
Recent Advances in CRISPR/Cas9-Based Genome Editing Tools for Cardiac Diseases

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
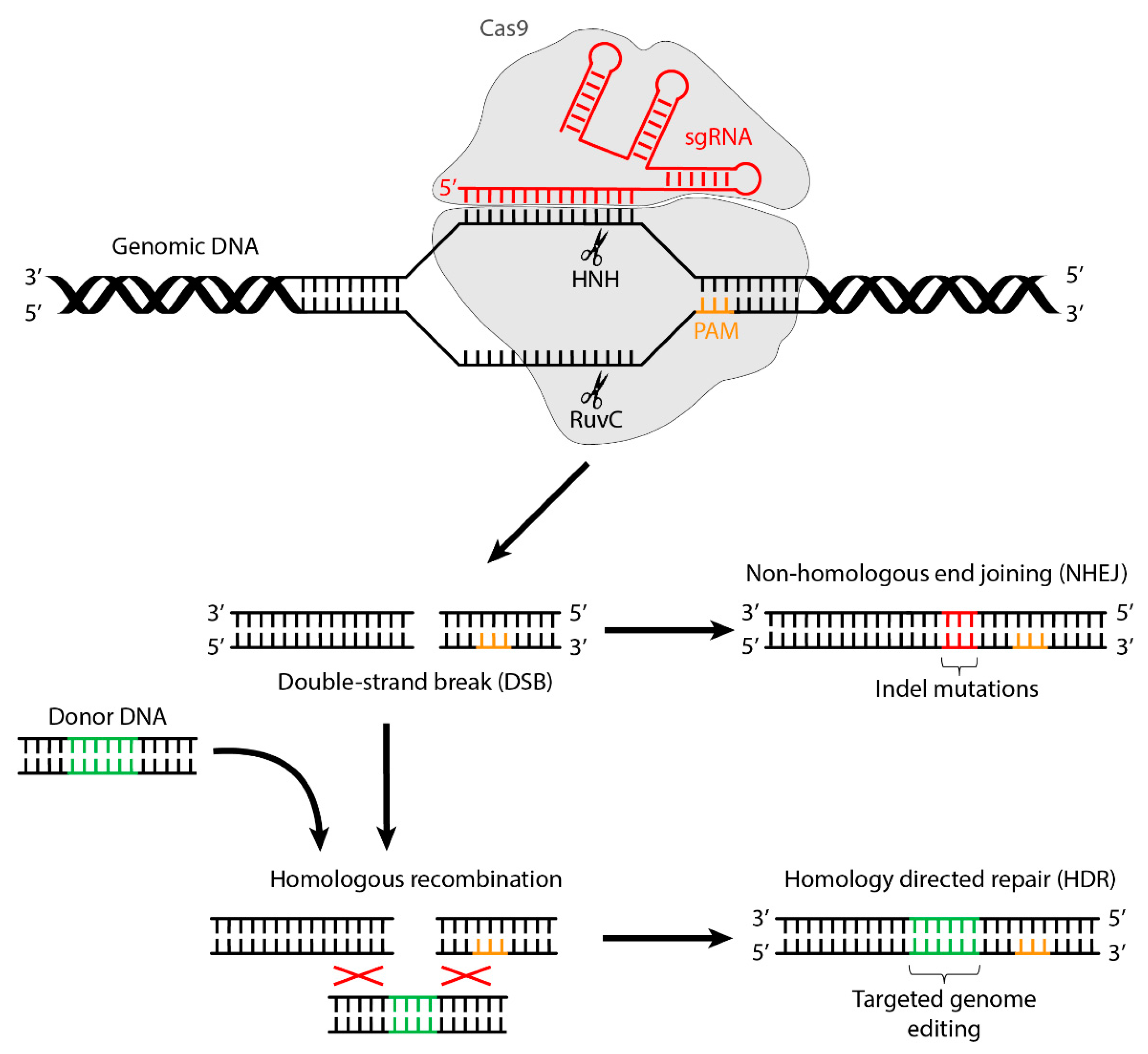
:1. Introduction
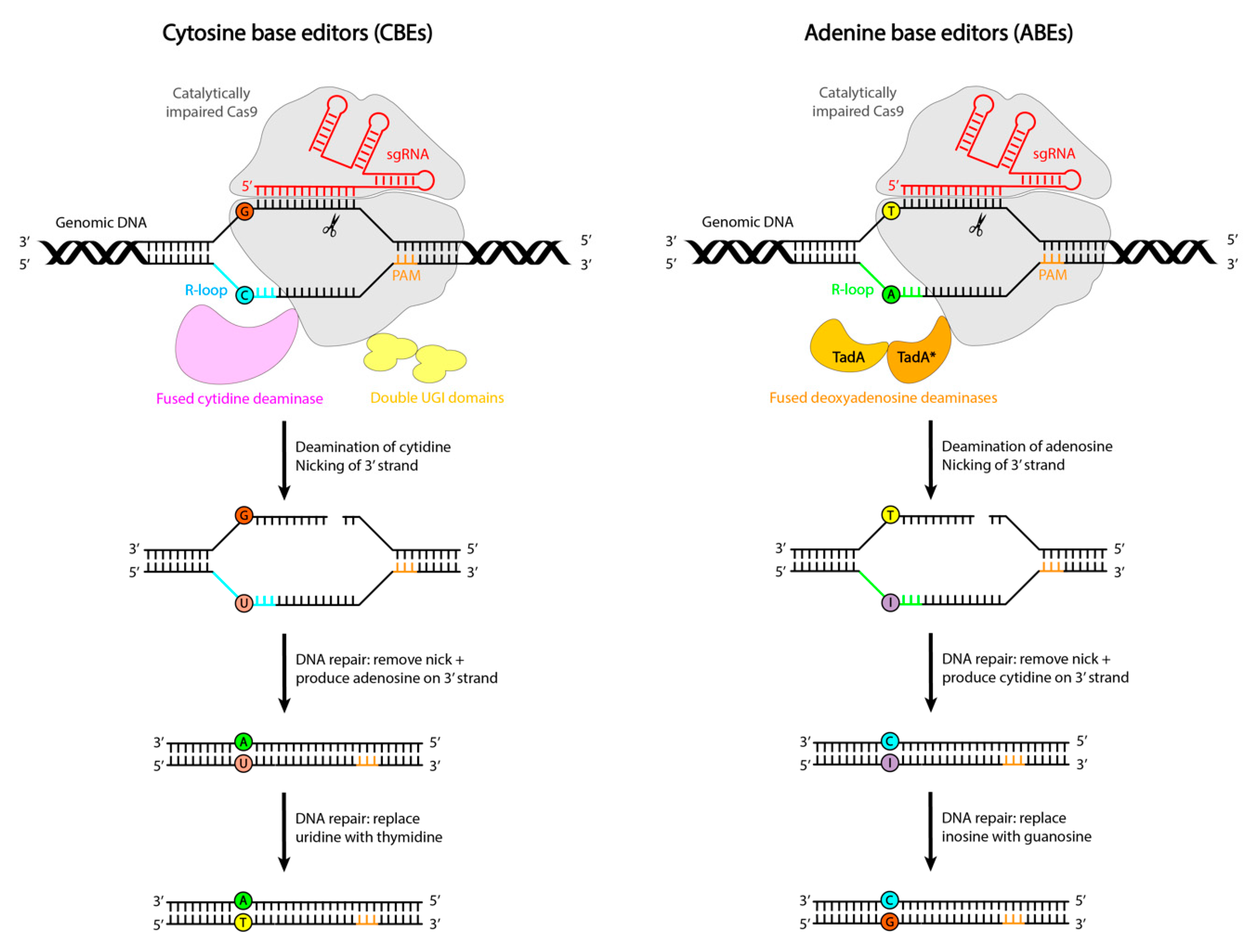
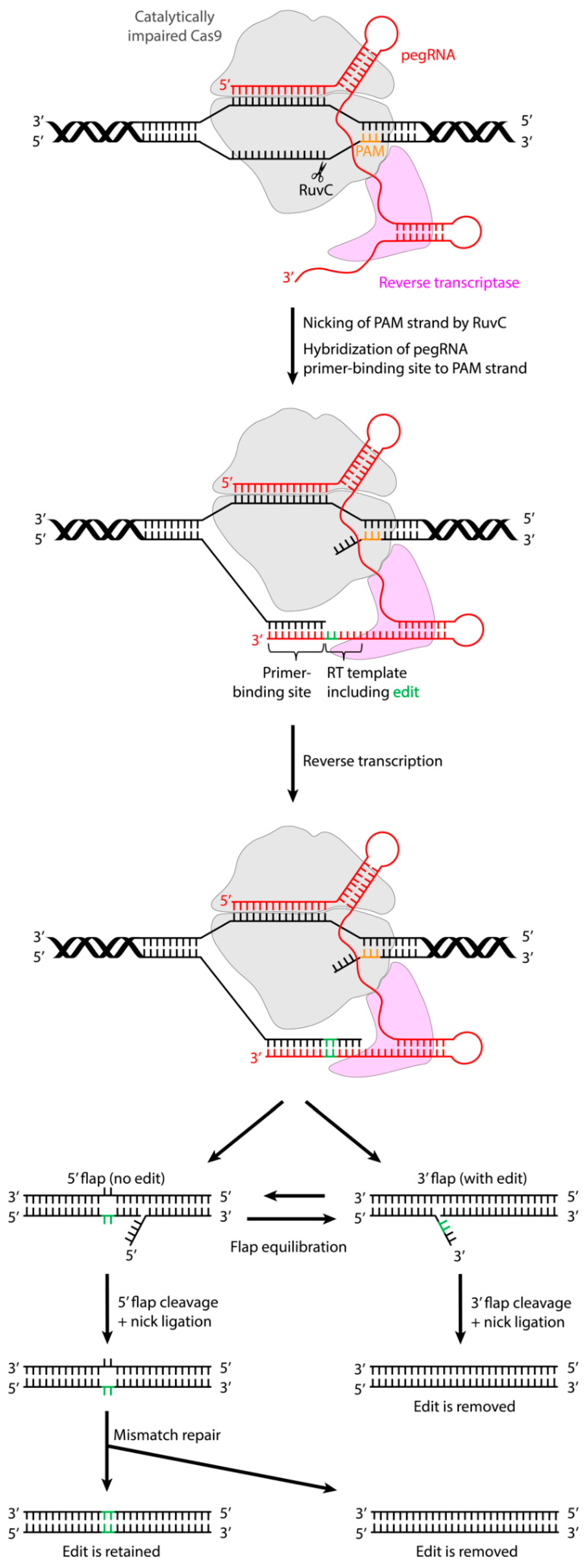
A New Generation of Genome Editing Tools: Base Editing and Prime Editing
2. Applications of CRISPR/Cas9-Based Genome Editing Systems in Cardiac Disease Models
2.1. In Vitro Models
2.2. In Vivo Models
3. Limitations of Genome Editors
3.1. Off-Targets
3.2. Flexibility of the PAM Sequence
3.3. Cell Fitness Effects
3.4. Genome Editing in Post-Mitotic Tissues
3.5. Delivery
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Scherer, S.; Davis, R.W. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 4951–4955. [Google Scholar] [CrossRef] [Green Version]
- Smithies, O.; Gregg, R.G.; Boggs, S.S.; Koralewski, M.A.; Kucherlapati, R.S. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 1985, 317, 230–234. [Google Scholar] [CrossRef]
- Capecchi, M.R. Altering the genome by homologous recombination. Science 1989, 244, 1288–1292. [Google Scholar] [CrossRef] [Green Version]
- Kucherlapati, R.S.; Eves, E.M.; Song, K.Y.; Morse, B.S.; Smithies, O. Homologous recombination between plasmids in mammalian cells can be enhanced by treatment of input DNA. Proc. Natl. Acad. Sci. USA 1984, 81, 3153–3157. [Google Scholar] [CrossRef] [Green Version]
- Kc, M.; Steer, C.J. A new era of gene editing for the treatment of human diseases. Swiss Med. Wkly. 2019, 149, w20021. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M. The genome editing revolution: Review. J. Genet. Eng. Biotechnol. 2020, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, E.; Jouve, C.; Hulot, J.S. CRISPR/Cas9 gene-editing strategies in cardiovascular cells. Cardiovasc. Res. 2020, 116, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Kondrateva, E.; Demchenko, A.; Lavrov, A.; Smirnikhina, S. An overview of currently available molecular Cas-tools for precise genome modification. Gene 2021, 769, 145225. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Yang, L.; Yang, B.; Chen, J. One Prime for All Editing. Cell 2019, 179, 1448–1450. [Google Scholar] [CrossRef]
- Lu, D.; Thum, T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 661–674. [Google Scholar] [CrossRef]
- Yeh, W.H.; Chiang, H.; Rees, H.A.; Edge, A.S.B.; Liu, D.R. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018, 9, 2184. [Google Scholar] [CrossRef]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C.; American Heart Association Council on Functional Genomics and Translational Biology; Council on Cardiovascular Disease in the Young; et al. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flugel, L.; Dorn, T.; Goedel, A.; Hohnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- My, I.; Di Pasquale, E. Genetic Cardiomyopathies: The Lesson Learned from hiPSCs. J. Clin. Med. 2021, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Termglinchan, V.; Cepeda, D.A.; Lee, J.; Diecke, S.; Hendel, A.; Itzhaki, I.; Ameen, M.; Shrestha, R.; Wu, H.; et al. A Comprehensive TALEN-Based Knockout Library for Generating Human-Induced Pluripotent Stem Cell-Based Models for Cardiovascular Diseases. Circ. Res. 2017, 120, 1561–1571. [Google Scholar] [CrossRef]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Vermglinchan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, J.Z.; Itzhaki, I.; Zhang, S.L.; Chen, H.; Haddad, F.; Kitani, T.; Wilson, K.D.; Tian, L.; Shrestha, R.; et al. Determining the Pathogenicity of a Genomic Variant of Uncertain Significance Using CRISPR/Cas9 and Human-Induced Pluripotent Stem Cells. Circulation 2018, 138, 2666–2681. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Oikonomopoulos, A.; Chen, H.; Li, Y.; Lam, C.K.; Sallam, K.; Perez, M.; Lux, R.L.; Sanguinetti, M.C.; Wu, J.C. Genome Editing of Induced Pluripotent Stem Cells to Decipher Cardiac Channelopathy Variant. J. Am. Coll. Cardiol. 2018, 72, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Makiyama, T.; Harita, T.; Sasaki, K.; Wuriyanghai, Y.; Hayano, M.; Nishiuchi, S.; Kohjitani, H.; Hirose, S.; Chen, J.; et al. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum. Mol. Genet. 2017, 26, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Sacchetto, C.; Vitiello, L.; de Windt, L.J.; Rampazzo, A.; Calore, M. Modeling Cardiovascular Diseases with hiPSC-Derived Cardiomyocytes in 2D and 3D Cultures. Int. J. Mol. Sci. 2020, 21, 3404. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, O.; Merten, A.L.; Schneidereit, D.; Guo, Y.; Schurmann, S.; Martinac, B. Stretch in Focus: 2D Inplane Cell Stretch Systems for Studies of Cardiac Mechano-Signaling. Front. Bioeng. Biotechnol. 2019, 7, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czirok, A.; Isai, D.G.; Kosa, E.; Rajasingh, S.; Kinsey, W.; Neufeld, Z.; Rajasingh, J. Optical-flow based non-invasive analysis of cardiomyocyte contractility. Sci. Rep. 2017, 7, 10404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Sun, N.; Bruce, M.A.; Wu, J.C.; Butte, M.J. Atomic force mechanobiology of pluripotent stem cell-derived cardiomyocytes. PLoS ONE 2012, 7, e37559. [Google Scholar] [CrossRef] [Green Version]
- Sapp, V.; Aguirre, A.; Mainkar, G.; Ding, J.; Adler, E.; Liao, R.; Sharma, S.; Jain, M. Genome-wide CRISPR/Cas9 screening in human iPS derived cardiomyocytes uncovers novel mediators of doxorubicin cardiotoxicity. Sci. Rep. 2021, 11, 13866. [Google Scholar] [CrossRef]
- Chemello, F.; Chai, A.C.; Li, H.; Rodriguez-Caycedo, C.; Sanchez-Ortiz, E.; Atmanli, A.; Mireault, A.A.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 2021, 7, eabg4910. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; He, S.; Huang, S.; Li, C.; Chen, Y.; Liu, Z.; Huang, X.; Wang, X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020, 6, 27. [Google Scholar] [CrossRef]
- Jang, H.; Jo, D.H.; Cho, C.S.; Shin, J.H.; Seo, J.H.; Yu, G.; Gopalappa, R.; Kim, D.; Cho, S.R.; Kim, J.H.; et al. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat. Biomed. Eng. 2021. [Google Scholar] [CrossRef]
- Xie, C.; Zhang, Y.P.; Song, L.; Luo, J.; Qi, W.; Hu, J.; Lu, D.; Yang, Z.; Zhang, J.; Xiao, J.; et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 2016, 26, 1099–1111. [Google Scholar] [CrossRef] [Green Version]
- El Refaey, M.; Xu, L.; Gao, Y.; Canan, B.D.; Adesanya, T.M.A.; Warner, S.C.; Akagi, K.; Symer, D.E.; Mohler, P.J.; Ma, J.; et al. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circ. Res. 2017, 121, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, C.; Li, H.; Wang, P.; Gao, Y.; Mokadam, N.A.; Ma, J.; Arnold, W.D.; Han, R. Efficient precise in vivo base editing in adult dystrophic mice. Nat. Commun. 2021, 12, 3719. [Google Scholar] [CrossRef]
- Bose, S.K.; White, B.M.; Kashyap, M.V.; Dave, A.; De Bie, F.R.; Li, H.; Singh, K.; Menon, P.; Wang, T.; Teerdhala, S.; et al. In utero adenine base editing corrects multi-organ pathology in a lethal lysosomal storage disease. Nat. Commun. 2021, 12, 4291. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.J.; Makarewich, C.A.; McAnally, J.; Anderson, D.M.; Zentilin, L.; Liu, N.; Giacca, M.; Bassel-Duby, R.; Olson, E.N. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc. Natl. Acad. Sci. USA 2016, 113, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, A.K.; Molenaar, B.; Versteeg, D.; Leitoguinho, A.R.; Demkes, C.; Spanjaard, B.; de Ruiter, H.; Akbari Moqadam, F.; Kooijman, L.; Zentilin, L.; et al. Postnatal Cardiac Gene Editing Using CRISPR/Cas9 With AAV9-Mediated Delivery of Short Guide RNAs Results in Mosaic Gene Disruption. Circ. Res. 2017, 121, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, S.J.; van Rooij, E. CRISPR Craze to Transform Cardiac Biology. Trends Mol. Med. 2019, 25, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef]
- Motta, B.M.; Pramstaller, P.P.; Hicks, A.A.; Rossini, A. The Impact of CRISPR/Cas9 Technology on Cardiac Research: From Disease Modelling to Therapeutic Approaches. Stem Cells Int. 2017, 2017, 8960236. [Google Scholar] [CrossRef] [Green Version]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [Green Version]
- Stemmer, M.; Thumberger, T.; Del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musunuru, K. The Hope and Hype of CRISPR-Cas9 Genome Editing: A Review. JAMA Cardiol. 2017, 2, 914–919. [Google Scholar] [CrossRef]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, J.M.; Cervantes, O.; Clement, M.K.; Wu, Y.; Zeng, J.; Bauer, D.E.; Pinello, L.; Joung, J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 2018, 36, 977–982. [Google Scholar] [CrossRef]
- Molla, K.A.; Yang, Y. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. 2019, 37, 1121–1142. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Moon, S.B.; Ko, J.H.; Kim, Y.S.; Kim, D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res. 2020, 48, 10576–10589. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Manghwar, H.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas System: Recent Advances and Future Prospects for Genome Editing. Trends Plant Sci. 2019, 24, 1102–1125. [Google Scholar] [CrossRef] [Green Version]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Khan, S.H. Genome-Editing Technologies: Concept, Pros, and Cons of Various Genome-Editing Techniques and Bioethical Concerns for Clinical Application. Mol. Ther. Nucleic Acids 2019, 16, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef]
- Rossidis, A.C.; Stratigis, J.D.; Chadwick, A.C.; Hartman, H.A.; Ahn, N.J.; Li, H.; Singh, K.; Coons, B.E.; Li, L.; Lv, W.; et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018, 24, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, I.G. Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front. Genet. 2020, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome Editing: Past, Present, and Future. Yale J. Biol. Med. 2017, 90, 653–659. [Google Scholar]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Kariya, T.; Ishikawa, K. Targeted delivery of therapeutic agents to the heart. Nat. Rev. Cardiol. 2021, 18, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.; Tiefenbacher, S.; Robinson, M.; Huang, M.; Srimani, J.; Mackenzie, D.; Christianson, T.; Pasi, K.J.; Rangarajan, S.; Symington, E.; et al. Activity of transgene-produced B-domain-deleted factor VIII in human plasma following AAV5 gene therapy. Blood 2020, 136, 2524–2534. [Google Scholar] [CrossRef] [PubMed]
- George, L.A.; Ragni, M.V.; Rasko, J.E.J.; Raffini, L.J.; Samelson-Jones, B.J.; Ozelo, M.; Hazbon, M.; Runowski, A.R.; Wellman, J.A.; Wachtel, K.; et al. Long-Term Follow-Up of the First in Human Intravascular Delivery of AAV for Gene Transfer: AAV2-hFIX16 for Severe Hemophilia B. Mol. Ther. 2020, 28, 2073–2082. [Google Scholar] [CrossRef]
- Fischer, M.D.; Michalakis, S.; Wilhelm, B.; Zobor, D.; Muehlfriedel, R.; Kohl, S.; Weisschuh, N.; Ochakovski, G.A.; Klein, R.; Schoen, C.; et al. Safety and Vision Outcomes of Subretinal Gene Therapy Targeting Cone Photoreceptors in Achromatopsia: A Nonrandomized Controlled Trial. JAMA Ophthalmol. 2020, 138, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Babalis, D.; Morley-Smith, A.C.; Hedger, M.; Suarez Barrientos, A.; Foldes, G.; Couch, L.S.; Chowdhury, R.A.; Tzortzis, K.N.; Peters, N.S.; et al. Investigation of the safety and feasibility of AAV1/SERCA2a gene transfer in patients with chronic heart failure supported with a left ventricular assist device—The SERCA-LVAD TRIAL. Gene Ther. 2020, 27, 579–590. [Google Scholar] [CrossRef]
- Van Haasteren, J.; Li, J.; Scheideler, O.J.; Murthy, N.; Schaffer, D.V. The delivery challenge: Fulfilling the promise of therapeutic genome editing. Nat. Biotechnol. 2020, 38, 845–855. [Google Scholar] [CrossRef]
- Levy, J.M.; Yeh, W.H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020, 4, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, E.J.; Zdechlik, A.C.; Ruis, B.L.; Rogers, C.B.; Lemmex, A.L.; Nelson, A.T.; Hendrickson, E.A.; Schmidt, D.; Gordon, W.R. Split Staphylococcus aureus prime editor for AAV delivery. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.Y.; VandenDriessche, T.; Chuah, M.K. Gene therapy for cardiovascular disease: Advances in vector development, targeting, and delivery for clinical translation. Cardiovasc. Res. 2015, 108, 4–20. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreurs, J.; Sacchetto, C.; Colpaert, R.M.W.; Vitiello, L.; Rampazzo, A.; Calore, M. Recent Advances in CRISPR/Cas9-Based Genome Editing Tools for Cardiac Diseases. Int. J. Mol. Sci. 2021, 22, 10985. https://doi.org/10.3390/ijms222010985
Schreurs J, Sacchetto C, Colpaert RMW, Vitiello L, Rampazzo A, Calore M. Recent Advances in CRISPR/Cas9-Based Genome Editing Tools for Cardiac Diseases. International Journal of Molecular Sciences. 2021; 22(20):10985. https://doi.org/10.3390/ijms222010985
Chicago/Turabian StyleSchreurs, Juliët, Claudia Sacchetto, Robin M. W. Colpaert, Libero Vitiello, Alessandra Rampazzo, and Martina Calore. 2021. "Recent Advances in CRISPR/Cas9-Based Genome Editing Tools for Cardiac Diseases" International Journal of Molecular Sciences 22, no. 20: 10985. https://doi.org/10.3390/ijms222010985
APA StyleSchreurs, J., Sacchetto, C., Colpaert, R. M. W., Vitiello, L., Rampazzo, A., & Calore, M. (2021). Recent Advances in CRISPR/Cas9-Based Genome Editing Tools for Cardiac Diseases. International Journal of Molecular Sciences, 22(20), 10985. https://doi.org/10.3390/ijms222010985