
Engineering Pyrrolysyl-tRNA Synthetase for the Incorporation of Non-Canonical Amino Acids with Smaller Side Chains




Abstract
:1. Introduction
2. Results and Discussion
2.1. General MbPylRS and ncAA Incorporation Readout Setup
2.2. Testing MbSacRS for Aliphatic Substrates
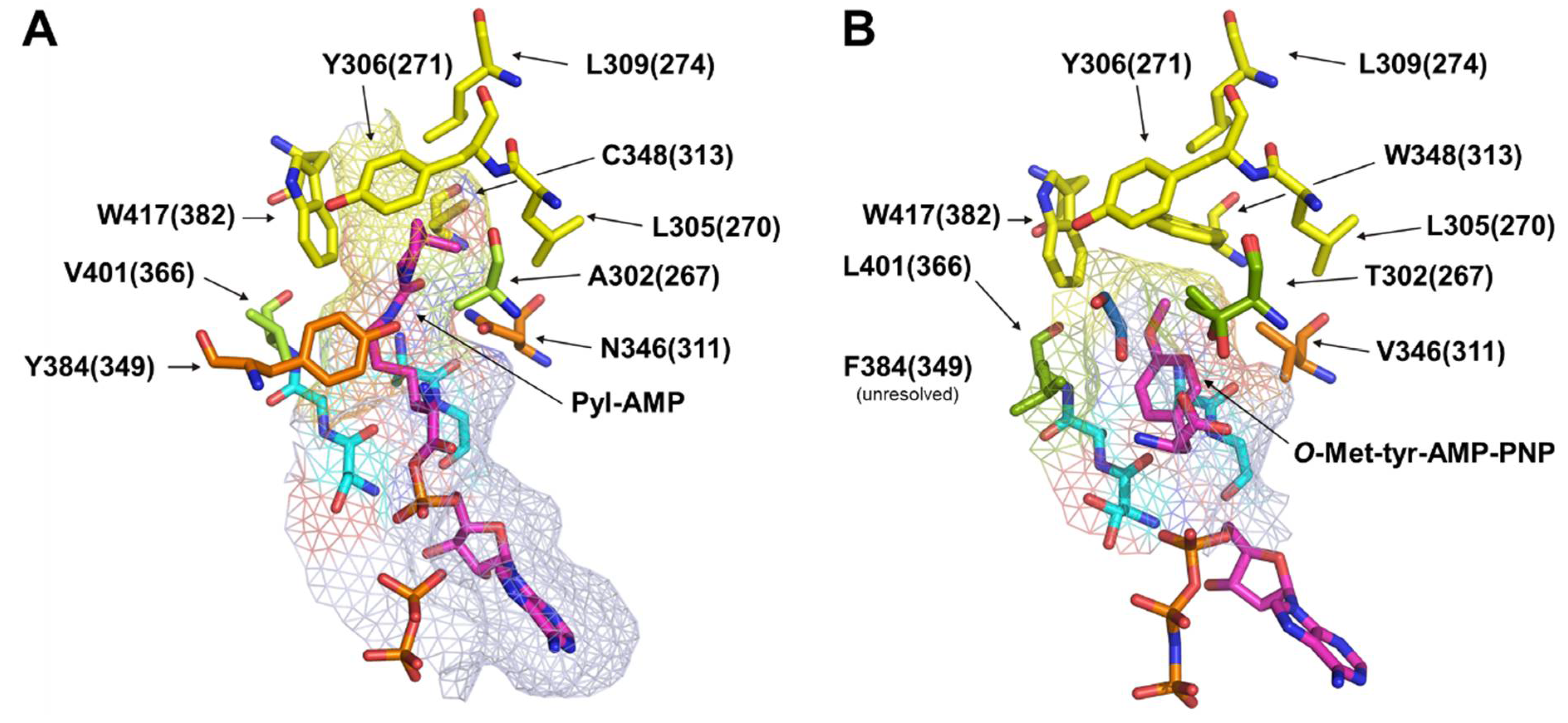
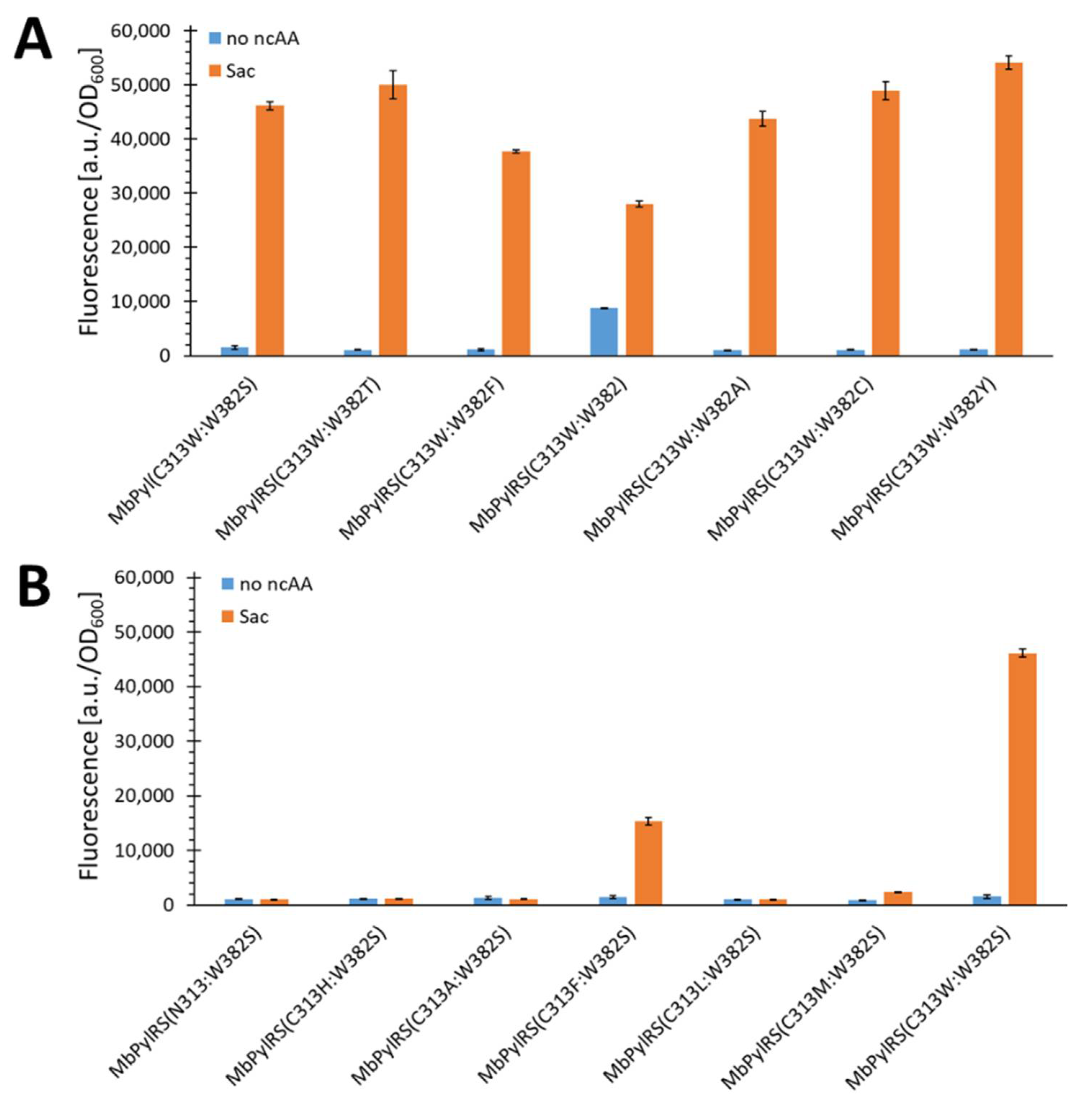
2.3. Elucidating the Structure–Activity Relationships of MbSacRS via Rational Mutation Studies
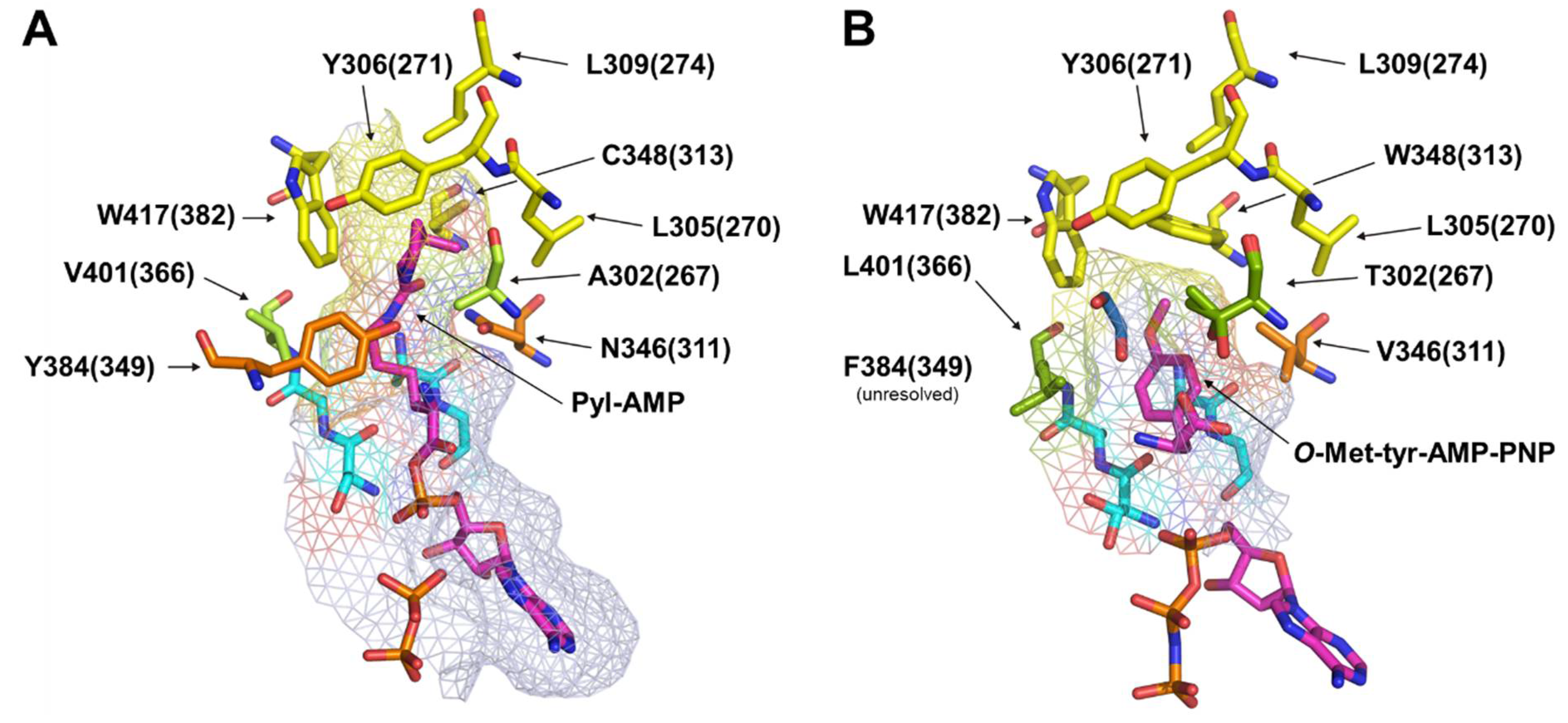
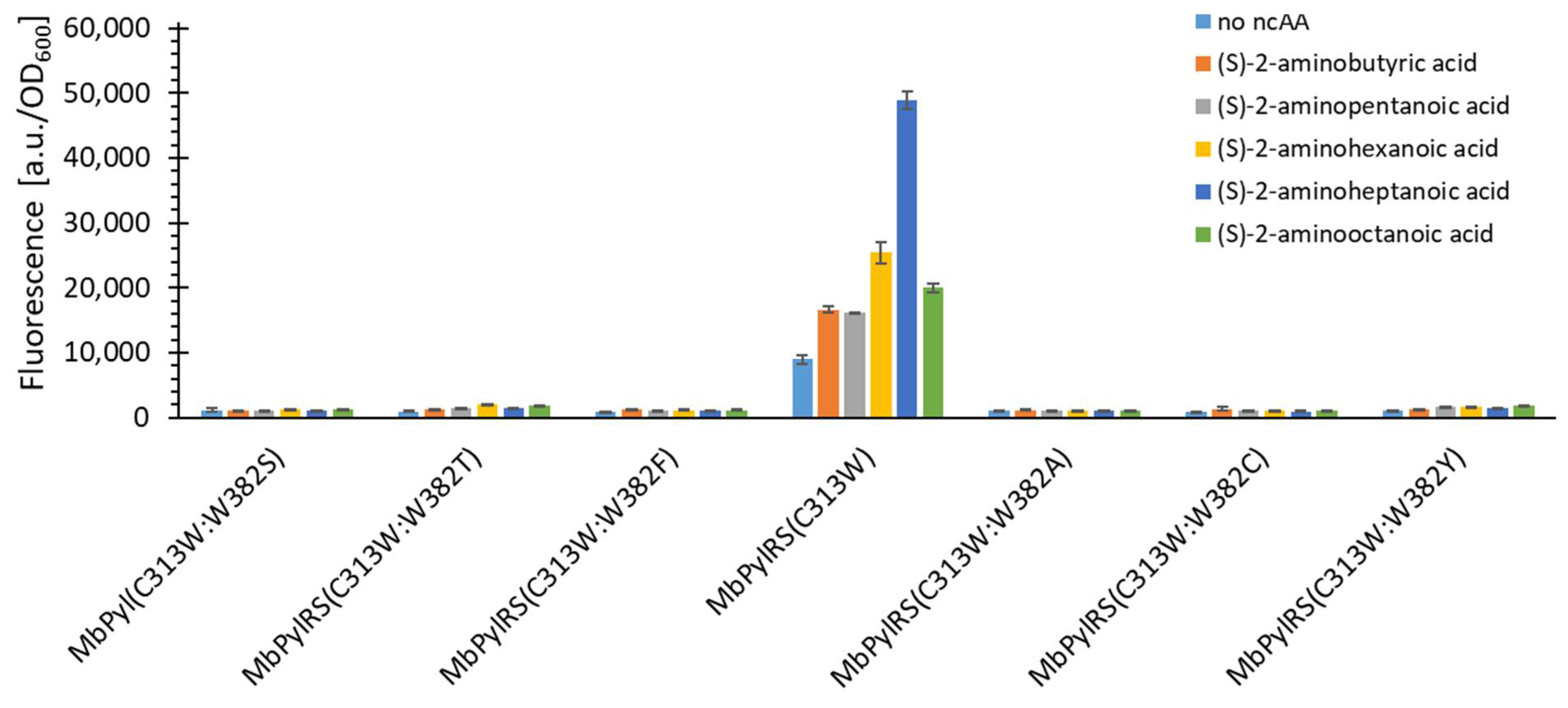
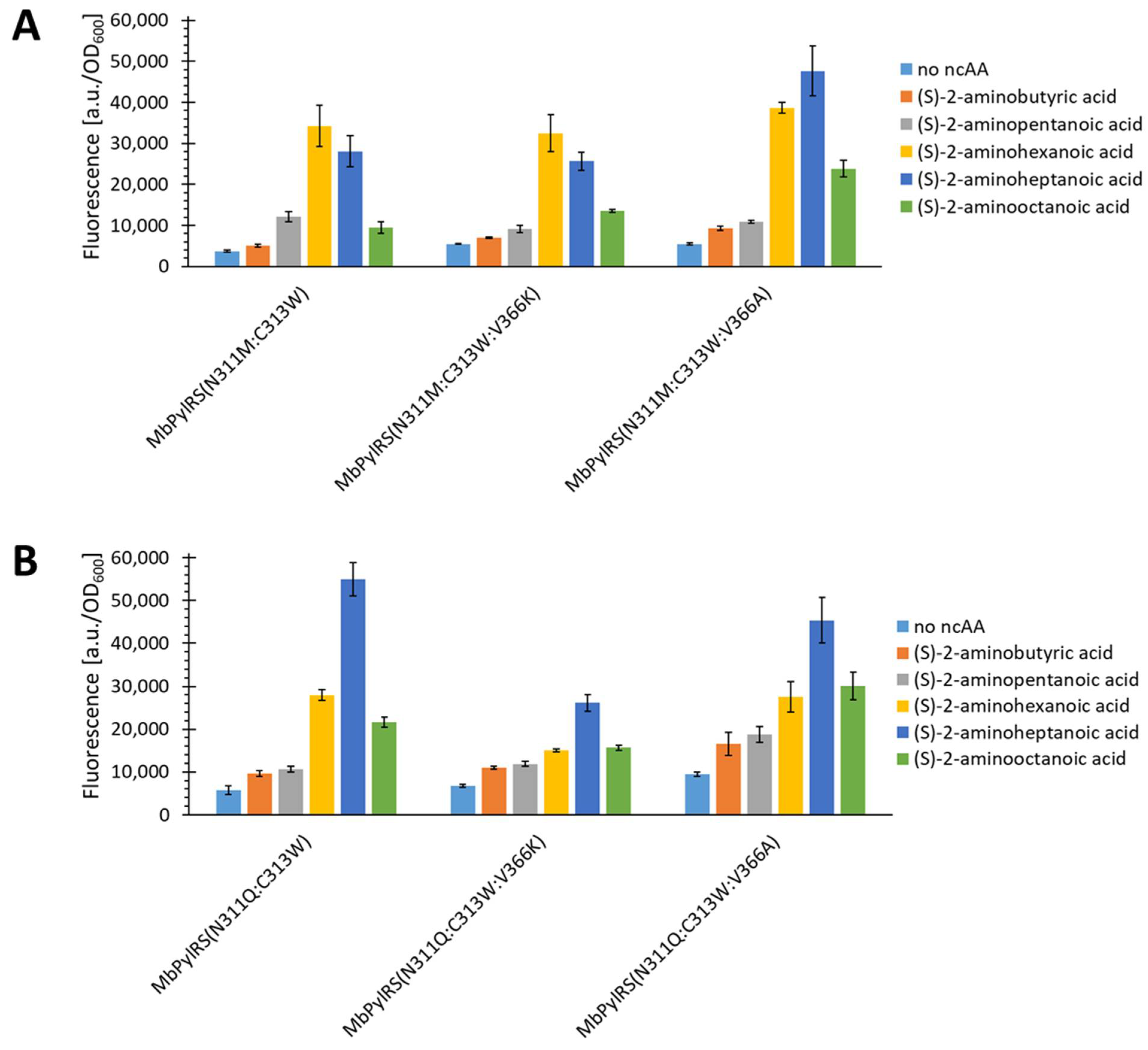
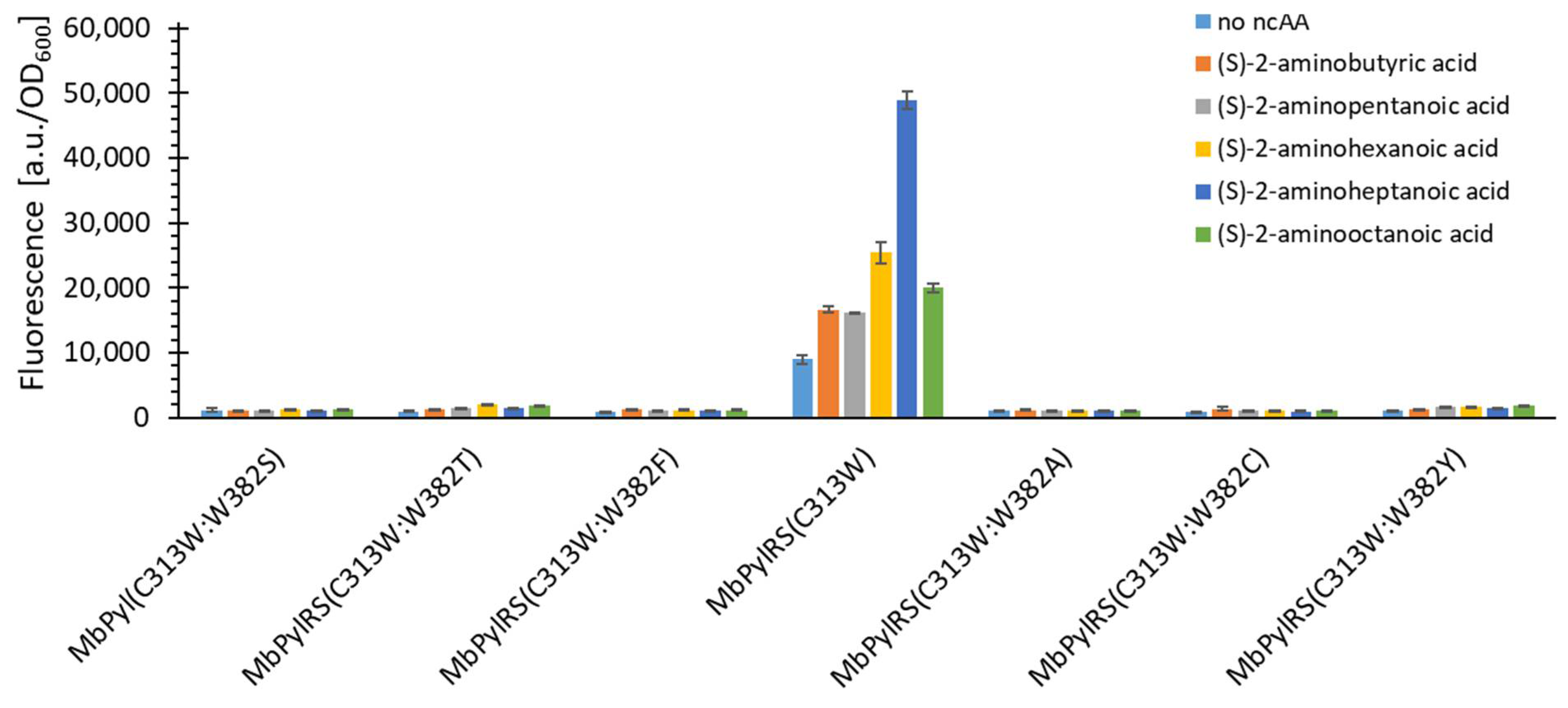
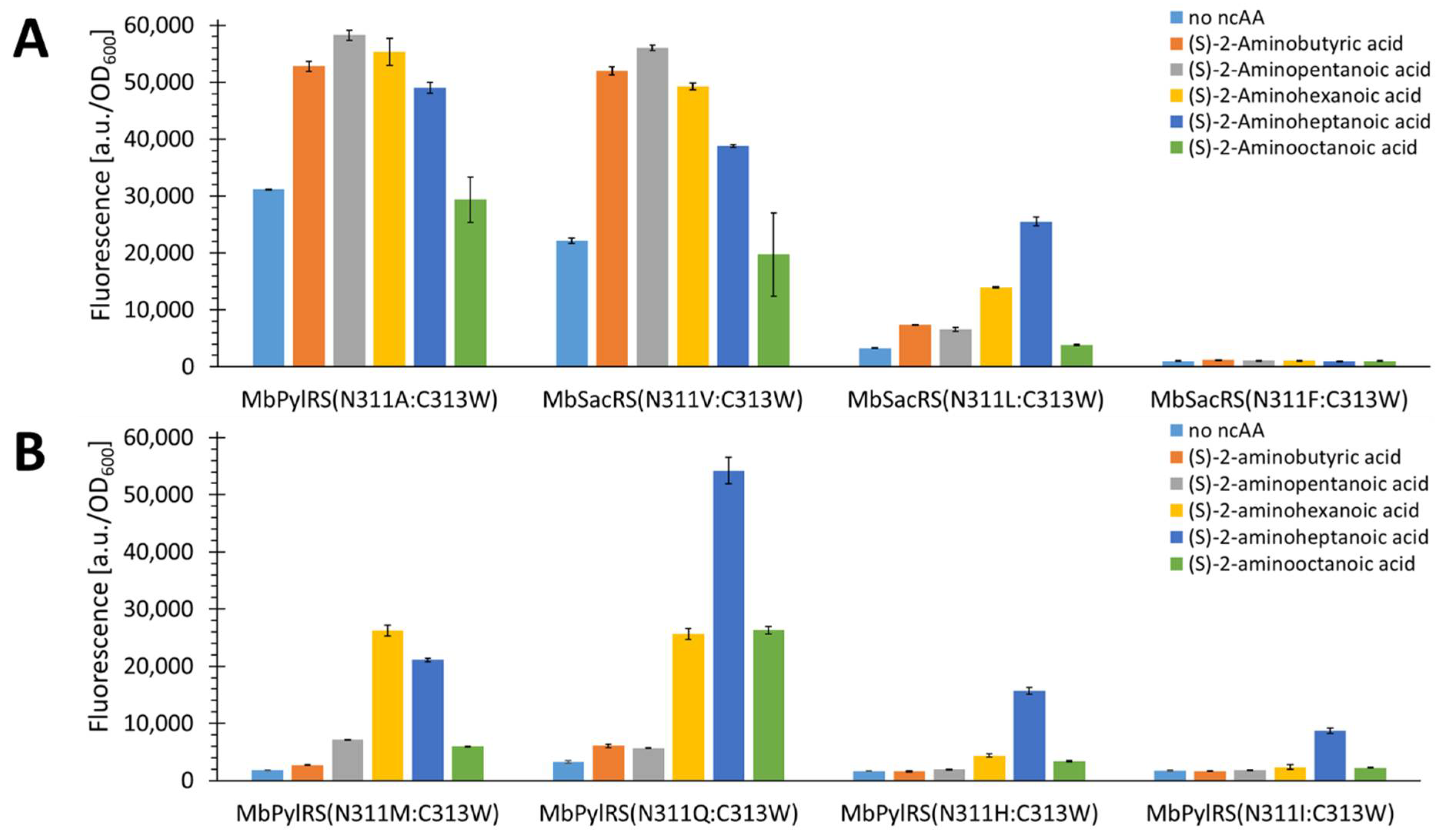
2.4. Rationalizing Sac Incorporation Data and Creating Aliphatic Substrate Activating MbPylRS Variants
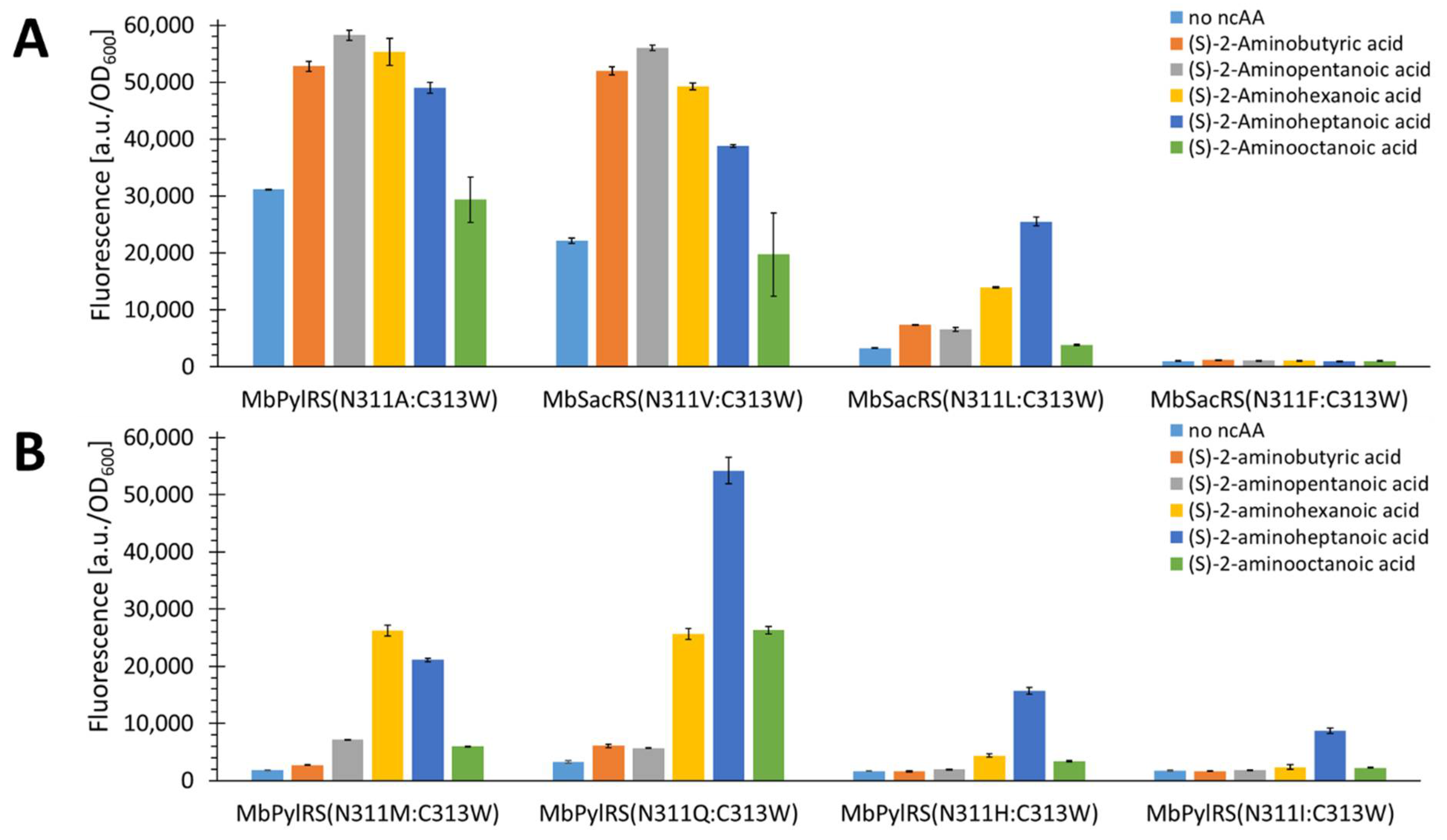
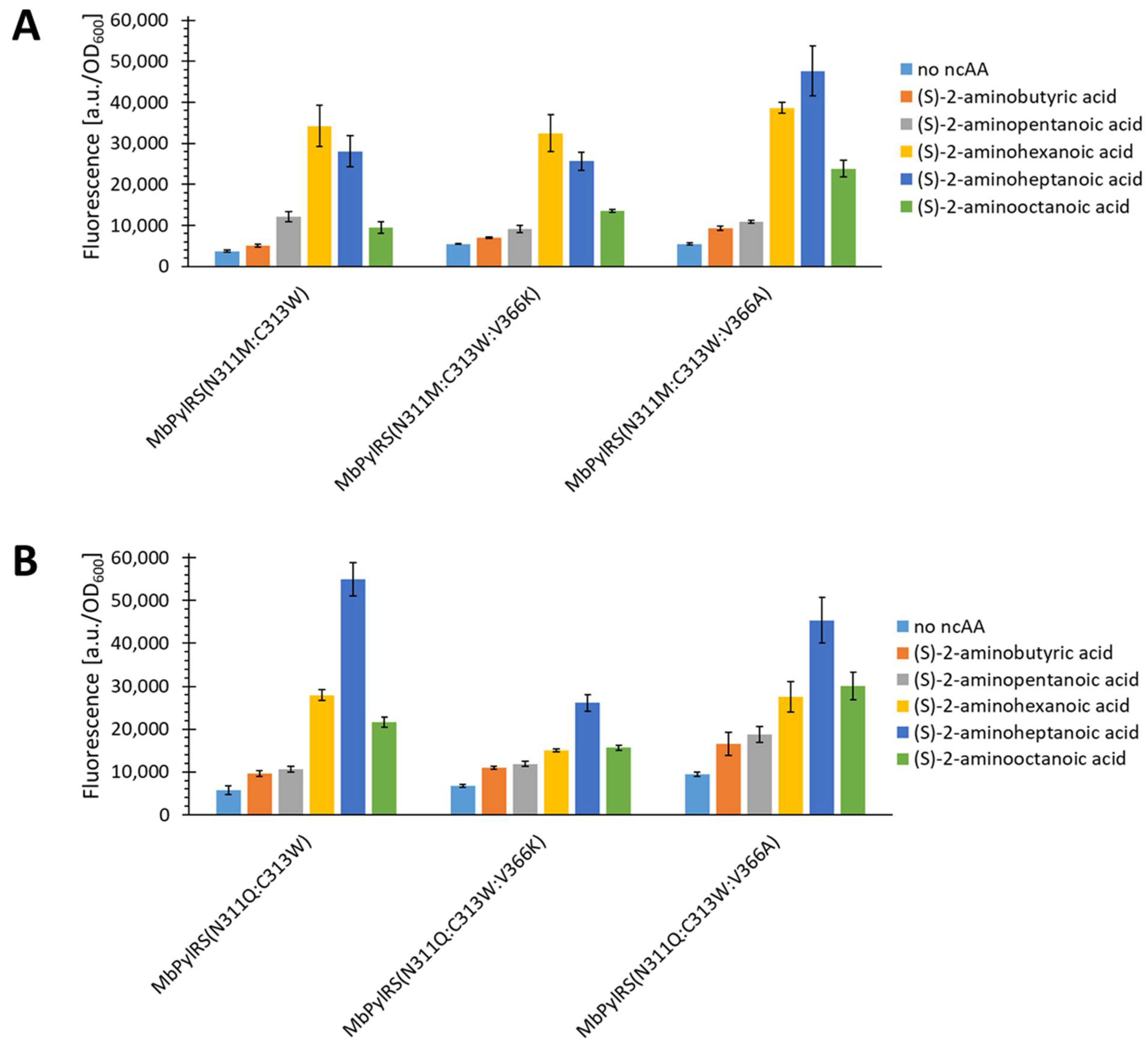
2.5. Semi-Rational Engineering of PylRS Constructs for Small Aliphatic Substrate Incorporation
2.6. Evaluating the Incorporation of Biochemically Useful Aliphatic ncAA Analogs
2.7. Analytics of Canonical/Non-Canonical Amino Acids Incorporation
3. Materials and Methods
3.1. Canonical and Non-Canonical Amino Acids
3.2. Plasmid Vector Construction
3.3. Site-Directed and Site-Saturation Mutagenesis
3.4. Analysis of SUMO-sfGFP Expression by Intact Cell Fluorescence.
3.5. Library Screening
3.6. Protein Expression
3.7. Protein Purification
3.8. ESI-MS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | amino acid |
| AMP | adenosine monophosphate |
| AMP-PNP | adenosine-5′-[(β,γ)-imido]triphosphate |
| aaRS | aminoacyl-tRNA synthetase |
| cAA | canonical amino acid |
| ESI-MS | electrospray ionization mass spectrometry |
| FTIR | Fourier-transform infrared spectroscopy |
| MjTyrRS | Methanocaldococcus jannaschii tyrosyl-tRNA synthetase |
| MmPylRS/MbPylRS | Methanosarcina mazei/barkeri pyrrolysyl-tRNA synthetase |
| ncAA | non-canonical amino a |
| NMR | nuclear magnetic resonance spectroscopy |
| OTS | orthogonal translation system |
| Pyl | pyrrolysine |
| PylRS | pyrrolysyl-tRNA synthetase |
| Sac | S-allyl-l-cysteine |
| sfGFP | superfolder GFP |
| SmbP | small metal-binding protein |
| RF1 | release factor 1 |
Appendix A
Appendix A.1. General Features and Perspectives of ncAAs Used in This Study
References
- Pagar, A.D.; Patil, M.D.; Flood, D.T.; Yoo, T.H.; Dawson, P.E.; Yun, H. Recent Advances in Biocatalysis with Chemical Modification and Expanded Amino Acid Alphabet. Chem. Rev. 2021, 121, 6173–6245. [Google Scholar] [CrossRef] [PubMed]
- Groff, D.; Thielges, M.C.; Cellitti, S.; Schultz, P.G.; Romesberg, F.E. Efforts toward the direct experimental characterization of enzyme microenvironments: Tyrosine 100 in dihydrofolate reductase. Angew. Chem. Int. Ed. 2009, 48, 3478–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, T.; Hauf, M.; Schildhauer, F.; Eberl, K.B.; Durkin, P.M.; Deniz, E.; Löffler, J.G.; Acevedo-Rocha, C.G.; Jaric, J.; Martins, B.M.; et al. Site-Resolved Observation of Vibrational Energy Transfer Using a Genetically Encoded Ultrafast Heater. Angew. Chem. Int. Ed. 2019. [Google Scholar] [CrossRef] [PubMed]
- Minnihan, E.C.; Young, D.D.; Schultz, P.G.; Stubbe, J. Incorporation of fluorotyrosines into ribonucleotide reductase using an evolved, polyspecific aminoacyl-tRNA synthetase. J. Am. Chem. Soc. 2011, 133, 15942–15945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.C.; Nastertorabi, F.; Xuan, W.; Han, G.W.; Stevens, R.C.; Schultz, P.G. A Single Reactive Noncanonical Amino Acid Is Able to Dramatically Stabilize Protein Structure. ACS Chem. Biol. 2019, 14, 1150–1153. [Google Scholar] [CrossRef]
- Agostini, F.; Völler, J.S.; Koksch, B.; Acevedo-Rocha, C.G.; Kubyshkin, V.; Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew. Chem. Int. Ed. 2017, 56, 9680–9703. [Google Scholar] [CrossRef]
- Drienovská, I.; Mayer, C.; Dulson, C.; Roelfes, G. A designer enzyme for hydrazone and oxime formation featuring an unnatural catalytic aniline residue. Nat. Chem. 2018, 10, 946–952. [Google Scholar] [CrossRef]
- Burke, A.J.; Lovelock, S.L.; Frese, A.; Crawshaw, R.; Ortmayer, M.; Dunstan, M.; Levy, C.; Green, A.P. Design and evolution of an enzyme with a non-canonical organocatalytic mechanism. Nature 2019, 570, 219–223. [Google Scholar] [CrossRef]
- Liu, C.C.; Schultz, P.G. Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [Green Version]
- Mukai, T.; Lajoie, M.J.; Englert, M.; Söll, D. Rewriting the Genetic Code. Annu. Rev. Microbiol. 2017, 71, 557–577. [Google Scholar] [CrossRef] [Green Version]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Tharp, J.M.; Liu, W.R. Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep Engineering of Pyrrolysyl-tRNA Synthetase to Genetically Encode Nε-(o-Azidobenzyloxycarbonyl) lysine for Site-Specific Protein Modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, T.; Kuratani, M.; Seki, E.; Hino, N.; Sakamoto, K.; Yokoyama, S. Structural Basis for Genetic-Code Expansion with Bulky Lysine Derivatives by an Engineered Pyrrolysyl-tRNA Synthetase. Cell Chem. Biol. 2019, 26, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.T.; Wang, Y.S.; Nakamura, A.; Eiler, D.; Kavran, J.M.; Wong, M.; Kiessling, L.L.; Steitz, T.A.; O’Donoghue, P.; Söll, D. Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 16724–16729. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.S.; Fang, X.; Wallace, A.L.; Wu, B.; Liu, W.R. A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J. Am. Chem. Soc. 2012, 134, 2950–2953. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.; Baumann, T.; Sun, H.; Wang, Y.; Ignatova, Z.; Budisa, N. Expanding the Scope of Orthogonal Translation with Pyrrolysyl-tRNA Synthetases Dedicated to Aromatic Amino Acids. Molecules 2020, 25, 4418. [Google Scholar] [CrossRef]
- Xiao, H.; Peters, F.B.; Yang, P.Y.; Reed, S.; Chittuluru, J.R.; Schultz, P.G. Genetic incorporation of histidine derivatives using an engineered pyrrolysyl-tRNA synthetase. ACS Chem. Biol. 2014, 9, 1092–1096. [Google Scholar] [CrossRef]
- Völler, J.S.; Biava, H.; Hildebrandt, P.; Budisa, N. An expanded genetic code for probing the role of electrostatics in enzyme catalysis by vibrational Stark spectroscopy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3053–3059. [Google Scholar] [CrossRef]
- Kavran, J.M.; Gundllapalli, S.; O’Donoghue, P.; Englert, M.; Söll, D.; Steitz, T.A. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. USA 2007, 104, 11268–11273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudeau, D.L.; Tawfik, D.S. Protein engineers turned evolutionists—the quest for the optimal starting point. Curr. Opin. Biotechnol. 2019, 60, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed. 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuriki, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef]
- Grasso, K.T.; Yeo, M.J.R.; Hillenbrand, C.M.; Ficaretta, E.D.; Italia, J.S.; Huang, R.L.; Chatterjee, A. Structural Robustness Affects the Engineerability of Aminoacyl-tRNA Synthetases for Genetic Code Expansion. Biochemistry 2021, 60, 489–493. [Google Scholar] [CrossRef]
- Hu, L.; Qin, X.; Huang, Y.; Cao, W.; Wang, C.; Wang, Y.; Ling, X.; Chen, H.; Wu, D.; Lin, Y.; et al. Thermophilic Pyrrolysyl-tRNA Synthetase Mutants for Enhanced Mammalian Genetic Code Expansion. ACS Synth. Biol. 2020, 9, 2723–2736. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Crystallographic Studies on Multiple Conformational States of Active-site Loops in Pyrrolysyl-tRNA Synthetase. J. Mol. Biol. 2008, 378, 634–652. [Google Scholar] [CrossRef]
- Vargas-Cortez, T.; Morones-Ramirez, J.R.; Balderas-Renteria, I.; Zarate, X. Expression and purification of recombinant proteins in Escherichia coli tagged with a small metal-binding protein from Nitrosomonas europaea. Protein Expr. Purif. 2016, 118, 49–54. [Google Scholar] [CrossRef]
- Ko, J.H.; Wang, Y.S.; Nakamura, A.; Guo, L.T.; Söll, D.; Umehara, T. Pyrrolysyl-tRNA synthetase variants reveal ancestral aminoacylation function. FEBS Lett. 2013, 587, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Baumann, T.; Hauf, M.; Richter, F.; Albers, S.; Möglich, A.; Ignatova, Z.; Budisa, N. Computational aminoacyl-tRNA synthetase library design for photocaged tyrosine. Int. J. Mol. Sci. 2019, 20, 2343. [Google Scholar] [CrossRef] [Green Version]
- Owens, A.E.; Grasso, K.T.; Ziegler, C.A.; Fasan, R. Two-Tier Screening Platform for Directed Evolution of Aminoacyl–tRNA Synthetases with Enhanced Stop Codon Suppression Efficiency. ChemBioChem 2017, 18, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, J.K.; Dellas, N.; Noel, J.P.; Wang, L. Stereochemical Basis for Engineered Pyrrolysyl-tRNA Synthetase and the Efficient in Vivo Incorporation of Structurally Divergent Non-native Amino Acids. ACS Chem. Biol. 2011, 6, 733–743. [Google Scholar] [CrossRef]
- Exner, M.P.; Kuenzl, T.; To, T.M.T.; Ouyang, Z.; Schwagerus, S.; Hoesl, M.G.; Hackenberger, C.P.R.; Lensen, M.C.; Panke, S.; Budisa, N. Design of S-Allylcysteine in Situ Production and Incorporation Based on a Novel Pyrrolysyl-tRNA Synthetase Variant. ChemBioChem 2017, 18, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Russell, W.K.; Wang, Z.; Wan, W.; Dodd, L.E.; Pai, P.J.; Russell, D.H.; Liu, W.R. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of l-phenylalanine and its derivatives. Mol. Biosyst. 2011, 7, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, R.; Wu, H.; Li, S.; Wang, P.G.; DeGrado, W.F.; Rozovsky, S.; Wang, L. Building and Breaking Bonds via a Compact S-Propargyl-Cysteine to Chemically Control Enzymes and Modify Proteins. Angew. Chem. Int. Ed. 2018, 57, 12702–12706. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.F.; Xu, J.; Shen, Z.; Takimoto, J.K.; Schultz, M.D.; Schmitz, R.J.; Xiang, Z.; Ecker, J.R.; Briggs, S.P.; Wang, L. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 2011, 7, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, T.; Hoshi, H.; Ohtake, K.; Takahashi, M.; Yamaguchi, A.; Hayashi, A.; Yokoyama, S.; Sakamoto, K. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep. 2015, 5, 9699. [Google Scholar] [CrossRef] [Green Version]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, P.; Prat, L.; Heinemann, I.U.; Ling, J.; Odoi, K.; Liu, W.R.; Söll, D. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012, 586, 3931–3937. [Google Scholar] [CrossRef] [Green Version]
- Ai, H.W.; Shen, W.; Brustad, E.; Schultz, P.G. Genetically encoded alkenes in yeast. Angew. Chem. Int. Ed. 2010, 49, 935–937. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Cai, W.; Tan, L.; Yu, Y.; Han, B.; Li, Y.; Xie, Y.; Su, Y.; Luo, X.; et al. Expanding the Structural Diversity of Protein Building Blocks with Noncanonical Amino Acids Biosynthesized from Aromatic Thiols. Angew. Chem. 2021, 133, 10128–10136. [Google Scholar] [CrossRef]
- Bryson, D.I.; Fan, C.; Guo, L.T.; Miller, C.; Söll, D.; Liu, D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, C.N.; Andrews, D.W. Site-Directed Mutagenesis by Inverse PCR. In E. coli Plasmid Vectors; Humana Press: Totowa, NJ, USA, 2003; pp. 209–224. [Google Scholar]
- Nov, Y. When second best is good enough: Another probabilistic look at saturation mutagenesis. Appl. Environ. Microbiol. 2012, 78, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchand, J.A.; Neugebauer, M.E.; Ing, M.C.; Lin, C.-I.; Pelton, J.G.; Chang, M.C.Y. Discovery of a pathway for terminal-alkyne amino acid biosynthesis. Nature 2019, 567, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J.L. Advances in chemical protein modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-N.; Kuppan, K.V.; Lee, M.D.; Jaudzems, K.; Huber, T.; Otting, G. O-tert -Butyltyrosine, an NMR Tag for High-Molecular-Weight Systems and Measurements of Submicromolar Ligand Binding Affinities. J. Am. Chem. Soc. 2015, 137, 4581–4586. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.K.; Yoo, C.; Hong, E.; Rhee, H.W. Photo-crosslinking: An Emerging Chemical Tool for Investigating Molecular Networks in Live Cells. ChemBioChem 2020, 21, 924–932. [Google Scholar] [CrossRef]
- Kim, G.; Weiss, S.J.; Levine, R.L. Methionine oxidation and reduction in proteins. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 901–905. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Lodder, M.; Zhou, J.; Baird, T.T.; Brown, K.C.; Craik, C.S.; Hecht, S.M. Chemically Mediated Site-Specific Cleavage of Proteins. J. Am. Chem. Soc. 2000, 122, 7402–7403. [Google Scholar] [CrossRef]
- Baird, T.; Wang, B.; Lodder, M.; Hecht, S.M.; Craik, C.S. Generation of Active Trypsin by Chemical Cleavage. Tetrahedron 2000, 56, 9477–9485. [Google Scholar] [CrossRef]
- Wang, B.; Brown, K.C.; Lodder, M.; Craik, C.S.; Hecht, S.M. Chemically Mediated Site-Specific Proteolysis. Alteration of Protein−Protein Interaction†. Biochemistry 2002, 41, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Liutkus, M.; Fraser, S.A.; Caron, K.; Stigers, D.J.; Easton, C.J. Peptide Synthesis through Cell-Free Expression of Fusion Proteins Incorporating Modified Amino Acids as Latent Cleavage Sites for Peptide Release. ChemBioChem 2016, 17, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Nolsøe, J.M.J.; Hansen, T.V. Asymmetric Iodolactonization: An Evolutionary Account. Eur. J. Org. Chem. 2014, 2014, 3051–3065. [Google Scholar] [CrossRef]
- Kristianslund, R.; Tungen, J.E.; Hansen, T.V. Catalytic enantioselective iodolactonization reactions. Org. Biomol. Chem. 2019, 17, 3079–3092. [Google Scholar] [CrossRef] [PubMed]
- Lodder, M.; Golovine, S.; Laikhter, A.L.; Karginov, V.A.; Hecht, S.M. Misacylated Transfer RNAs Having a Chemically Removable Protecting Group. J. Org. Chem. 1998, 63, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Witkop, B. Nonenzymatic cleavage of peptide bonds: The methionine residues in bovine pancreatic ribonuclease. J. Biol. Chem. 1962, 237, 1856–1860. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, X.; Chen, X.; Aramsangtienchai, P.; Tong, Z.; Lin, H. Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chem. Rev. 2018, 118, 919–988. [Google Scholar] [CrossRef]
- Chen, J.J.; Boehning, D. Protein lipidation as a regulator of apoptotic calcium release: Relevance to cancer. Front. Oncol. 2017, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Coleman, D.T.; Gray, A.L.; Kridel, S.J.; Cardelli, J.A. Palmitoylation regulates the intracellular trafficking and stability of c-Met. Oncotarget 2016, 7, 32664–32677. [Google Scholar] [CrossRef]
- Hang, H.C.; Linder, M.E. Exploring Protein Lipidation with Chemical Biology. Chem. Rev. 2011, 111, 6341–6358. [Google Scholar] [CrossRef] [Green Version]
- Bech, E.M.; Pedersen, S.L.; Jensen, K.J. Chemical Strategies for Half-Life Extension of Biopharmaceuticals: Lipidation and Its Alternatives. ACS Med. Chem. Lett. 2018, 9, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Acevedo-Rocha, C.G.; Geiermann, A.-S.; Budisa, N.; Merkel, L. Design of protein congeners containing β-cyclopropylalanine. Mol. Biosyst. 2012, 8, 2719. [Google Scholar] [CrossRef]
- Budisa, N.; Steipe, B.; Demange, P.; Eckerskorn, C.; Kellermann, J.; Huber, R. High-level Biosynthetic Substitution of Methionine in Proteins by its Analogs 2-Aminohexanoic Acid, Selenomethionine, Telluromethionine and Ethionine in Escherichia coli. Eur. J. Biochem. 1995, 230, 788–796. [Google Scholar] [CrossRef]
- Kiick, K.L.; Saxon, E.; Tirrell, D.A.; Bertozzi, C.R. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. USA 2002, 99, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Tanrikulu, I.C.; Schmitt, E.; Mechulam, Y.; Goddard, W.A.; Tirrell, D.A. Discovery of Escherichia coli methionyl-tRNA synthetase mutants for efficient labeling of proteins with azidonorleucine in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 15285–15290. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.A.; Kurra, Y.; Wang, X.; Zeng, Y.; Lee, Y.; Sharma, V.; Lin, H.; Dai, S.Y.; Liu, W.R. A Versatile Approach for Site-Specific Lysine Acylation in Proteins. Angew. Chem. Int. Ed. 2017, 56, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Salaün, J. Cyclopropane Derivatives and their Diverse Biological Activities. Top. Curr. Chem. 2000, 207, 1–67. [Google Scholar]
- Geigert, J.; Neidleman, S.L.; Dalietos, D.J. Novel haloperoxidase substrates. Alkynes and cyclopropanes. J. Biol. Chem. 1983, 258, 2273–2277. [Google Scholar] [CrossRef]
- Dalton, H.; Golding, B.T.; Waters, B.W.; Higgins, R.; Taylor, J.A. Oxidations of cyclopropane, methylcyclopropane, and arenes with the mono-oxygenase system from Methylococcus capsulatus. J. Chem. Soc. Chem. Commun. 1981, 28, 482. [Google Scholar] [CrossRef]
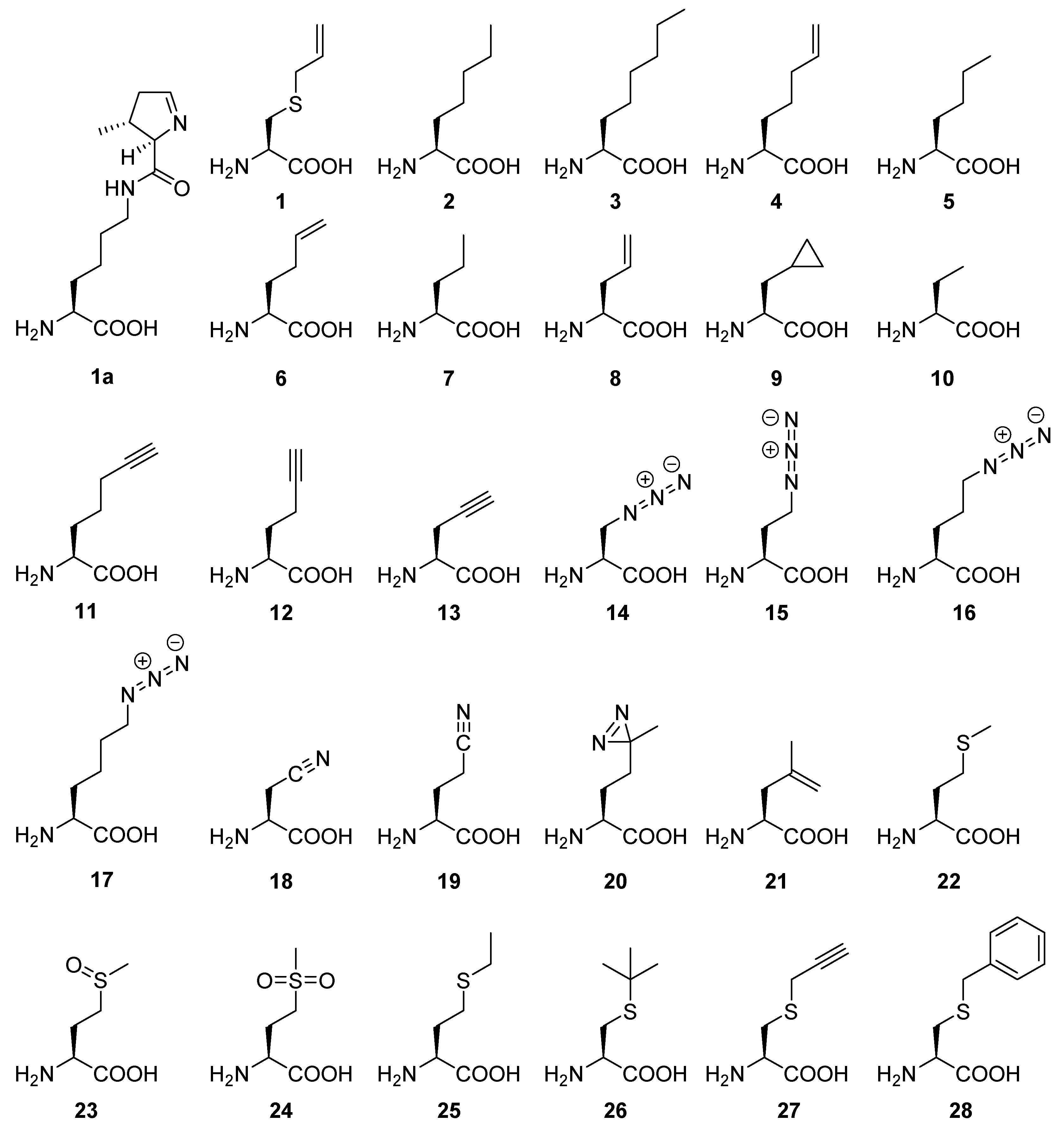


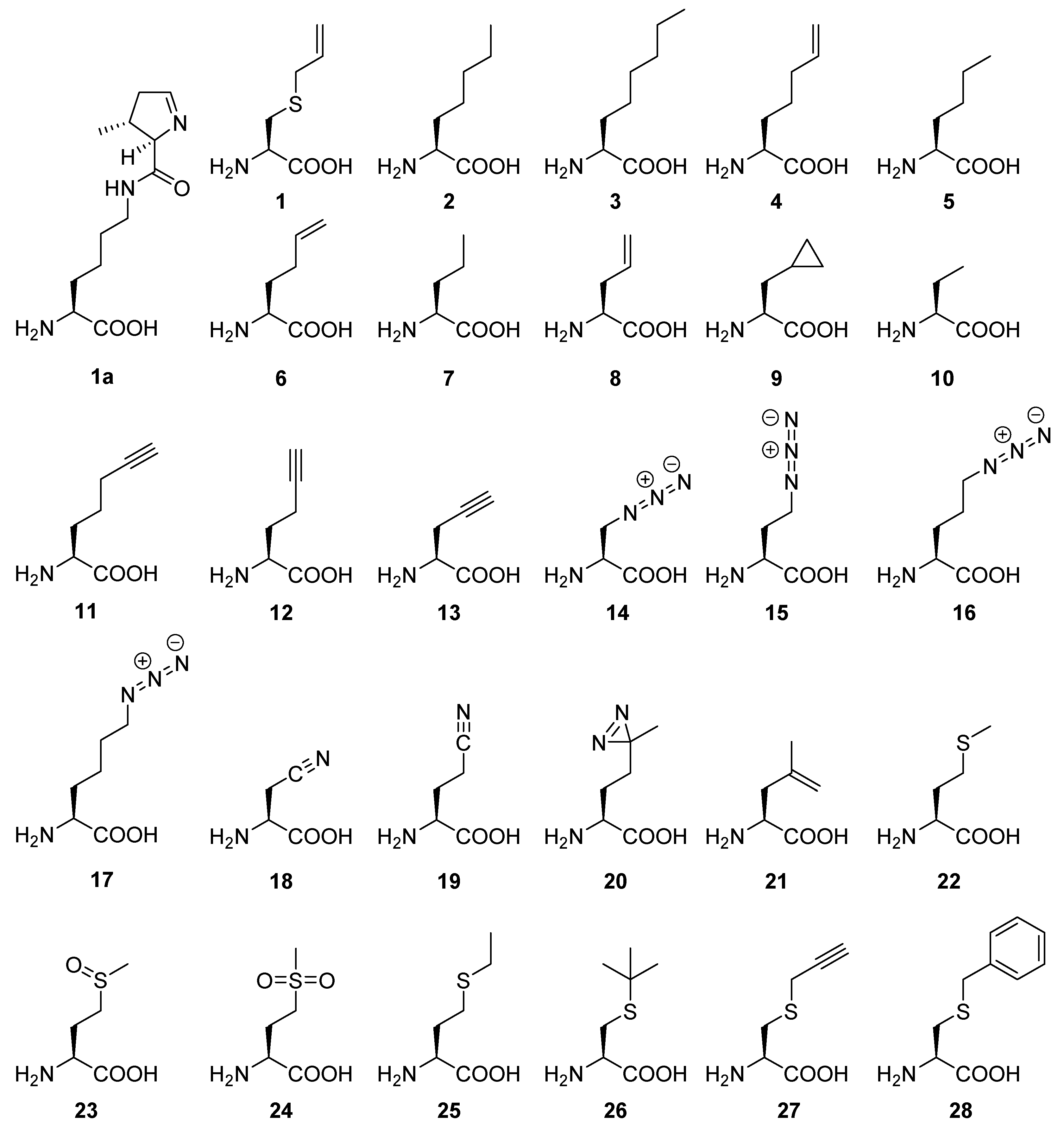{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | E. coli Strains 1 | MbPylRS Construct | Reporter Construct | Calculated Mass [Da] | Observed Mass [Da] | Δ Mass [Da] | Protein Yield [mg∙L−1] 2 |
|---|---|---|---|---|---|---|---|
| 1 | BL21 | N311M:C313W:V366A | a | 40,194.9 | 40,196 | 1.1 | 10.8 |
| 2 | BL21 | N311Q:C313W | a | 40,178.8 | 40,180 | 1.2 | 5.1 |
| 3 | BL21 | N311M:C313W:V366A | a | 40,192.9 | 40,194 | 1.1 | 1.6 |
| 4 | BL21 | N311M:C313W:V366A | a | 40,176.8 | 40,179 | 2.2 | 1.7 |
| 5 | BL21 | N311M:C313W | a | 40,164.8 | 40,166 | 1.2 | 1.9 |
| 6 | BL21 | N311M:C313W:V366K | a | 40,162.8 | 40,164 | 1.2 | 1.4 |
| 7 | BL21 | N311M:C313W | a | 40,150.8 | 40,153 | 2.2 | 1.2 |
| 8 | BL21 | N311M:C313W | a | 40,148.8 | 40,150 | 1.2 | 0.7 |
| 9 | BL21 | N311M:C313W | a | 40,162.8 | 40,164 | 1.2 | 1.4 |
| 10 | BL21 | N311M:C313W:V366A | a | 40,136.8 | 40,195 | 58.2 | 0.8 |
| 11 | C321.ΔA.exp | N311M:C313W | b | 38,990.9 | 38,992 | 1.1 | 5.1 |
| 12 | C321.ΔA.exp | N311M:C313W | b | 38,976.9 | 38,979 | 2.1 | 4.9 |
| 13 | C321.ΔA.exp | N311M:C313W | b | 38,962.8 | 38,965 | 2.2 | 11.3 |
| 14 | JX33 | N311M:C313W | b | 38,979.8 | 38,996 | 16.2 | 4.3 |
| 15 | C321.ΔA.exp | N311M:C313W | b | 38,993.8 | 38,994 | 0.2 | 14.2 |
| 16 | C321.ΔA.exp | N311M:C313W | b 4 | 39,007.9 | 39,007 | 0.9 | 5.3 |
| 17 | C321.ΔA.exp | N311M:C313W | b | 39,021.9 | 38,997 | 24.9 | 6.4 |
| 18 | C321.ΔA.exp | N311Q:C313W:V366K | b | 38,963.8 | 39,015 | 51.2 | 19 |
| 19 | C321.ΔA.exp | N311Q:C313W:V366K | b | 38,977.8 | 39,014 | 36.2 | 19.9 |
| 20 | BL21 | N311M:C313W:V366K | a | 40,190.8 | 40,194 | 3.2 | 4.8 |
| 21 | BL21 | N311M:C313W | b | 38,978.9 | 38,982 | 3.1 | 2.6 |
| 22 | BL21 | N311M:C313W | b | 38,998.9 | 38,998 | 0.9 | 3.6 |
| 23 | BL21 | N311Q:C313W | b | 39,014.9 | 39,012 | 2.9 | 4.6 |
| 24 | BL21 | N311Q:C313W:V366K | b | 39,030.9 | 39,015 | 15.9 | 10.7 |
| 25 | BL21 | N311Q:C313W | b | 39,013 | 39,014 | 1 | 9.8 |
| 26 | BL21 | N311M:C313W:V366A | a | 40,210.9 | 40,211 | 0.1 | 21 |
| 27 | BL21 | N311M:C313W:V366A | a 3 | - | - | - | - |
| 28 | BL21 | N311M:C313W:V366A: W382N | b | 39,061 | 39,063 | 2 | 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, N.G.; Goettig, P.; Rappsilber, J.; Budisa, N. Engineering Pyrrolysyl-tRNA Synthetase for the Incorporation of Non-Canonical Amino Acids with Smaller Side Chains. Int. J. Mol. Sci. 2021, 22, 11194. https://doi.org/10.3390/ijms222011194
Koch NG, Goettig P, Rappsilber J, Budisa N. Engineering Pyrrolysyl-tRNA Synthetase for the Incorporation of Non-Canonical Amino Acids with Smaller Side Chains. International Journal of Molecular Sciences. 2021; 22(20):11194. https://doi.org/10.3390/ijms222011194
Chicago/Turabian StyleKoch, Nikolaj G., Peter Goettig, Juri Rappsilber, and Nediljko Budisa. 2021. "Engineering Pyrrolysyl-tRNA Synthetase for the Incorporation of Non-Canonical Amino Acids with Smaller Side Chains" International Journal of Molecular Sciences 22, no. 20: 11194. https://doi.org/10.3390/ijms222011194
APA StyleKoch, N. G., Goettig, P., Rappsilber, J., & Budisa, N. (2021). Engineering Pyrrolysyl-tRNA Synthetase for the Incorporation of Non-Canonical Amino Acids with Smaller Side Chains. International Journal of Molecular Sciences, 22(20), 11194. https://doi.org/10.3390/ijms222011194