
TMEM16A, a Homoharringtonine Receptor, as a Potential Endogenic Target for Lung Cancer Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. TCGA Data Mining
2.2. Cell Culture
2.3. Western Blot Analysis
2.4. Electrophysiology
2.5. Molecular Docking
2.6. Site-Directed Mutagenesis
2.7. CCK-8 Assay
2.8. Wound Healing Assay
2.9. Annexin V Assay
2.10. Tumor Xenografts in Mice
2.11. Data Analysis
3. Results
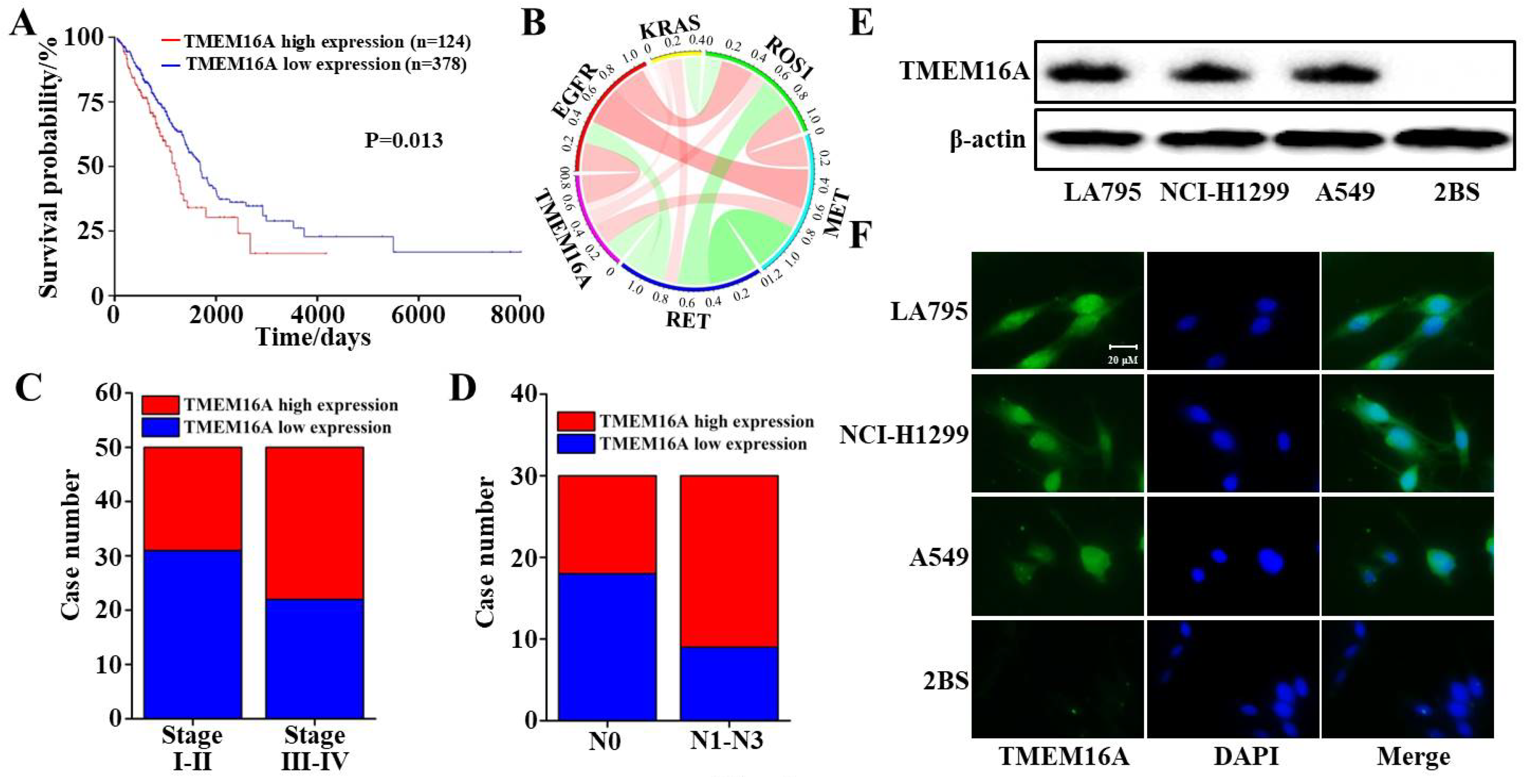
3.1. TMEM16A Is Highly Expressed in Lung Adenocarcinoma Cells
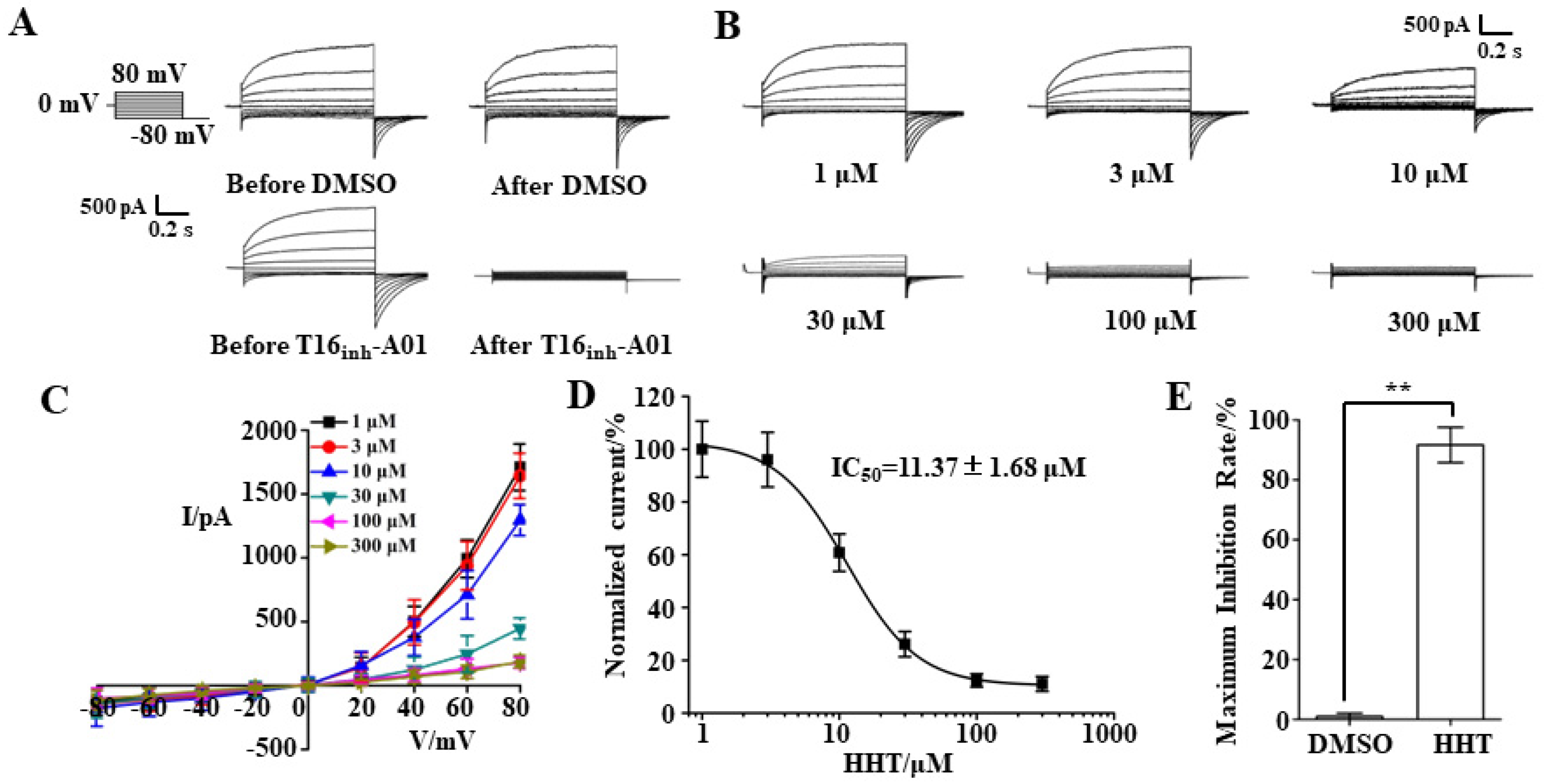
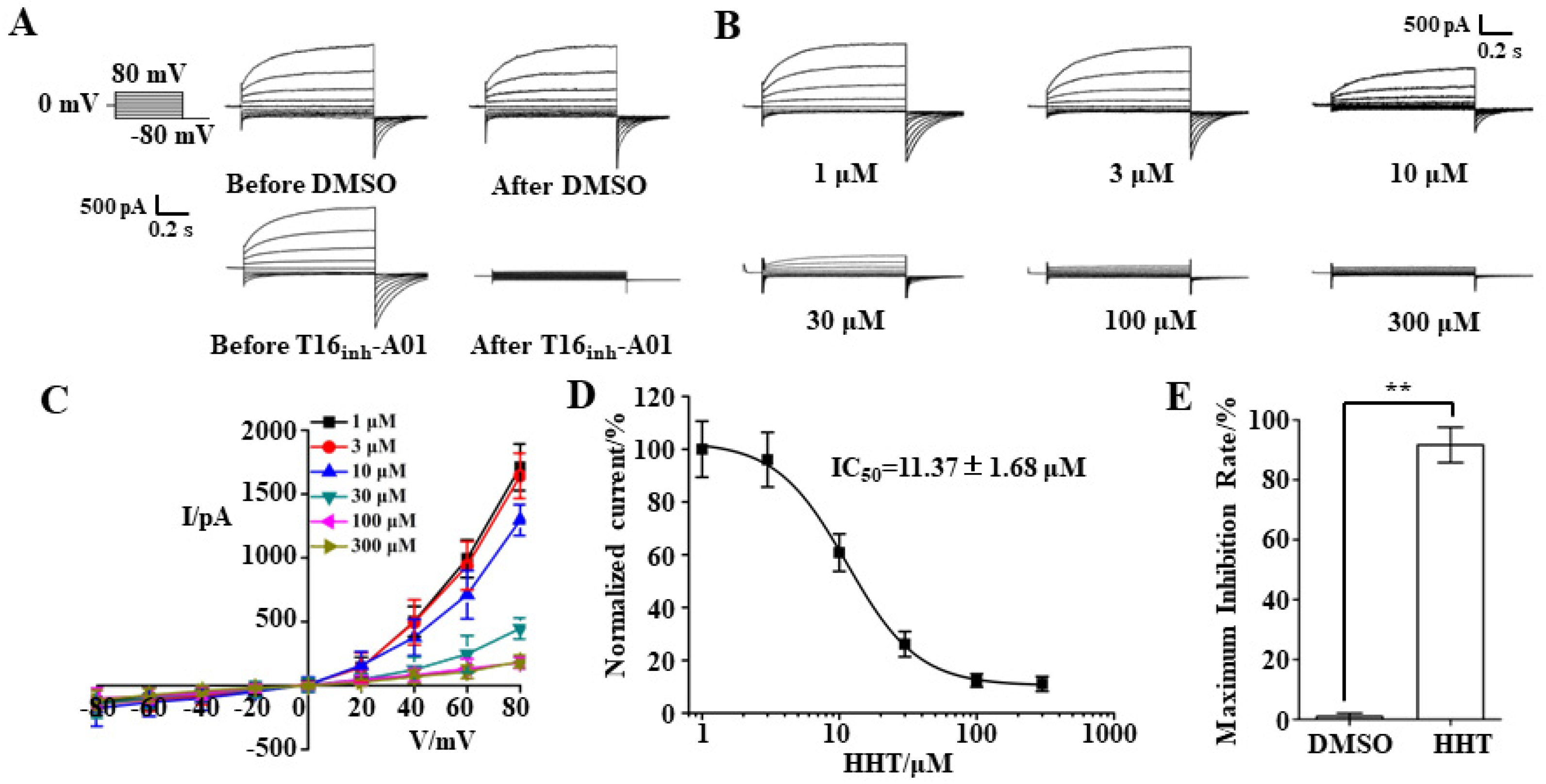
3.2. TMEM16A Currents Are Inhibited by HHT in a Concentration-Dependent Manner
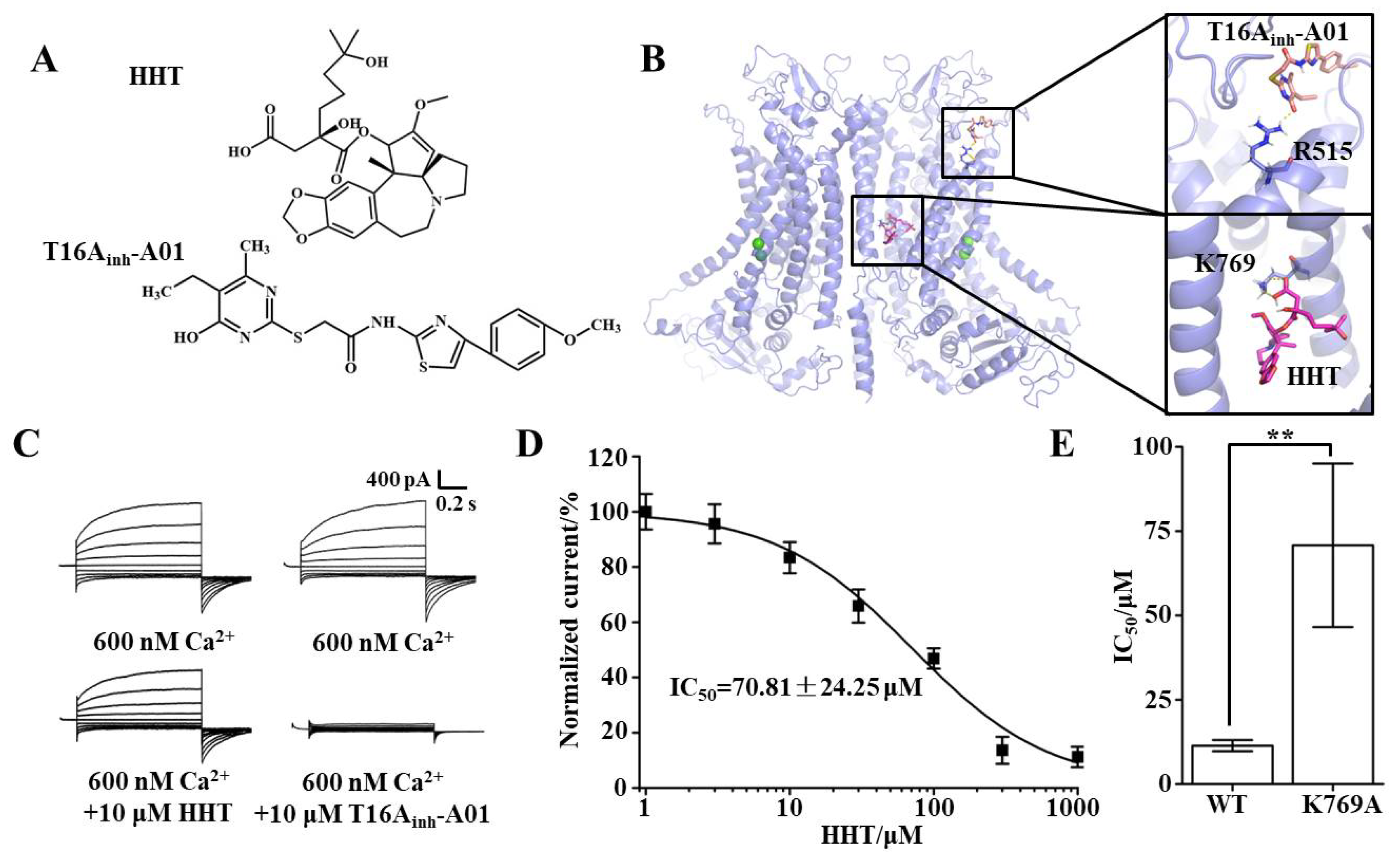
3.3. Key Binding Site of HHT and TMEM16A
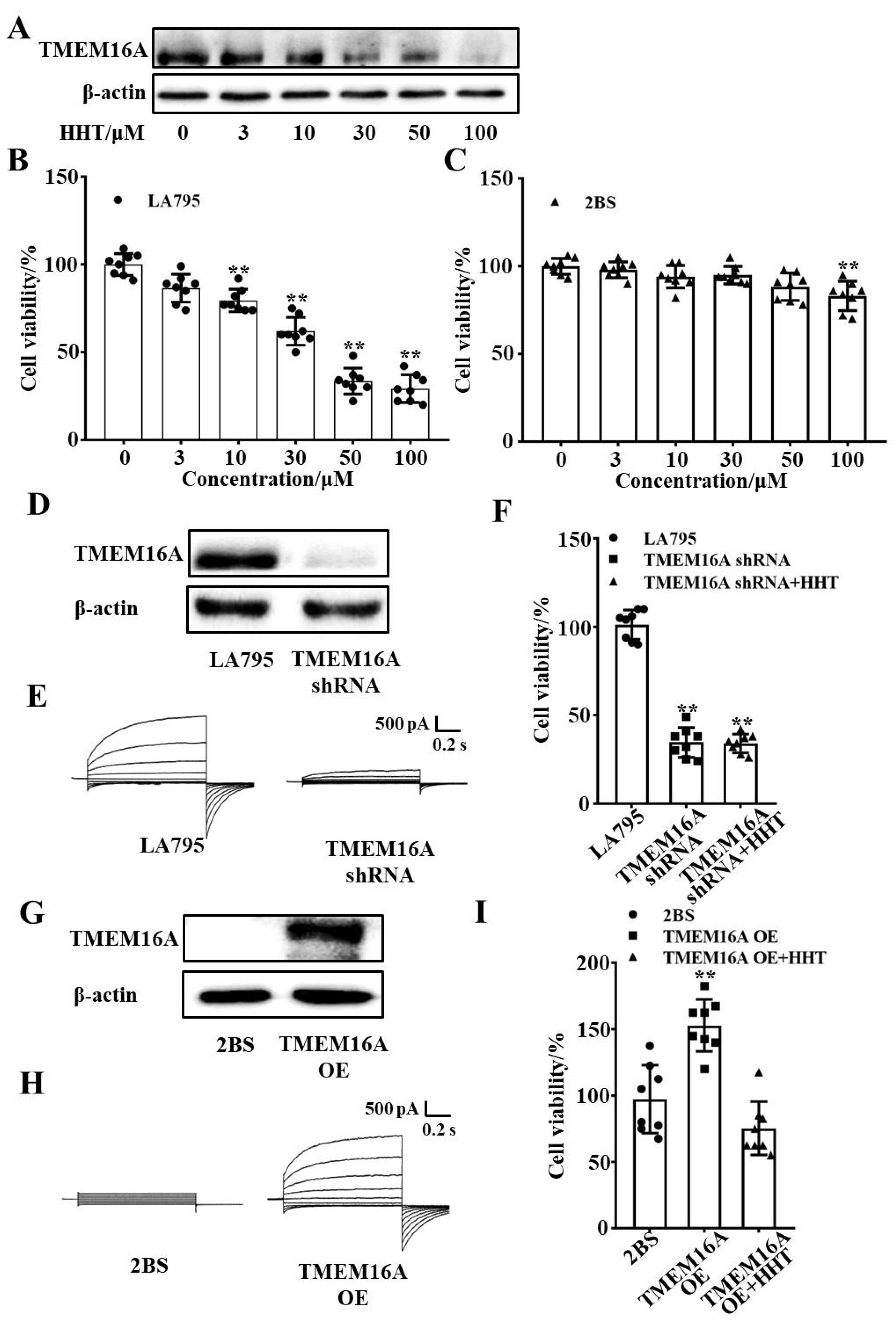
3.4. TMEM16A Is a Potential Drug Target of HHT That Inhibits Lung Cancer Cell Proliferation
3.5. TMEM16A Is a Potential Drug Target of HHT That Inhibits Lung Cancer Cell Migration
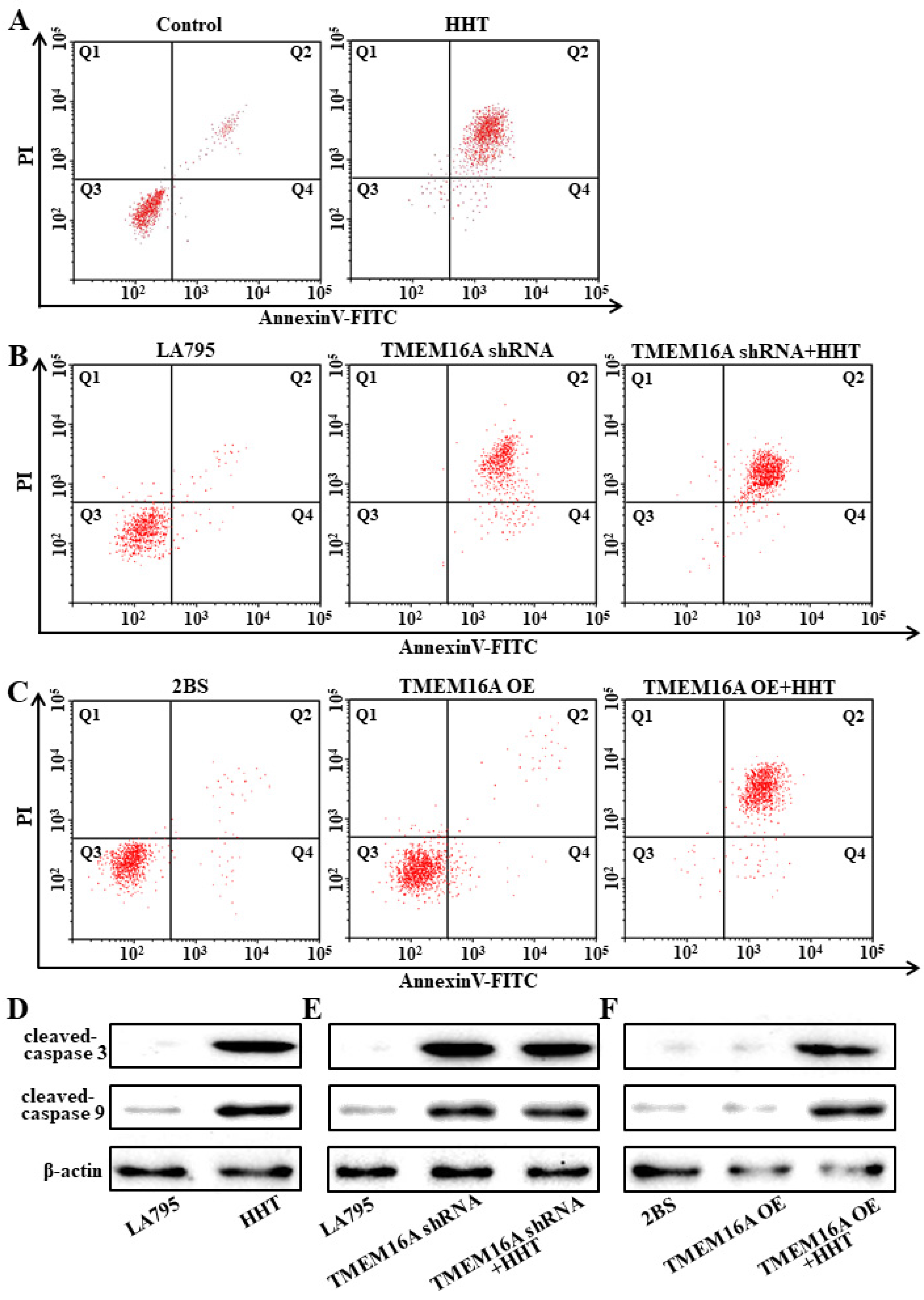
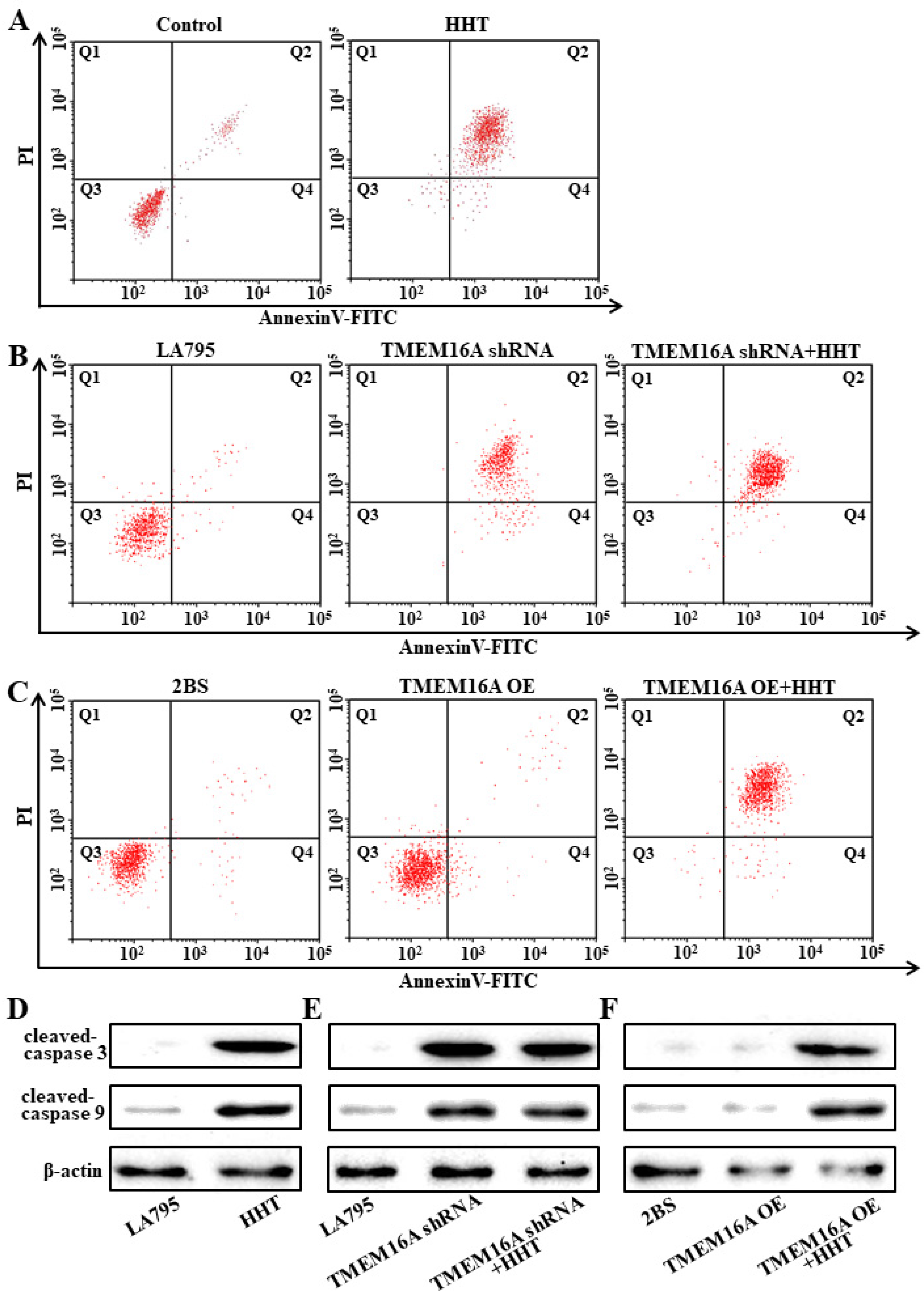
3.6. TMEM16A Is a Drug Target of HHT That Promotes Lung Cancer Cell Apoptosis
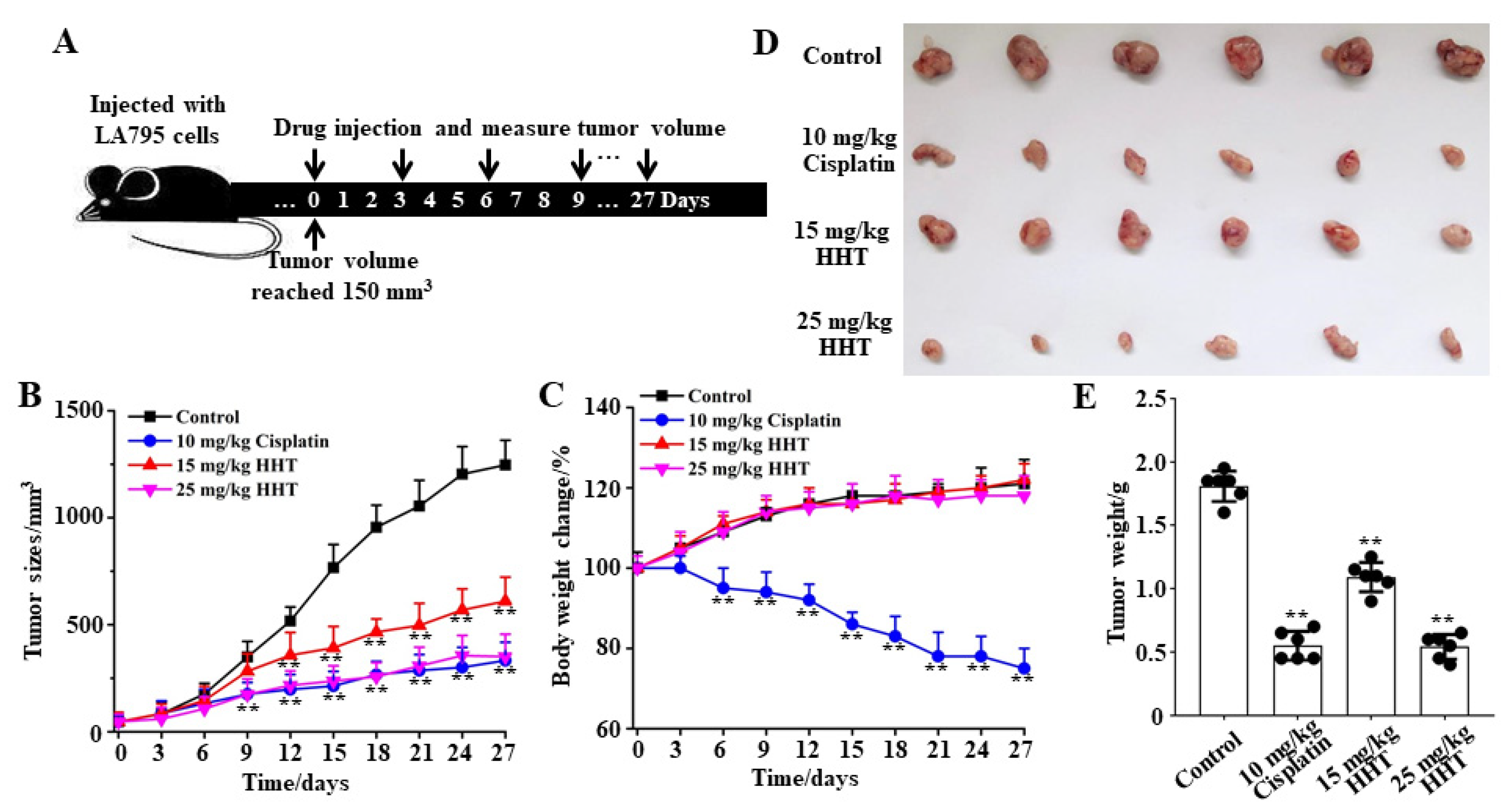
3.7. HHT Inhibits Lung Cancer Growth In Vivo
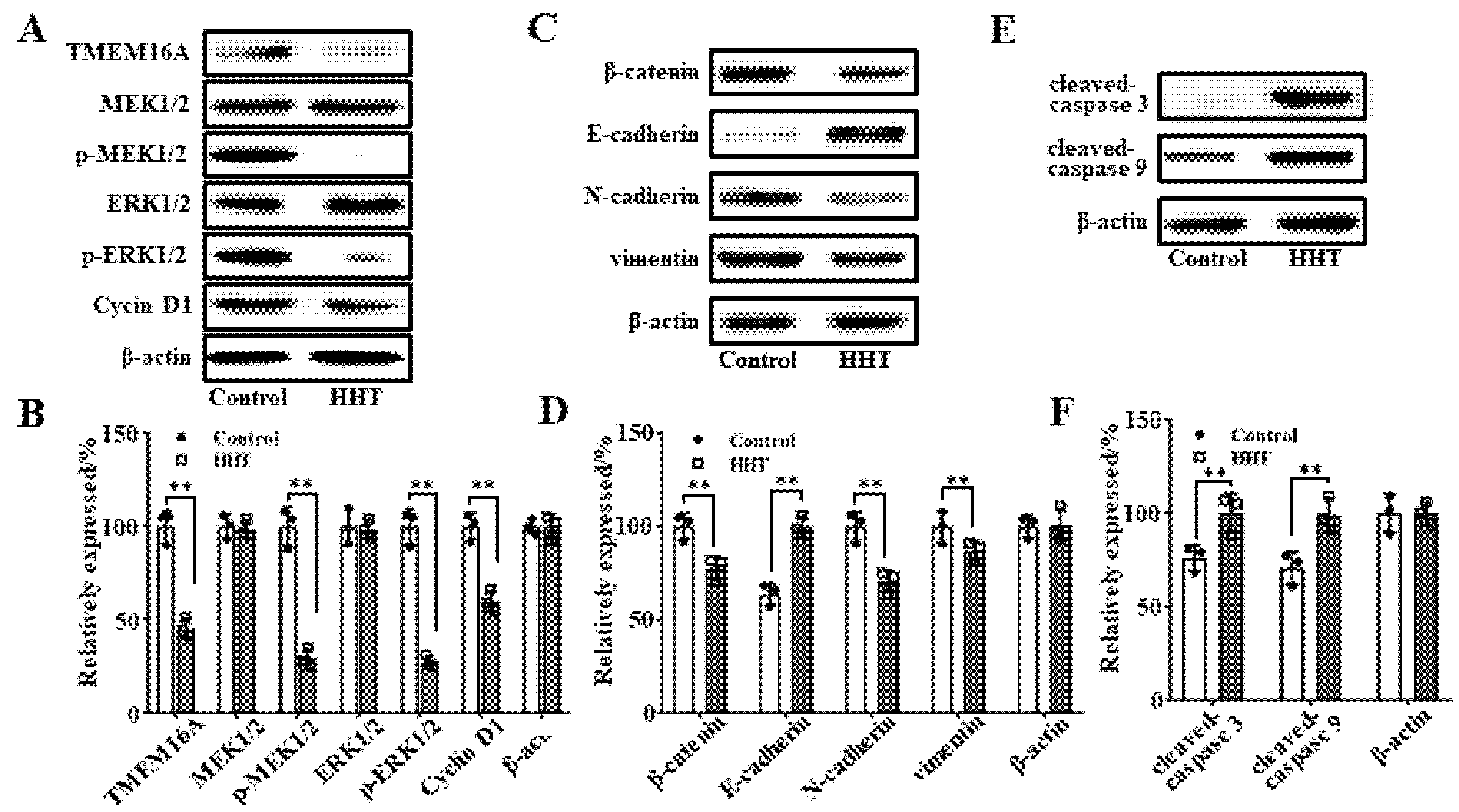
3.8. Molecular Mechanism Underlying HHT-Mediated Inhibition of Tumor Growth In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung Cancer. Med. Clin. N. Am. 2019, 103, 463–473. [Google Scholar] [CrossRef]
- Li, T.; Pan, K.; Ellinwood, A.K.; Cress, R.D. Survival Trends of Metastatic Lung Cancer in California by Age at Diagnosis, Gender, Race/Ethnicity, and Histology, 1990–2014. Clin. Lung Cancer 2020, 22, e602–e611. [Google Scholar] [CrossRef]
- He, S.; Li, H.; Cao, M.; Sun, D.; Lei, L.; Li, N.; Peng, J.; Chen, W. Trends and risk factors of lung cancer in China. Chin. J. Cancer Res. 2020, 32, 683–694. [Google Scholar] [CrossRef]
- Bernard, A.; Pages, P.B.; Mariet, A.S.; Pforr, A.; Cottenet, J.; Quantin, C. Evaluation of surgical practice in the treatment of lung cancer in France from the PMSI national database. Rev. Mal. Respir. 2019, 36, 31–38. [Google Scholar] [CrossRef]
- Hoy, H.; Lynch, T.; Beck, M. Surgical Treatment of Lung Cancer. Crit. Care Nurs. Clin. 2019, 31, 303–313. [Google Scholar] [CrossRef]
- Nagasaka, M.; Gadgeel, S.M. Role of chemotherapy and targeted therapy in early-stage non-small cell lung cancer. Expert Rev. Anticancer Ther. 2018, 18, 63–70. [Google Scholar] [CrossRef]
- Baker, S.; Dahele, M.; Lagerwaard, F.J.; Senan, S. A critical review of recent developments in radiotherapy for non-small cell lung cancer. Radiat. Oncol. 2016, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer epidemiology, biomarkers & prevention: A publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar]
- Tan, W.L.; Jain, A.; Takano, A.; Newell, E.W.; Iyer, N.G.; Lim, W.T.; Tan, E.H.; Zhai, W.; Hillmer, A.M.; Tam, W.L.; et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016, 17, e347–e362. [Google Scholar] [CrossRef]
- Guo, S.; Chen, Y.F.; Shi, S.; Pang, C.L.; Wang, X.Z.; Zhang, H.L.; Zhan, Y.; An, H.L. The Molecular Mechanism of Ginsenoside Analogs Activating TMEM16A. Biophys. J. 2020, 118, 262–272. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Pang, C.; Ren, X.; Li, J.; Wang, X.; Shi, S.; Qi, J.; Zhang, H.; Zhan, Y.; et al. Entering the spotlight: Chitosan oligosaccharides as novel activators of CaCCs/TMEM16A. Pharmacol. Res. 2019, 146, 104323. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, K. Exploiting the Diversity of Ion Channels: Modulation of Ion Channels for Therapeutic Indications. Handbook of experimental pharmacology. In Handbook of Experimental Pharmacology; Springer Nature: Cham, Switzerland, 2019; Volume 260, pp. 187–205. [Google Scholar] [PubMed]
- Guo, S.; Chen, Y.; Pang, C.; Wang, X.; Shi, S.; Zhang, H.; An, H.; Zhan, Y. Matrine is a novel inhibitor of the TMEM16A chloride channel with antilung adenocarcinoma effects. J. Cell Physiol. 2019, 234, 8698–8708. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zou, L.; Ma, K.; Yu, J.; Wu, H.; Wei, M.; Xiao, Q. Cell-specific mechanisms of TMEM16A Ca(2+)-activated chloride channel in cancer. Mol. Cancer 2017, 16, 152. [Google Scholar] [CrossRef] [PubMed]
- Crottes, D.; Jan, L.Y. The multifaceted role of TMEM16A in cancer. Cell Calcium. 2019, 82, 102050. [Google Scholar] [CrossRef]
- Guo, S.; Bai, X.; Liu, Y.F.; Shi, S.; Wang, X.Z.; Zhan, Y.; Kang, X.J.; Chen, Y.F.; An, H.L. Inhibition of TMEM16A by Natural Product Silibinin: Potential Lead Compounds for Treatment of Lung Adenocarcinoma. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Wanitchakool, P.; Wolf, L.; Koehl, G.E.; Sirianant, L.; Schreiber, R.; Kulkarni, S.; Duvvuri, U.; Kunzelmann, K. Role of Anoctamins in Cancer and Apoptosis. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130096. [Google Scholar] [CrossRef]
- Guo, S.; Chen, Y.; Shi, S.; Wang, X.; Zhang, H.; Zhan, Y.; An, H. Arctigenin, a novel TMEM16A inhibitor for lung adenocarcinoma therapy. Pharmacol. Res. 2020, 155, 104721. [Google Scholar] [CrossRef]
- Oh, U.; Jung, J. Cellular functions of TMEM16/anoctamin. Pflug. Arch. Eur. J. Physiol. 2016, 468, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Li, W.; Yuan, M.; Li, C.; Liu, S.; Jiang, C.; Wu, Y.; Cai, K.; Liu, Y. Homoharringtonine production by endophytic fungus isolated from Cephalotaxus hainanensis Li. World J. Microbiol. Biotechnol. 2016, 32, 110. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Kantarjian, H.; Cortes, J. Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009. Cancer 2009, 115, 5382–5393. [Google Scholar] [CrossRef]
- Li, C.; Dong, L.; Su, R.; Bi, Y.; Qing, Y.; Deng, X.; Zhou, Y.; Hu, C.; Yu, M.; Huang, H.; et al. Homoharringtonine exhibits potent anti-tumor effect and modulates DNA epigenome in acute myeloid leukemia by targeting SP1/TET1/5hmC. Haematologica 2020, 105, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Zhang, Q.; Yuan, X.; Chen, Y.; Wu, Y. Synergistic killing effects of homoharringtonine and arsenic trioxide on acute myeloid leukemia stem cells and the underlying mechanisms. J. Exp. Clin. Cancer Res. CR 2019, 38, 308. [Google Scholar] [CrossRef] [Green Version]
- Pal, I.; Safari, M.; Jovanovic, M.; Bates, S.E.; Deng, C. Targeting Translation of mRNA as a Therapeutic Strategy in Cancer. Curr. Hematol. Malig. Rep. 2019, 14, 219–227. [Google Scholar] [CrossRef]
- Wang, L.B.; Wang, D.N.; Wu, L.G.; Cao, J.; Tian, J.H.; Liu, R.; Ma, R.; Yu, J.J.; Wang, J.; Huang, Q.; et al. Homoharringtonine inhibited breast cancer cells growth via miR-18a-3p/AKT/mTOR signaling pathway. Int. J. Biol. Sci. 2021, 17, 995–1009. [Google Scholar] [CrossRef]
- Wang, F.F.; Huang, J.C.; Guo, T.T.; Zheng, Y.H.; Zhang, L.; Zhang, D.; Wang, F.J.; Naren, D.L.; Cui, Y.S.; Liu, X.Y.; et al. Homoharringtonine synergizes with quizartinib in FLT3-ITD acute myeloid leukemia by targeting FLT3-AKT-c-Myc pathway. Biochem. Pharmacol. 2021, 188, 114538. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Chen, Y.; Pang, C.; Wang, X.; Qi, J.; Mo, L.; Zhang, H.; An, H.; Zhan, Y. Ginsenoside Rb1, a novel activator of the TMEM16A chloride channel, augments the contraction of guinea pig ileum. Pflug. Arch. Eur. J. Physiol. 2017, 469, 681–692. [Google Scholar] [CrossRef]
- Paulino, C.; Kalienkova, V.; Lam, A.K.M.; Neldner, Y.; Dutzler, R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 2017, 552, 421–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling Protein Tertiary and Quaternary Structure Using Evolutionary Information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Na, W.J.; Ma, B.; Shi, S.; Chen, Y.F.; Zhang, H.L.; Zhan, Y.; An, H.L. Procyanidin B1, a novel and specific inhibitor of Kv10.1 channel, suppresses the evolution of hepatoma. Biochem. Pharmacol. 2020, 178, 114089. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Qiu, L.; Chen, Y.; Wang, X.; Ma, B.; Qu, C.; Cui, J.; Zhang, H.; Xing, C.; Zhan, Y.; et al. TMEM16A-inhibitor loaded pH-responsive nanoparticles: A novel dual-targeting antitumor therapy for lung adenocarcinoma. Biochem. Pharmacol. 2020, 178, 114062. [Google Scholar] [CrossRef]
- Ji, Q.; Guo, S.; Wang, X.; Pang, C.; Zhan, Y.; Chen, Y.; An, H. Recent advances in TMEM16A: Structure, function, and disease. J. Cell. Physiol. 2019, 234, 7856–7873. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Han, Z.; Cui, X.; Peng, D.; Xing, Y. Inhibition of TMEM16A suppresses growth and induces apoptosis in hepatocellular carcinoma. Int. J. Clin. Oncol. 2020, 25, 1145–1154. [Google Scholar] [CrossRef]
- Tang, J.F.; Li, G.L.; Zhang, T.; Du, Y.M.; Huang, S.Y.; Ran, J.H.; Li, J.; Chen, D.L. Homoharringtonine inhibits melanoma cells proliferation in vitro and vivo by inducing DNA damage, apoptosis, and G2/M cell cycle arrest. Arch. Biochem. Biophys. 2021, 700, 108774. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Wu, J.; Wei, B.; Shi, Y.; Lu, X.; Ding, Y.; Wang, C.; Li, Y. Homoharringtonine enhances the effect of imatinib on chronic myelogenous leukemia cells by downregulating ZFX. Mol. Med. Rep. 2019, 20, 3233–3239. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ding, W.; Ding, Y.; Ma, J.; Qian, Z.; Shao, J.; Li, Y. Homoharringtonine suppresses imatinib resistance via the Bcl-6/p53 pathway in chronic myeloid leukemia cell lines. Oncotarget 2017, 8, 37594–37604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.Y.; Tang, J.Y.; Wang, Y.P. Homoharringtonine: A new treatment option for myeloid leukemia. Hematology 2004, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhu, M.; Gong, Z.; Yang, T.; Yu, R.; Wang, J.; Zhang, Y. Homoharringtonine suppresses LoVo cell growth by inhibiting EphB4 and the PI3K/AKT and MAPK/EKR1/2 signaling pathways. Food Chem. Toxicol. 2020, 136, 110960. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, Y.; Chen, J.; Chen, R.; Gu, L.; Xue, H.; Pan, C.; Tang, J.; Shen, S. Homoharringtonine is a safe and effective substitute for anthracyclines in children younger than 2 years old with acute myeloid leukemia. Front. Med. 2019, 13, 378–387. [Google Scholar] [CrossRef]
- Lichota, A.; Gwozdzinski, K. Anticancer Activity of Natural Compounds from Plant and Marine Environment. Int. J. Mol. Sci. 2018, 19, 3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.G.; Nam, H.G.; Kim, J.H.; Mun, S. Optimal Design of a Four-Zone Simulated Moving Bed Process for Separation of Homoharringtonine and Harringtonine. Can. J. Chem. Eng. 2011, 89, 304–313. [Google Scholar] [CrossRef]
- Kim, W.K.; Chae, H.J.; Kim, J.H. Microwave-assisted Extraction of Homoharringtonine from Cephalotaxus koreana. Biotechnol. Bioproc. Eng. 2010, 15, 481–487. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, R.G.; Jiang, D.P. TMEM16A as a Potential Biomarker in the Diagnosis and Prognosis of Lung Cancer. Arch. Iran. Med. 2019, 22, 32–38. [Google Scholar] [PubMed]
- Qu, F.; Zhou, Y.; Yu, W. A review of research progress on mechanisms and overcoming strategies of acquired osimertinib resistance. Anti-Cancer Drugs 2021. [Google Scholar] [CrossRef]
- Koopman, B.; Groen, H.J.M.; Schuuring, E.; Hiltermann, T.J.N.; Timens, W.; den Dunnen, W.F.A.; van den Berg, A.; Ter Elst, A.; van Kruchten, M.; Kluiver, J.L.; et al. Actionability of on-target ALK Resistance Mutations in Patients with Non-Small Cell Lung Cancer: Local Experience and Review of the Literature. Clin. Lung Cancer 2021, 31, S1525–S7304. [Google Scholar]
- Shi, Y.; Ye, J.; Yang, Y.; Zhao, Y.; Shen, H.; Ye, X.; Xie, W. The Basic Research of the Combinatorial Therapy of ABT-199 and Homoharringtonine on Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 692497. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, S.; Bai, X.; Shi, S.; Deng, Y.; Kang, X.; An, H. TMEM16A, a Homoharringtonine Receptor, as a Potential Endogenic Target for Lung Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 10930. https://doi.org/10.3390/ijms222010930
Guo S, Bai X, Shi S, Deng Y, Kang X, An H. TMEM16A, a Homoharringtonine Receptor, as a Potential Endogenic Target for Lung Cancer Treatment. International Journal of Molecular Sciences. 2021; 22(20):10930. https://doi.org/10.3390/ijms222010930
Chicago/Turabian StyleGuo, Shuai, Xue Bai, Sai Shi, Yawen Deng, Xianjiang Kang, and Hailong An. 2021. "TMEM16A, a Homoharringtonine Receptor, as a Potential Endogenic Target for Lung Cancer Treatment" International Journal of Molecular Sciences 22, no. 20: 10930. https://doi.org/10.3390/ijms222010930
APA StyleGuo, S., Bai, X., Shi, S., Deng, Y., Kang, X., & An, H. (2021). TMEM16A, a Homoharringtonine Receptor, as a Potential Endogenic Target for Lung Cancer Treatment. International Journal of Molecular Sciences, 22(20), 10930. https://doi.org/10.3390/ijms222010930