteli, T. Systematic identification of pathological lamin A interactors. Mol. Biol. Cell 2014, 25, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [CrossRef]
- Ponder, J.W.; Wu, C.; Ren, P.; Pande, V.S.; Chodera, J.D.; Schnieders, M.J.; Haque, I.; Mobley, D.L.; Lambrecht, D.S.; DiStasio, R.A.; et al. Current status of the AMOEBA polarizable force field. J. Phys. Chem. B 2010, 114, 2549–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Xia, Z.; Zhang, J.; Best, R.; Wu, C.; Ponder, J.W.; Ren, P. The Polarizable Atomic Multipole-based AMOEBA Force Field for Proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Becane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Di Barletta, M.R.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Scharner, J.; Brown, C.A.; Bower, M.; Iannaccone, S.T.; Khatri, I.A.; Escolar, D.; Gordon, E.; Felice, K.; Crowe, C.A.; Grosmann, C.; et al. Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum. Mutat. 2011, 32, 152–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vytopil, M.; Benedetti, S.; Ricci, E.; Galluzzi, G.; Dello Russo, A.; Merlini, L.; Boriani, G.; Gallina, M.; Morandi, L.; Politano, L.; et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J. Med. Genet. 2003, 40, e132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quijano-Roy, S.; Mbieleu, B.; Bönnemann, C.G.; Jeannet, P.Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.T.; Liu, X.; Zhang, W.; Liu, J.; Zuo, Y.H.; Xiao, J.X.; Zhu, Y.; Yuan, Y.; Wang, Z.X. Muscle Magnetic Resonance Imaging in Patients with Various Clinical Subtypes of. Chin. Med. J. 2018, 131, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Flannery, K.M.; Zirbel, L.N.; Nagy, P.L.; Mathews, K.D.; Moore, S.A.; Wallrath, L.L. LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Hum. Mol. Genet. 2012, 21, 1544–1556. [Google Scholar] [CrossRef] [Green Version]
- Dialynas, G.; Shrestha, O.K.; Ponce, J.M.; Zwerger, M.; Thiemann, D.A.; Young, G.H.; Moore, S.A.; Yu, L.; Lammerding, J.; Wallrath, L.L. Myopathic lamin mutations cause reductive stress and activate the nrf2/keap-1 pathway. PLoS Genet. 2015, 11, e1005231. [Google Scholar] [CrossRef] [Green Version]
- Duffy, J.B. GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genesis 2002, 34, 1–15. [Google Scholar] [CrossRef]
- Koh, Y.H.; Popova, E.; Thomas, U.; Griffith, L.C.; Budnik, V. Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell 1999, 98, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Bryantsev, A.L.; Baker, P.W.; Lovato, T.L.; Jaramillo, M.S.; Cripps, R.M. Differential requirements for Myocyte Enhancer Factor-2 during adult myogenesis in Drosophila. Dev. Biol. 2012, 361, 191–207. [Google Scholar] [CrossRef] [Green Version]
- Schejter, E.D.; Baylies, M.K. Born to run: Creating the muscle fiber. Curr. Opin. Cell Biol. 2010, 22, 566–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tixier, V.; Bataille, L.; Jagla, K. Diversification of muscle types: Recent insights from Drosophila. Exp. Cell Res. 2010, 316, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Schulman, V.K.; Dobi, K.C.; Baylies, M.K. Morphogenesis of the somatic musculature in Drosophila melanogaster. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 313–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortier, T.M.; Vasa, P.P.; Woodard, C.T. Orphan nuclear receptor betaFTZ-F1 is required for muscle-driven morphogenetic events at the prepupal-pupal transition in Drosophila melanogaster. Dev. Biol. 2003, 257, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Dialynas, G.; Speese, S.; Budnik, V.; Geyer, P.K.; Wallrath, L.L. The role of Drosophila Lamin C in muscle function and gene expression. Development 2010, 137, 3067–3077. [Google Scholar] [CrossRef] [Green Version]
- Becker, R.; Leone, M.; Engel, F.B. Microtubule Organization in Striated Muscle Cells. Cells 2020, 9, 1395. [Google Scholar] [CrossRef]
- Bugnard, E.; Zaal, K.J.; Ralston, E. Reorganization of microtubule nucleation during muscle differentiation. Cell Motil. Cytoskelet. 2005, 60, 1–13. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Vigoreaux, J.O. Genetics of the Drosophila flight muscle myofibril: A window into the biology of complex systems. Bioessays 2001, 23, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Suggs, J.A.; Wang, B.J.; Han, A.; Bhide, S.; Cryderman, D.E.; Moore, S.A.; Bernstein, S.I.; Wallrath, L.L.; Melkani, G.C. Suppression of myopathic lamin mutations by muscle-specific activation of AMPK and modulation of downstream signaling. Hum. Mol. Genet. 2019, 28, 351–371. [Google Scholar] [CrossRef]
- Ben Yaou, R.; Yun, P.; Dabaj, I.; Norato, G.; Donkervoort, S.; Xiong, H.; Nascimento, A.; Maggi, L.; Sarkozy, A.; Monges, S.; et al. International retrospective natural history study of LMNA-related congenital muscular dystrophy. Brain Commun. 2021, 3, fcab075. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.; Kozlov, S.; Burke, B.; Stewart, C.L. The laminopathies: Nuclear structure meets disease. Curr. Opin. Genet. Dev. 2003, 13, 223–230. [Google Scholar] [CrossRef]
- Vermeire, P.J.; Stalmans, G.; Lilina, A.V.; Fiala, J.; Novak, P.; Herrmann, H.; Strelkov, S.V. Molecular Interactions Driving Intermediate Filament Assembly. Cells 2021, 10, 2457. [Google Scholar] [CrossRef]
- Tan, D.; Yang, H.; Yuan, Y.; Bonnemann, C.; Chang, X.; Wang, S.; Wu, Y.; Wu, X.; Xiong, H. Phenotype-Genotype Analysis of Chinese Patients with Early-Onset LMNA-Related Muscular Dystrophy. PLoS ONE 2015, 10, e0129699. [Google Scholar] [CrossRef] [PubMed]
- Steele-Stallard, H.B.; Pinton, L.; Sarcar, S.; Ozdemir, T.; Maffioletti, S.M.; Zammit, P.S.; Tedesco, F.S. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic. Front. Physiol. 2018, 9, 1332. [Google Scholar] [CrossRef] [Green Version]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior—Life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Krimm, I.; Ostlund, C.; Gilquin, B.; Couprie, J.; Hossenlopp, P.; Mornon, J.P.; Bonne, G.; Courvalin, J.C.; Worman, H.J.; Zinn-Justin, S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002, 10, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Strelkov, S.V.; Schumacher, J.; Burkhard, P.; Aebi, U.; Herrmann, H. Crystal structure of the human lamin A coil 2B dimer: Implications for the head-to-tail association of nuclear lamins. J. Mol. Biol. 2004, 343, 1067–1080. [Google Scholar] [CrossRef]
- Lilina, A.V.; Chernyatina, A.A.; Guzenko, D.; Strelkov, S.V. Lateral A. J. Struct. Biol. 2020, 209, 107404. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, M.R.; Litman, J.M.; Qi, G.; O’Connell, C.E.; Wipfler, M.J.; Marini, R.J.; Bernabe, H.V.; Tollefson, W.T.A.; Braun, T.A.; Casavant, T.L.; et al. Structural Insights into Hearing Loss Genetics from Polarizable Protein Repacking. Biophys. J. 2019, 117, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, C.D.; Wuller, J.M.; Elgin, S.C. Raising large quantities of Drosophila for biochemical experiments. Methods Cell Biol. 1994, 44, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Zhong, Y.; Wu, C.F. Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J. Neurosci. 1990, 10, 3754–3768. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient # | DNA Mutation | Gene Affected | Amino Acid Substitution | Age of Onset | Symptoms (Age of Symptoms) | CK Levels (Age Taken) | Clinical Diagnosis (Age of Diagnosis) |
|---|---|---|---|---|---|---|---|
| 1 | c.746G>A | LMNA | R249Q | 2 years | Lordosis, toe walking, abnormal gait, lack of reflex, lower extremity weakness (2 yo) Low resting heart rate (24 yo) Atrial fibrillation (27 yo) | 582 U/L (27 yo) | Polymyositis (2 yo) AD-EDMD (27 yo) |
| 2 | c.745C>T | LMNA | R249W | 5 months | Hyperlordosis, dropped head, poor feeding (5 mo) | 1697 U/L (5 mo) | L-CMD (5 mo) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinz, B.E.; Walker, S.G.; Xiong, A.; Gogal, R.A.; Schnieders, M.J.; Wallrath, L.L. In Silico and In Vivo Analysis of Amino Acid Substitutions That Cause Laminopathies. Int. J. Mol. Sci. 2021, 22, 11226. https://doi.org/10.3390/ijms222011226
Hinz BE, Walker SG, Xiong A, Gogal RA, Schnieders MJ, Wallrath LL. In Silico and In Vivo Analysis of Amino Acid Substitutions That Cause Laminopathies. International Journal of Molecular Sciences. 2021; 22(20):11226. https://doi.org/10.3390/ijms222011226
Chicago/Turabian StyleHinz, Benjamin E., Sydney G. Walker, Austin Xiong, Rose A. Gogal, Michael J. Schnieders, and Lori L. Wallrath. 2021. "In Silico and In Vivo Analysis of Amino Acid Substitutions That Cause Laminopathies" International Journal of Molecular Sciences 22, no. 20: 11226. https://doi.org/10.3390/ijms222011226
APA StyleHinz, B. E., Walker, S. G., Xiong, A., Gogal, R. A., Schnieders, M. J., & Wallrath, L. L. (2021). In Silico and In Vivo Analysis of Amino Acid Substitutions That Cause Laminopathies. International Journal of Molecular Sciences, 22(20), 11226. https://doi.org/10.3390/ijms222011226