Impact of SMTP Targeting Plasminogen and Soluble Epoxide Hydrolase on Thrombolysis, Inflammation, and Ischemic Stroke


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Origin of SMTP
2.1. Background: Thrombotic and Thromboembolic Disorders and Treatment
2.2. Search for a Bioactive Compound that Enhances Physiological Thrombolysis
2.3. Discovery of SMTP
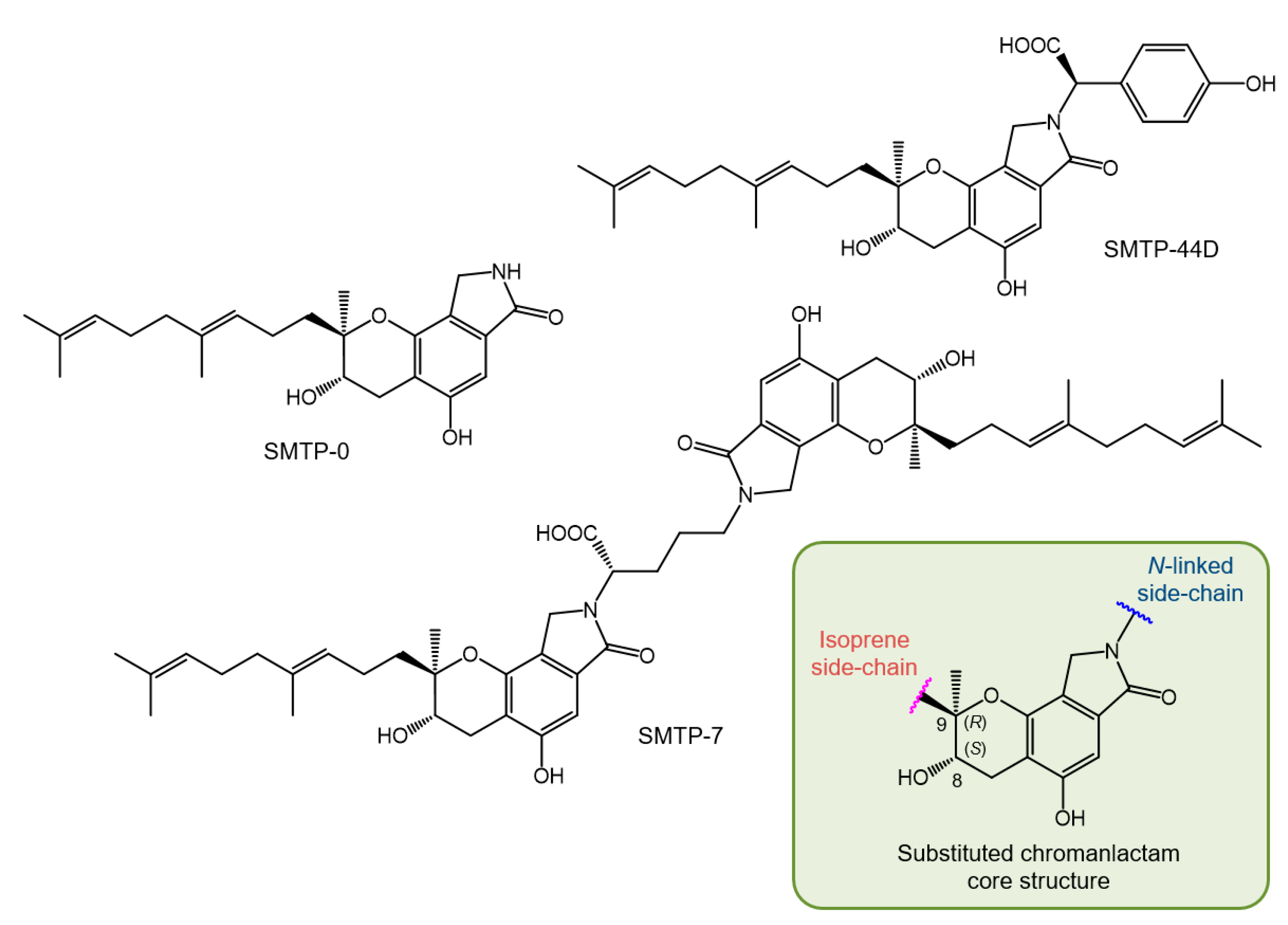
2.4. SMTP Congeners
2.5. Structure of SMTP
2.6. Biosynthesis of SMTP
2.7. Other Triprenyl Phenols
3. Biochemical Actions of SMTP
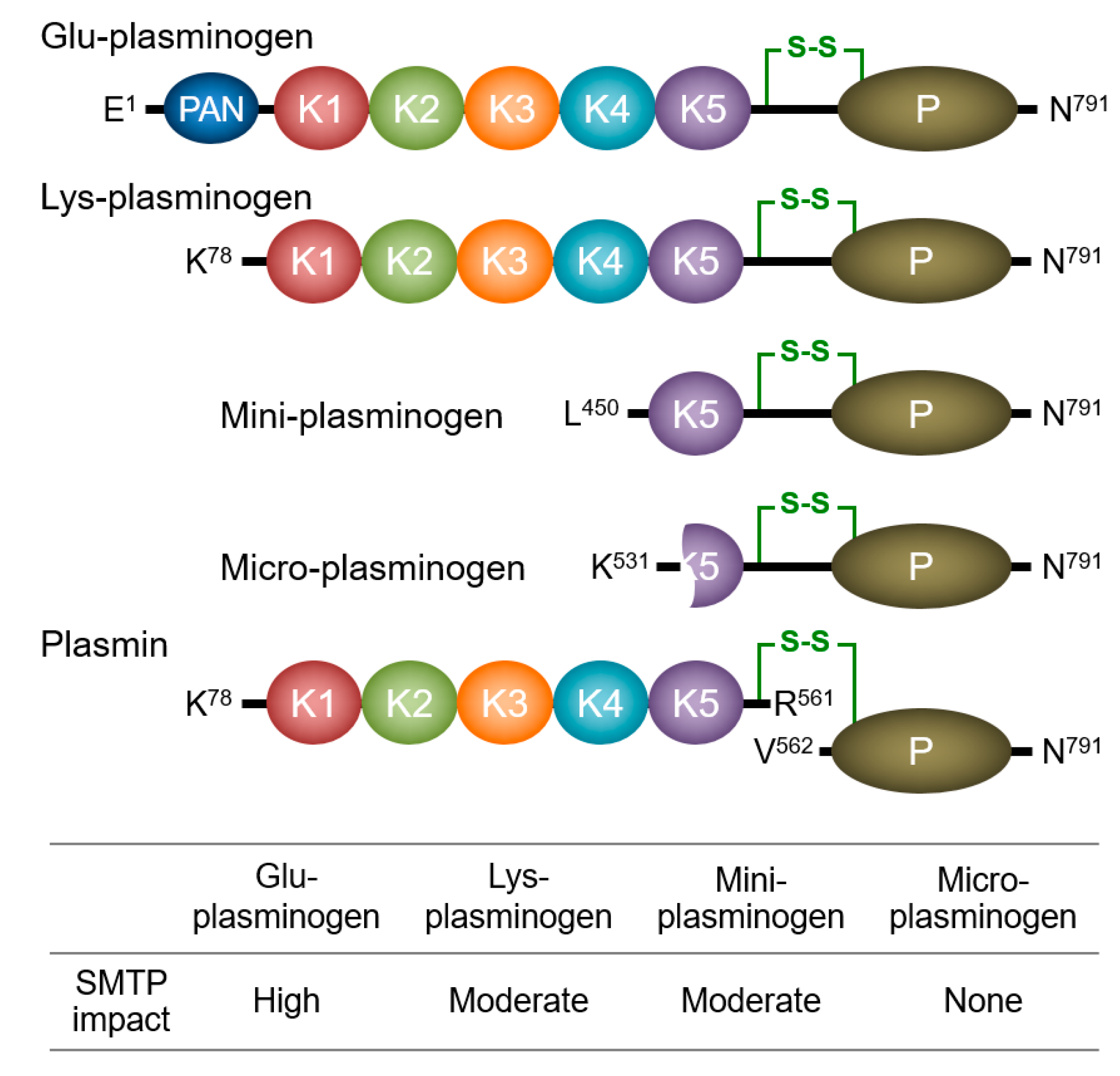
3.1. Enhancement of Plasminogen–Fibrin Binding and Plasminogen Activation
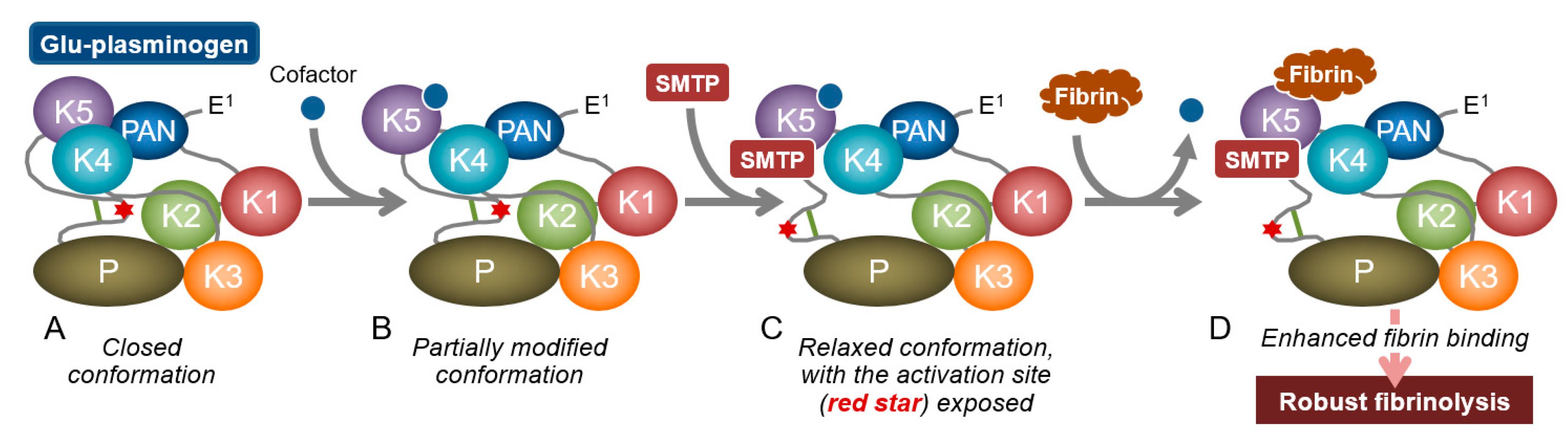
3.2. Modulation of Plasminogen Conformation
3.3. Concept of Zymogen Modulation
3.4. Unexpected Anti-Inflammatory Action of SMTP
3.5. Soluble Epoxide Hydrolase as an Anti-Inflammatory Target of SMTP
3.6. Antioxidative Action
3.7. Structure–Activity Relationship
4. Pharmacological Activity of SMTP
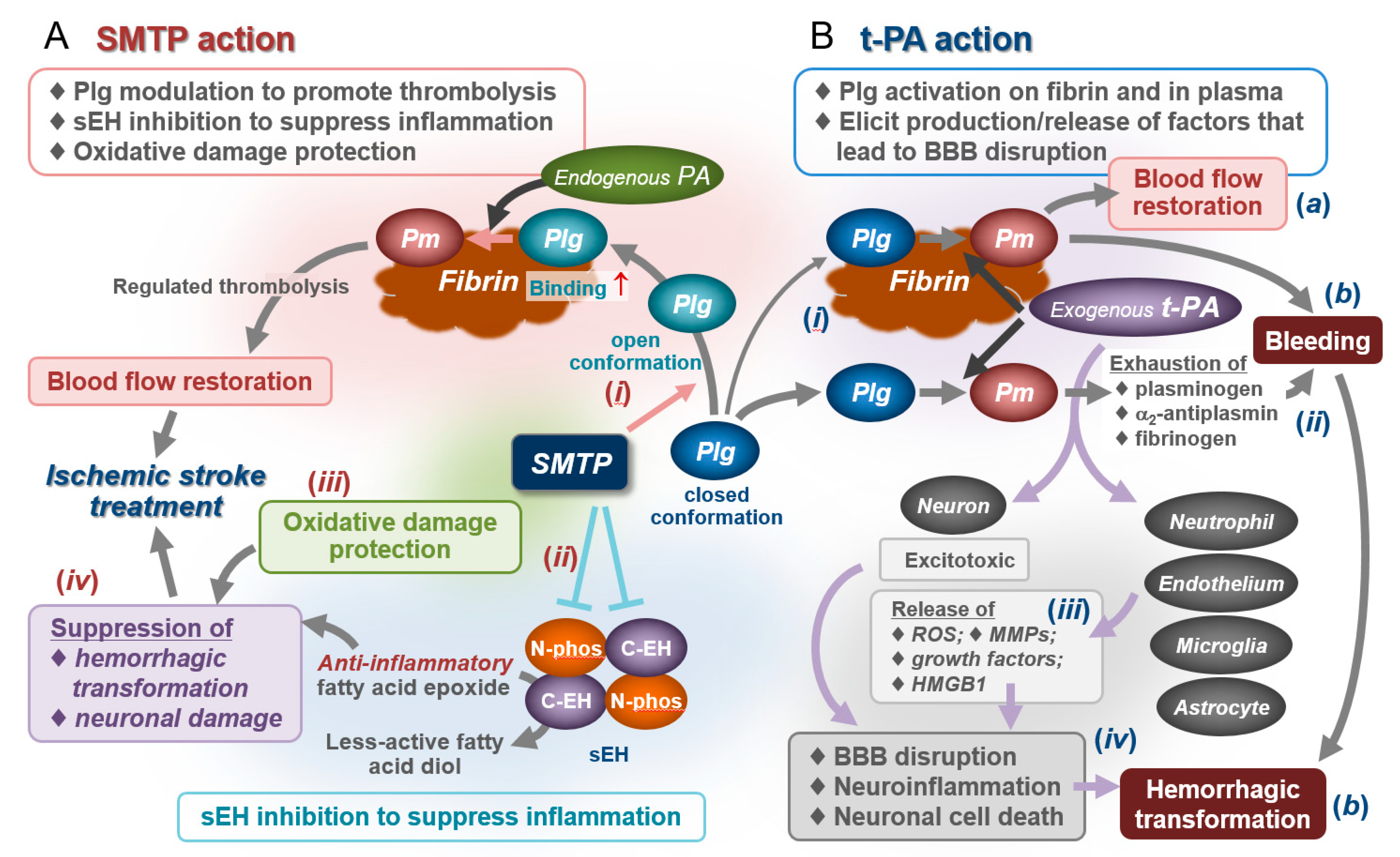
4.1. Ischemic Stroke and Treatment Strategy
4.2. Efficacy in Embolic Stroke Models
4.3. Efficacy in Thrombotic Stroke Models
4.4. Efficacy in Mechanical Cerebral Ischemia Models
4.5. Effects on Bleeding and Hemorrhagic Transformation: Roles for Anti-Inflammatory/Antioxidative Actions
5. Development of SMTP
5.1. Chemistry, Manufacturing, and Controls
5.2. Nonclinical Studies
5.3. Phase 1 Clinical Study
5.4. Phase 2 Clinical Study
6. Conclusions and Perspectives
7. Patents
- Hasumi, K.; Hu, W.; Ohyama, S.; Narasaki, R. Method for Selectively Producing Triprenyl Phenol Compound and Utilization of the Same Compound as Medicine. JP2000306840, filed 31 August 2000.
- Hasumi, K.; Ohyama, S.; Harada, T.; Hu, W. Plasminogen Fragment Having Neovascularization-Inhibiting Activity and Method for Searching Compound Which Induce Production of the Plasminogen Fragment. JP2001328411, filed 19 September 2001.
- Hasumi, K.; Hu, W. New Triprenylphenol Compound. JP2003015279, filed 23 January 2003.
- Hasumi, K.; Hu, W. Medical Composition for Preventing and Treating Angiogenesis-associated Disease. JP2003015279, filed 23 January 2003.
- Hasumi, K.; Maeda, F.; Mitsumori, K. Agent for Improvement of Hepatic Function. PCT/JP2006/302796, filed 17 February 2006.
- Hasumi, K.; Yagasaki, K. Pharmaceutical Composition for Treatment or Prevention of Nephritis and Method for Producing Same. PCT/JP2006/318972, filed 25 September 2006.
- Hasumi; K.; Nomura, Y.; Narasaki, R.; Tometsuka, C. Skin Aging Inhibitor, Cosmetic and Skin Care Preparation. JP2007036638, filed 16 February 2007.
- Hasumi, K.; Kitano, Y.; Oish, H.; Koide, H.; Hasegawa, K.; Narasaki, R. Triprenyl Phenol Compound, Process for Production of Triprenyl Phenol Compound, and Thrombolysis Enhancer. PCT/JP2007/055749, filed 20 March 2007.
- Hasumi, K.; Koide, H.; Narasaki, R. Triprenylphenol Compound and Thrombus Dissolution Accelerator. JP2008060249, filed 10 March 2008.
- Hasumi, K.; Koide, H.; Narasaki, R. Triprenylphenol Compound and Thrombus Dissolution Accelerator. JP2008060250, filed 10 March 2008.
- Hasumi, K.; Koide, H.; Hasegawa, K.; Nishimura, N. Antioxidant. JP2010019735, filed 29 January 2010.
- Honda, K.; Hashimoto, T.; Shibata, K.; Hasegawa, K.; Hasumi, K. Cytoprotective Agent. PCT/JP2010/051711, filed 5 February 2010.
- Hasumi, K.; Ishikawa, M.; Chikanishi, T.; Nishimura, N.; Hasegawa, K. Pharmaceutical Composition for Metabolic Syndrome, Obesity, Hyperglycemia, Hyperlipidemia and/or Fatty Liver. PCT/JP2010/053545, filed 4 April 2010.
- Ishikawa, M.; Tanaka, I.; Shirafuji, T.; Hasumi, K. Prophylactic or Therapeutic Agent for Inflammatory Bowel Diseases or Autoimmune Peripheral Neuropathy. PCT/JP2011/058405, filed 1 April 2011.
- Hasumi, K.; Suzuki, E.; Ogawa, N.; Otake, S.; Kitano, Y.; Hasegawa, K.; Nishimura; N. Soluble Epoxide Hydrolase Inhibitor. PCT/JP2012/054472, filed 23 February 2012.
- Hasumi, K.; Suzuki, E.; Nishimura, Y.; Kitano, Y.; Hasegawa, K.; Nishimura; N.; Tsujihara, K. Chroman Derivative. PCT/JP2013/055729, filed 1 March 2013.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUDA | 12-(3-adamantan-1-yl-ureido) dodecanoic acid |
| BBB | blood–brain barrier |
| DHET | dihydroxyeicosatrienoic acid |
| C-EH | epoxide hydrolase of the C-terminal domain of soluble epoxide hydrolase |
| EET | epoxyeicosatrienoic acid |
| LDL | low-density lipoprotein |
| MMP | matrix metalloproteinase |
| N-phos | phosphatase of the N-terminal domain of soluble epoxide hydrolase |
| PA | plasminogen activator |
| PAI-1 | plasminogen activator inhibitor-1 |
| PAN | Plasminogen–apple–nematode |
| ROS | reactive oxygen species |
| scu-PA | single-chain urokinase-type plasminogen activator |
| sEH | soluble epoxide hydrolase |
| t-PA | tissue-type plasminogen activator |
| u-PA | urokinase-type plasminogen activator |
References
- Versteeg, H.H.; Heemskerk, J.W.M.; Levi, M.; Reitsma, P.H. New Fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How it all starts: Initiation of the clotting cascade. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, K.; Yamamichi, S.; Harada, T. Small-molecule modulators of zymogen activation in the fibrinolytic and coagulation systems: Review Article. FEBS J. 2010, 277. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Castellino, F.J.; Suzuki, Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 2018, 16, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Garagiola, I.; Biguzzi, E. Advances in the treatment of bleeding disorders. J. Thromb. Haemost. 2016, 14, 2095–2106. [Google Scholar] [CrossRef]
- Kolev, K.; Longstaff, C. Bleeding related to disturbed fibrinolysis. Br. J. Haematol. 2016, 175, 12–23. [Google Scholar] [CrossRef]
- Chang, Y.; Dabiri, G.; Damstetter, E.; Baiyee Ebot, E.; Powers, J.G.; Phillips, T. Coagulation disorders and their cutaneous presentations: Pathophysiology. J. Am. Acad. Dermatol. 2016, 74, 783–792. [Google Scholar] [CrossRef]
- Hoffman, M.; Pawlinski, R. Hemostasis: Old system, new players, new directions. Thromb. Res. 2014, 133. [Google Scholar] [CrossRef]
- Mega, J.L.; Simon, T. Pharmacology of antithrombotic drugs: An assessment of oral antiplatelet and anticoagulant treatments. Lancet 2015, 386, 281–291. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Peter, K. Novel Antithrombotic Drugs on the Horizon. Circ. Res. 2017, 121, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Bivard, A.; Lin, L.; Parsonsb, M.W. Review of Stroke Thrombolytics. J. Stroke 2013, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Sobolewski, K.; Bridgeman, P.; Boutsikaris, D. Systemic thrombolysis for pulmonary embolism: A review. Pharm. Ther. 2016, 41, 770–775. [Google Scholar]
- Ibrahim, H.; Rondina, M.; Welt, F.G.P. Antithrombotic drugs in cardiovascular medicine: A year in review. Curr. Opin. Cardiol. 2018, 33, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.N.; Marrero, M.; Masrur, S.; Pervez, M.; Barrocas, A.M.; Abdullah, A.; Oleinik, A.; Rosand, J.; Smith, E.E.; Dzik, W.H.; et al. Management of thrombolysis-associated symptomatic intracerebral hemorrhage. Arch. Neurol. 2010, 67, 965–969. [Google Scholar] [CrossRef]
- Piran, S.; Schulman, S. Treatment of bleeding complications in patients on anticoagulant therapy. Blood 2019, 133, 425–435. [Google Scholar] [CrossRef]
- De Andrade, N.K.; Motta, R.H.L.; Bergamaschi, C.D.C.; Oliveira, L.B.; Guimarães, C.C.; Araújo, J.D.O.; Lopes, L.C. Bleeding risk in patients using oral anticoagulants undergoing surgical procedures in dentistry: A systematic review and meta-analysis. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Macman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Hu, W.; Narasaki, R.; Nishimura, N.; Hasumi, K. SMTP (Stachybotrys microspora triprenyl phenol) enhances clot clearance in a pulmonary embolism model in rats. Thromb. J. 2012, 10. [Google Scholar] [CrossRef]
- Matsumoto, N.; Suzuki, E.; Tsujihara, K.; Nishimura, Y.; Hasumi, K. Structure-activity relationships of the plasminogen modulator SMTP with respect to the inhibition of soluble epoxide hydrolase. J. Antibiot. (Tokyo) 2015, 68, 685–690. [Google Scholar] [CrossRef]
- Shibata, K.; Hashimoto, T.; Nobe, K.; Hasumi, K.; Honda, K. A novel finding of a low-molecular-weight compound, SMTP-7, having thrombolytic and anti-inflammatory effects in cerebral infarction of mice. Naunyn. Schmiedebergs. Arch. Pharmacol. 2010, 382. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Nishimura, N.; Suzuki, E.; Zhuang, J.; Hasegawa, K.; Takamatsu, H.; Honda, K.; Hasumi, K. SMTP-7, a novel small-molecule thrombolytic for ischemic stroke: A study in rodents and primates. J. Cereb. Blood Flow Metab. 2014, 34. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Niizuma, K.; Shimizu, H.; Fujimura, M.; Hasumi, K.; Tominaga, T. SMTP-7, a new thrombolytic agent, decreases hemorrhagic transformation after transient middle cerebral artery occlusion under warfarin anticoagulation in mice. Brain Res. 2014, 1578. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ohta, Y.; Shang, J.; Li, X.; Liu, X.; Shi, X.; Feng, T.; Yamashita, T.; Sato, K.; Takemoto, M.; et al. Reduction of Ischemia Reperfusion-Related Brain Hemorrhage by Stachybotrys Microspora Triprenyl Phenol-7 in Mice With Antioxidant Effects. J. Stroke Cerebrovasc. Dis. 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Suzuki, E.; Ishikawa, M.; Shirafuji, T.; Hasumi, K. Soluble epoxide hydrolase as an anti-inflammatory target of the thrombolytic stroke drug SMTP-7. J. Biol. Chem. 2014, 289, 35826–35838. [Google Scholar] [CrossRef]
- Hashimoto, T.; Shibata, K.; Nobe, K.; Hasumi, K.; Honda, K. A novel embolic model of cerebral infarction and evaluation of Stachybotrys microspora triprenyl phenol-7 (SMTP-7), a novel fungal triprenyl phenol metabolite. J. Pharmacol. Sci. 2010, 114. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Saito, A.; Fujimura, M.; Shimizu, H.; Mekawy, M.; Hasumi, K.; Tominaga, T. Stachybotrys microspora triprenyl phenol-7, a novel fibrinolytic agent, suppresses superoxide production, matrix metalloproteinase-9 expression, and thereby attenuates ischemia/reperfusion injury in rat brain. Neurosci. Lett. 2011, 503. [Google Scholar] [CrossRef]
- Suzuki, E.; Nishimura, N.; Yoshikawa, T.; Kunikiyo, Y.; Hasegawa, K.; Hasumi, K. Efficacy of SMTP-7, a small-molecule anti-inflammatory thrombolytic, in embolic stroke in monkeys. Pharmacol. Res. Perspect. 2018, 6. [Google Scholar] [CrossRef]
- Shibata, K.; Hashimoto, T.; Hasumi, K.; Honda, K.; Nobe, K. Evaluation of the effects of a new series of SMTPs in the acetic acid-induced embolic cerebral infarct mouse model. Eur. J. Pharmacol. 2018, 818. [Google Scholar] [CrossRef]
- Shi, X.; Ohta, Y.; Shang, J.; Morihara, R.; Nakano, Y.; Fukui, Y.; Liu, X.; Feng, T.; Huang, Y.; Sato, K.; et al. Neuroprotective effects of SMTP-44D in mice stroke model in relation to neurovascular unit and trophic coupling. J. Neurosci. Res. 2018, 96. [Google Scholar] [CrossRef]
- Cushman, M. Epidemiology and Risk Factors for Venous Thrombosis. Semin. Hematol. 2007, 44, 62–69. [Google Scholar] [CrossRef]
- Previtali, E.; Bucciarelli, P.; Passamonti, S.M.; Martinelli, I. Risk factors for venous and arterial thrombosis. Blood Transfus. 2011, 9, 120–138. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Otsuka, F.; Yasuda, S.; Noguchi, T.; Ishibashi-Ueda, H. Pathology of coronary atherosclerosis and thrombosis. Cardiovasc. Diagn. Ther. 2016, 6, 396–408. [Google Scholar] [CrossRef]
- Rijken, D.C.; Lijnen, H.R. New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Majithia, A.; Bhatt, D.L. Novel Antiplatelet Therapies for Atherothrombotic Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Yamada, N.; Ito, M. Novel Anticoagulant Therapy of Venous Thromboembolism: Current Status and Future Directions. Ann. Vasc. Dis. 2017, 10, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without aspirin in stable cardiovascular disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Steffel, J.; Eikelboom, J.W.; Anand, S.S.; Shestakovska, O.; Yusuf, S.; Fox, K.A.A. The COMPASS Trial. Circulation 2020, 142, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Simpson, J.R.; Silver, B.; Silver, B. Safety of Thrombolysis in Acute Ischemic Stroke: A Review of Complications, Risk Factors, and Newer Technologies. Neurohospitalist 2011, 1, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Demaerschalk, B.M.; Cheng, N.T.; Kim, A.S. Intravenous Thrombolysis for Acute Ischemic Stroke Within 3 Hours Versus Between 3 and 4.5 Hours of Symptom Onset. Neurohospitalist 2015, 5, 101–109. [Google Scholar] [CrossRef]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Hajar, R. Statins: Past and Present. Hear. Views 2011, 12, 121. [Google Scholar] [CrossRef]
- Anglés-Cano, E. Overview on fibrinolysis: Plasminogen activation pathways on fibrin and cell surfaces. Chem. Phys. Lipids 1994, 67–68, 353–362. [Google Scholar] [CrossRef]
- McLean, J.W.; Tomlinson, J.E.; Kuang, W.J.; Eaton, D.L.; Chen, E.Y.; Fless, G.M.; Scanu, A.M.; Lawn, R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987, 330, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A.; Gavishi, D.; Breslow, J.L.; Nachman, R.L. Lipoprotein(a) modulation of endothelial cell surface fibrinolysis and its potential role in atherosclerosis. Nature 1989, 339, 303–305. [Google Scholar] [CrossRef]
- Tachikawa, K.; Hasumi, K.; Endo, A. Enhancement of plasminogen binding to U937 cells and fibrin by complestatin. Thromb. Haemost. 1997, 77. [Google Scholar] [CrossRef]
- Tachikawa, K.; Hasumi, K.; Endo, A. Enhancement of plasminogen binding and fibrinolysis by chloropeptin I. Thromb. Res. 1997, 87. [Google Scholar] [CrossRef]
- Shinohara, C.; Hasumi, K.; Hatsumi, W.; Endo, A. Staplabin, a novel fungal triprenyl phenol which stimulates the binding of plasminogen to fibrin and U937 cells. J. Antibiot. (Tokyo) 1996, 49. [Google Scholar] [CrossRef]
- Kohyama, T.; Hasumi, K.; Hamanaka, A.; Endo, A. SMTP-1 and -2, novel analogs of staplabin produced by Stachybotrys microspora IFO30018. J. Antibiot. (Tokyo) 1997, 50. [Google Scholar] [CrossRef]
- Hasumi, K.; Ohyama, S.; Kohyama, T.; Ohsaki, Y.; Takayasu, R.; Endo, A. Isolation of SMTP-3, 4, 5 and -6, novel analogs of staplabin, and their effects on plasminogen activation and fibrinolysis. J. Antibiot. (Tokyo) 1998, 51. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Ohyama, S.; Hasumi, K. Activation of fibrinolysis by SMTP-7 and -8, novel staplabin analogs with a pseudosymmetric structure. J. Antibiot. (Tokyo) 2000, 53. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Narasaki, R.; Ohyama, S.; Hasumi, K. Selective production of staplabin and SMTPs in cultures of Stachybotrys microspora Fed with precursor amines. J. Antibiot. (Tokyo) 2001, 54. [Google Scholar] [CrossRef]
- Hu, W.; Kitano, Y.; Hasumi, K. SMTP-4D, -5D, -7D and -8D, a new series of the non-lysine-analog plasminogen modulators with a D-amino acid moiety. J. Antibiot. (Tokyo) 2003, 56. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, K.; Hasegawa, K.; Kitano, Y. Isolation and absolute configuration of SMTP-0, a simplest congener of the SMTP family nonlysine-analog plasminogen modulators. J. Antibiot. (Tokyo) 2007, 60. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Koide, H.; Hu, W.; Nishimura, N.; Narasaki, R.; Kitano, Y.; Hasumi, K. Structure-activity relationships of 11 new congeners of the SMTP plasminogen modulator. J. Antibiot. (Tokyo) 2010, 63. [Google Scholar] [CrossRef]
- Koide, H.; Hasegawa, K.; Nishimura, N.; Narasaki, R.; Hasumi, K. A new series of the SMTP plasminogen modulators with a phenylamine-based side chain. J. Antibiot. (Tokyo) 2012, 65. [Google Scholar] [CrossRef]
- Koide, H.; Narasaki, R.; Hasegawa, K.; Nishimura, N.; Hasumi, K. A new series of the SMTP plasminogen modulator with a phenylglycine-based side chain. J. Antibiot. (Tokyo) 2012, 65. [Google Scholar] [CrossRef]
- Nishimura, Y.; Suzuki, E.; Hasegawa, K.; Nishimura, N.; Kitano, Y.; Hasumi, K. Pre-SMTP, a key precursor for the biosynthesis of the SMTP plasminogen modulators. J. Antibiot. (Tokyo) 2012, 65. [Google Scholar] [CrossRef] [PubMed]
- Otake, S.; Ogawa, N.; Kitano, Y.; Hasumi, K.; Suzuki, E. Isoprene side-chain of SMTP is essential for soluble epoxide hydrolase inhibition and cellular localization. Nat. Prod. Commun. 2016, 11. [Google Scholar] [CrossRef]
- Jacolot, M.; Jean, M.; Tumma, N.; Bondon, A.; Chandrasekhar, S.; Van De Weghe, P. Synthesis of stachybotrin C and all of its stereoisomers: Structure revision. J. Org. Chem. 2013, 78, 7169–7175. [Google Scholar] [CrossRef]
- Kuroda, Y.; Hasegawa, K.; Noguchi, K.; Chiba, K.; Hasumi, K.; Kitano, Y. Confirmation of the absolute configuration of Stachybotrin C using single-crystal X-ray diffraction analysis of its 4-bromobenzyl ether derivative. J. Antibiot. (Tokyo) 2018, 71. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Matsuda, Y.; Gao, H.; Hu, D.; Yao, X.S.; Abe, I. Biosynthesis of LL-Z1272β: Discovery of a New Member of NRPS-like Enzymes for Aryl-Aldehyde Formation. ChemBioChem 2016, 17, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Semeiks, J.; Borek, D.; Otwinowski, Z.; Grishin, N.V. Comparative genome sequencing reveals chemotype-specific gene clusters in the toxigenic black mold Stachybotrys. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Fu, Q.; Wu, W.; Cai, M.; Zhou, X.; Zhang, Y. Producing novel fibrinolytic isoindolinone derivatives in marine fungus Stachybotrys longispora FG216 by the rational supply of amino compounds according to its biosynthesis pathway. Mar. Drugs 2017, 15, 214. [Google Scholar] [CrossRef]
- Ayer, W.A.; Miao, S. Secondary metabolites of the aspen fungus Stachybotrys cylindrospora. Can. J. Chem. 1993, 71, 487–493. [Google Scholar] [CrossRef]
- Alajarín, M.; Sánchez-Andrada, P.; López-Leonardo, C.; Álvarez, Á. On the mechanism of phthalimidine formation via o-phthalaldehyde monoimines. New [1,5]-H sigmatropic rearrangements in molecules with the 5-aza-2,4-pentadienal skeleton. J. Org. Chem. 2005, 70, 7617–7623. [Google Scholar] [CrossRef]
- Gyimesi-Forrás, K.; Leitner, A.; Akasaka, K.; Lindner, W. Comparative study on the use of ortho-phthalaldehyde, naphthalene-2,3- dicarboxaldehyde and anthracene-2,3-dicarboxaldehyde reagents for α-amino acids followed by the enantiomer separation of the formed isoindolin-1-one derivatives using quinine-type chir. J. Chromatogr. A 2005, 1083, 80–88. [Google Scholar] [CrossRef]
- Kawano, Y. Development of a Novel PAI-1 Inhibitor Using Beta-SMTP. Master’s Thesis, Tokyo University of Agriculture and Technology, Tokyo, Japan, March 2010. [Google Scholar]
- Takahashi, F.; Hasumi, K.; Endo, A. Modulation of the plasma cholesteryl ester transfer by stachybotramide. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1995, 1258, 70–74. [Google Scholar] [CrossRef]
- Wang, A.; Xu, Y.; Gao, Y.; Huang, Q.; Luo, X.; An, H.; Dong, J. Chemical and bioactive diversities of the genera Stachybotrys and Memnoniella secondary metabolites. Phytochem. Rev. 2015, 14, 623–655. [Google Scholar] [CrossRef]
- Miyazaki, W.; Izawa, T.; Nakano, Y.; Shinohara, M.; Hing, K.; Kinoshita, T.; Inoue, K. Effects of K-76 monocarboxylic acid, an anticomplementary agent, on various in vivo immunological reactions and on experimental glomerulonephritis. Complement 1984, 1, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Sawadjoon, S.; Kittakoop, P.; Isaka, M.; Kirtikara, K.; Madla, S.; Thebtaranonth, Y. Antiviral and antiplasmodial spirodihydrobenzofuran terpenes from the fungus Stachybotrys nephrospora. Planta Med. 2004, 70, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, R.; Dobashi, K.; Kojima, I.; Sakai, K.; Shibamoto, N.; Yoshioka, T.; Nishida, H.; Okamoto, R.; Akagawa, H.; Mizuno, S. Mer-NF5003B, E and F, novel sesquiterpenoids as avian myeloblastosis virus protease inhibitors produced by Stachybotrys sp. J. Antibiot. (Tokyo) 1994, 47, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.K.T.; Wichmann, C.F.; Meinz, M.S.; Guariglia, L.; Glacobbe, R.A.; Mochales, S.; Kong, L.; Honeycutt, S.S.; Zlnk, D.; Bills, G.F.; et al. A novel inositol mono-phosphatase inhibitor from Memnoniella echinata: Producing organism, fermentation, isolation, physico-chemical and in vitro biological properties. J. Antibiot. (Tokyo) 1992, 45, 1397–1402. [Google Scholar] [CrossRef][Green Version]
- Lin, T.W.; Chang, W.W.; Chen, C.C.; Tsai, Y.C. Stachybotrydial, a potent inhibitor of fucosyltransferase and sialyltransferase. Biochem. Biophys. Res. Commun. 2005, 331, 953–957. [Google Scholar] [CrossRef]
- Sakai, K.; Watanabe, K.; Masuda, K.; Tsuji, M.; Hasumi, K.; Endo, A. Isolation, Characterization and Biological Activities of Novel Triprenyl Phenols as Pancreatic Cholesterol Esterase Inhibitors Produced by Stachybotrys sp. F-1839. J. Antibiot. (Tokyo) 1995, 48, 447–456. [Google Scholar] [CrossRef]
- Nakamura, M.; Ito, Y.; Ogawa, K.; Michisuji, Y.; Sato, S.I.; Takada, M.; Hayashi, M.; Yaginuma, S.; Yamamoto, S. Stachybocins, Novel Endothelin Receptor Antagonists, Produced by Stachybotrys sp. M6222: I. Taxonomy, Fermentation, Isolation and Characterization. J. Antibiot. (Tokyo) 1995, 48, 1389–1395. [Google Scholar] [CrossRef]
- Vázquez, M.J.; Vega, A.; Rivera-Sagredo, A.; Jiménez-Alfaro, M.D.; Díez, E.; Hueso-Rodríguez, J.A. Novel sesquiterpenoids as tyrosine kinase inhibitors produced by Stachybotrys chortarum. Tetrahedron 2004, 60, 2379–2385. [Google Scholar] [CrossRef]
- Vértesy, L.; Kogler, H.; Markus, A.; Schiell, M.; Vogel, M.; Wink, J. Memnopeptide A, a novel terpene peptide from Memnoniella with an activating effect on SERCA2. J. Antibiot. (Tokyo) 2001, 54, 771–782. [Google Scholar] [CrossRef]
- Li, Y.; Wu, C.; Liu, D.; Proksch, P.; Guo, P.; Lin, W. Chartarlactams A-P, phenylspirodrimanes from the sponge-associated fungus stachybotrys chartarum with antihyperlipidemic activities. J. Nat. Prod. 2014, 77, 138–147. [Google Scholar] [CrossRef]
- Ma, X.; Li, L.; Zhu, T.; Ba, M.; Li, G.; Gu, Q.; Guo, Y.; Li, D. Phenylspirodrimanes with anti-HIV activity from the sponge-derived fungus Stachybotrys chartarum mxh-x73. J. Nat. Prod. 2013, 76, 2298–2306. [Google Scholar] [CrossRef]
- Wu, B.; Oesker, V.; Wiese, J.; Malien, S.; Schmaljohann, R.; Imhoff, J.F. Spirocyclic drimanes from the marine fungus Stachybotrys sp. strain MF347. Mar. Drugs 2014, 12, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; de Guzman, F.S.; Gloer, J.B.; Shearer, C.A. Stachybotrins A and B: Novel Bioactive Metabolites from a Brackish Water Isolate of the Fungus Stachybotrys sp. J. Org. Chem. 1992, 57, 6700–6703. [Google Scholar] [CrossRef]
- Nozawa, Y.; Yamamoto, K.; Ito, M.; Sakai, N.; Mizoue, K.; Mizobe, F.; Hanada, K. Stachybotrin C and parvisporin, novel neuritogenic compounds. I. Taxonomy, isolation, physico-chemical and biological properties. J. Antibiot. (Tokyo) 1997, 50, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Zink, D.L.; Williams, M.; Polishook, J.D.; Sanchez, M.; Silverman, K.C.; Lingham, R.B. Kampanols: Novel Ras farnesyl-protein transferase inhibitors from Stachybotrys kampalensis. Bioorganic Med. Chem. Lett. 1998, 8, 2071–2076. [Google Scholar] [CrossRef]
- Minagawa, K.; Kouzuki, S.; Nomura, K.; Yamaguchi, T.; Kawamura, Y.; Matsushima, K.; Tani, H.; Ishii, K.; Tanimoto, T.; Kamigauchi, T. Bisabosquals, novel squalene synthase inhibitors. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. (Tokyo) 2001, 54, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, K.; Kouzuki, S.; Yoshimoto, J.; Kawamura, Y.; Tani, H.; Iwata, T.; Terui, Y.; Nakai, H.; Yagi, S.; Hattori, N.; et al. Stachyflin and acetylstachyflin, novel anti-influenza A virus substances, produced by Stachybotrys sp. RF-7260. I. Isolation, structure elucidation and biological activities. J. Antibiot. (Tokyo) 2002, 55, 155–164. [Google Scholar] [CrossRef]
- Sasaoka, M.; Wada, Y.; Hasumi, K. Stachybotrydial selectively enhances fibrin binding and activation of Glu-plasminogen. J. Antibiot. (Tokyo) 2007, 60. [Google Scholar] [CrossRef]
- Takayasu, R.; Hasumi, K.; Shinohara, C.; Endo, A. Enhancement of fibrin binding and activation of plasminogen by staplabin through induction of a conformational change in plasminogen. FEBS Lett. 1997, 418. [Google Scholar] [CrossRef]
- Christensen, U. The AH-site of plasminogen and two C-terminal fragments. A weak lysine-binding site preferring ligands not carrying a free carboxylate function. Biochem. J. 1984, 223, 413–421. [Google Scholar] [CrossRef]
- Cockell, C.S.; Marshall, J.M.; Dawson, K.M.; Cederholm-Williams, S.A.; Ponting, C.P. Evidence that the conformation of unliganded human plasminogen is maintained via an intramolecular interaction between the lysine-binding site of kringle 5 and the N-terminal peptide. Biochem. J. 1998, 333, 99–105. [Google Scholar] [CrossRef] [PubMed]
- An, S.S.A.; Carreño, C.; Marti, D.N.; Schaller, J.; Albericio, F.; Llinas, M. Lysine-50 is a likely site for anchoring the plasminogen N-terminal peptide to lysine-binding kringles. Protein Sci. 1998, 7, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.P.; Caradoc-Davies, T.; Cowieson, N.; Horvath, A.J.; Quek, A.J.; Encarnacao, J.A.; Steer, D.; Cowan, A.; Zhang, Q.; Lu, B.G.C.; et al. The X-ray Crystal Structure of Full-Length Human Plasminogen. Cell Rep. 2012, 1, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Bodin, C.; Olsson, K. Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J. Thromb. Haemost. 2012, 10, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Medved, L.; Nieuwenhuizen, W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb. Haemost. 2003, 89, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, K.; Narasaki, R.; Yamamichi, S.; Suzuki, E.; Hasumi, K. Mechanism of the action of SMTP-7, a novel small-molecule modulator of plasminogen activation. Blood Coagul. Fibrinolysis 2014, 25. [Google Scholar] [CrossRef]
- Battistel, M.D.; Grishaev, A.; An, S.S.A.; Castellino, F.J.; Lliná, M. Solution structure and functional characterization of human plasminogen kringle 5. Biochemistry 2009, 48, 10208–10219. [Google Scholar] [CrossRef]
- Thorsen, S. The Mechanism of Plasminogen Activation and the Variability of the Fibrin Effector during Tissue-type Plasminogen Activator—Mediated Fibrinolysis. Ann. N. Y. Acad. Sci. 1992, 667, 52–63. [Google Scholar] [CrossRef]
- Ohyama, S.; Wada, Y.; Hasumi, K. Antibiotic A10255 (Thioplabin) enchances fibrin binding and activation of plasminogen. J. Antibiot. (Tokyo) 2002, 55. [Google Scholar] [CrossRef]
- Ohyama, S.; Harada, T.; Chikanishi, T.; Miura, Y.; Hasumi, K. Nonlysine-analog plasmmogen modulators promote autoproteolytic generation of plasmin(ogen) fragments with angiostatin-like activity. Eur. J. Biochem. 2004, 271. [Google Scholar] [CrossRef]
- Kikuchi, T.; Hasumi, K. Enhancement of plasminogen activation by surfactin C: Augmentation of fibrinolysis in vitro and in vivo. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2002, 1596. [Google Scholar] [CrossRef]
- Kikuchi, T.; Hasumi, K. Enhancement of reciprocal activation of prourokinase and plasminogen by the bacterial lipopeptide surfactins and iturin Cs. J. Antibiot. (Tokyo) 2003, 56. [Google Scholar] [CrossRef]
- Wu, W.; Narasaki, R.; Maeda, F.; Hasumi, K. Glucosyldiacylglycerol enhances reciprocal activation of prourokinase and plasminogen. Biosci. Biotechnol. Biochem. 2004, 68. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hasumi, K.; Kuniyasu, T.; Endo, A. Isolation of plactins A, B, C and D, novel cyclic pentapeptides that stimulate cellular fibrinolytic activity. J. Antibiot. (Tokyo) 1996, 49. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hasumi, K.; Sugimoto, M.; Endo, A. Enhancement of fibrinolysis by plactins: Structure-activity relationship and effects in human U937 cells and in mice. Thromb. Haemost. 1998, 79. [Google Scholar] [CrossRef]
- Harada, T.; Tsuruta, T.; Yamagata, K.; Inoue, T.; Hasumi, K. Dual modulation of prothrombin activation by the cyclopentapeptide plactin. FEBS J. 2009, 276. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Yamamichi, S.; Choi-Miura, N.-H.; Hasumi, K. The cyclopentapeptide plactin enhances cellular binding and autoactivation of the serine protease plasma hyaluronan-binding protein. Thromb. Res. 2010, 126. [Google Scholar] [CrossRef] [PubMed]
- Hu, W. Novel Fungal Triprenyl Phenols That Enhance Fibrinolysis and Suppress Tumor Growth. Ph.D. Thesis, Tokyo University of Agriculture and Technology, Tokyo, Japan, March 2003. [Google Scholar]
- Lund, L.R.; Green, K.A.; Stoop, A.A.; Ploug, M.; Almholt, K.; Lilla, J.; Nielsen, B.S.; Christensen, I.J.; Craik, C.S.; Werb, Z.; et al. Plasminogen activation independent of uPA and tPA maintains wound healing in gene-deficient mice. EMBO J. 2006, 25, 2686–2697. [Google Scholar] [CrossRef]
- Danø, K.; Behrendt, N.; Høyer-Hansen, G.; Johnsen, M.; Lund, L.R.; Ploug, M.; Rømer, J. Plasminogen activation and cancer. Thromb. Haemost. 2005, 93, 676–681. [Google Scholar] [CrossRef]
- Castellino, F.J.; Ploplis, V.A. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005, 93, 647–654. [Google Scholar] [CrossRef]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A.; Fang, X.; Snyder, G.D.; Weintraub, N.L. Epoxyeicosatrienoic acids (EETs): Metabolism and biochemical function. Prog. Lipid Res. 2004, 43, 55–90. [Google Scholar] [CrossRef]
- Morisseau, C.; Hammock, B.D. Impact of Soluble Epoxide Hydrolase and Epoxyeicosanoids on Human Health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.M.; McReynolds, C.B.; Schmidt, W.K.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Role of soluble epoxide hydrolase in metabolism of PUFAs in psychiatric and neurological disorders. Front. Pharmacol. 2019, 9. [Google Scholar] [CrossRef]
- Oguro, A.; Imaoka, S. Lysophosphatidic acids are new substrates for the phosphatase domain of soluble epoxide hydrolase. J. Lipid Res. 2012, 53, 505–512. [Google Scholar] [CrossRef]
- Morisseau, C.; Schebb, N.H.; Dong, H.; Ulu, A.; Aronov, P.A.; Hammock, B.D. Role of soluble epoxide hydrolase phosphatase activity in the metabolism of lysophosphatidic acids. Biochem. Biophys. Res. Commun. 2012, 419, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Proschak, E. Phosphatase activity of soluble epoxide hydrolase. Prostaglandins Other Lipid Mediat. 2017, 133, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Luria, A.; Bettaieb, A.; Xi, Y.; Shieh, G.J.; Liu, H.C.; Inoue, H.; Tsai, H.J.; Imig, J.D.; Haj, F.G.; Hammock, B.D. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 9038–9043. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y. Inhibition of soluble epoxide hydrolase for renal health. Front. Pharmacol. 2019, 9. [Google Scholar] [CrossRef]
- Ren, Q. Soluble epoxide hydrolase inhibitor: A novel potential therapeutic or prophylactic drug for psychiatric disorders. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Shyue, S.K.; Wu, C.H.; Chen, C.C.; Lin, C.C.; Chang, C.F.; Chen, S.F. Deletion or inhibition of soluble epoxide hydrolase protects against brain damage and reduces microglia-mediated neuroinflammation in traumatic brain injury. Oncotarget 2017, 8, 103236–103260. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Y.; Zhang, X.; Lv, H.; Pang, W.; Sun, X.; Gan, L.M.; Hammock, B.D.; Ai, D.; Zhu, Y. Inhibition of soluble epoxide hydrolase alleviated atherosclerosis by reducing monocyte infiltration in Ldlr−/− mice. J. Mol. Cell. Cardiol. 2016, 98, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Koerner, I.P.; Noppens, R.; Grafe, M.; Tsai, H.J.; Morisseau, C.; Luria, A.; Hammock, B.D.; Falck, J.R.; Alkayed, N.J. Soluble epoxide hydrolase: A novel therapeutic target in stroke. J. Cereb. Blood Flow Metab. 2007, 27, 1931–1940. [Google Scholar] [CrossRef]
- Zhang, W.; Otsuka, T.; Sugo, N.; Ardeshiri, A.; Alhadid, Y.K.; Iliff, J.J.; DeBarber, A.E.; Koop, D.R.; Alkayed, N.J. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke 2008, 39, 2073–2078. [Google Scholar] [CrossRef]
- Chang, L.H.; Lin, H.C.; Huang, S.S.; Chen, I.C.; Chu, K.W.; Chih, C.L.; Liang, Y.W.; Lee, Y.C.; Chen, Y.Y.; Lee, Y.H.; et al. Blockade of soluble epoxide hydrolase attenuates post-ischemic neuronal hyperexcitation and confers resilience against stroke with TrkB activation. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Liu, Y.; Dang, H.; Li, D.; Pang, W.; Hammock, B.D.; Zhu, Y. Inhibition of soluble epoxide hydrolase attenuates high-fat-diet-induced hepatic steatosis by reduced systemic inflammatory status in mice. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, A.L.; Liao, J.; Li, H.; Dong, H.; Chung, Y.T.; Bai, H.; Matkowskyj, K.A.; Hammock, B.D.; Yang, G.Y. Soluble epoxide hydrolase gene deficiency or inhibition attenuates chronic active inflammatory bowel disease in IL-10(2/2) mice. Dig. Dis. Sci. 2012, 57, 2580–2591. [Google Scholar] [CrossRef]
- Panigrahy, D.; Kalish, B.T.; Huang, S.; Bielenberg, D.R.; Le, H.D.; Yang, J.; Edin, M.L.; Lee, C.R.; Benny, O.; Mudge, D.K.; et al. Epoxyeicosanoids promote organ and tissue regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13528–13533. [Google Scholar] [CrossRef]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef]
- Shibata, K.; Hashimoto, T.; Nobe, K.; Hasumi, K.; Honda, K. Neuroprotective mechanisms of SMTP-7 in cerebral infarction model in mice. Naunyn. Schmiedebergs. Arch. Pharmacol. 2011, 384. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Krishnamurthi, R.V.; Parmar, P.; Norrving, B.; Mensah, G.; Bennett, D.A.; Barker-Collo, S.; Moran, A.E.; Sacco, R.L.; Truelsen, T.; et al. Update on the global burden of ischemic and hemorrhagic stroke in 1990-2013: The GBD 2013 study. Neuroepidemiology 2015, 45, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.P., Jr.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef]
- Arboix, A.; Alioc, J. Cardioembolic Stroke: Clinical Features, Specific Cardiac Disorders and Prognosis. Curr. Cardiol. Rev. 2010, 6, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.W. Lacunar infarction, mortality over time and mortality relative to other ischemic strokes. J. Insur. Med. 2014, 44, 32–37. [Google Scholar]
- Kamel, H.; Healey, J.S. Cardioembolic Stroke. Circ. Res. 2017, 120, 514–526. [Google Scholar] [CrossRef]
- Rothwell, P.M. Atherothrombosis and ischaemic stroke. Br. Med. J. 2007, 334, 379–380. [Google Scholar] [CrossRef]
- Palomeras Soler, E.; Casado Ruiz, V. Epidemiology and Risk Factors of Cerebral Ischemia and Ischemic Heart Diseases: Similarities and Differences. Curr. Cardiol. Rev. 2010, 6, 138–149. [Google Scholar] [CrossRef]
- Sacco, S.; Marini, C.; Totaro, R.; Russo, T.; Cerone, D.; Carolei, A. A population-based study of the incidence and prognosis of lacunar stroke. Neurology 2006, 66, 1335–1338. [Google Scholar] [CrossRef]
- Lastilla, M. Lacunar infarct. Clin. Exp. Hypertens. 2006, 28, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol. 2013, 12, 483–497. [Google Scholar] [CrossRef]
- Guadagno, J.V.; Calautti, C.; Baron, J.C. Progress in imaging stroke: Emerging clinical applications. Br. Med. Bull. 2003, 65, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.C. Treatments to promote neural repair after stroke. J. Stroke 2018, 20, 57–70. [Google Scholar] [CrossRef]
- Hassan, A. Recent Advances in the Management of Acute Ischemic Stroke; Springer: London, UK, 2010; pp. 61–68. [Google Scholar] [CrossRef]
- Roesly, H. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. J. Emerg. Med. 2018, 55, 151. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 h after stroke with a mismatch between deficit and infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef]
- Mican, J.; Toul, M.; Bednar, D.; Damborsky, J. Structural Biology and Protein Engineering of Thrombolytics. Comput. Struct. Biotechnol. J. 2019, 17, 917–938. [Google Scholar] [CrossRef]
- Shibata, K.; Hashimoto, T.; Miyazaki, T.; Miyazaki, A.; Nobe, K. Thrombolytic Therapy for Acute Ischemic Stroke: Past and Future. Curr. Pharm. Des. 2019, 25, 242–250. [Google Scholar] [CrossRef]
- Lin, H.; Xu, L.; Yu, S.; Hong, W.; Huang, M.; Xu, P. Therapeutics targeting the fibrinolytic system. Exp. Mol. Med. 2020, 52, 367–379. [Google Scholar] [CrossRef]
- Dela Peña, I.; Borlongan, C.; Shen, G.; Davis, W. Strategies to extend thrombolytic time window for ischemic stroke treatment: An unmet clinical need. J. Stroke 2017, 19, 50–60. [Google Scholar] [CrossRef]
- Bhogal, P.; Andersson, T.; Maus, V.; Mpotsaris, A.; Yeo, L. Mechanical Thrombectomy—A Brief Review of a Revolutionary new Treatment for Thromboembolic Stroke. Clin. Neuroradiol. 2018, 28, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.P.; Desai, S.M.; Kenmuir, C.L.; Rocha, M.; Starr, M.T.; Molyneaux, B.J.; Gross, B.A.; Jankowitz, B.T.; Jovin, T.G. Eligibility for endovascular trial enrollment in the 6-to 24-hour time window analysis of a single comprehensive stroke center. Stroke 2018, 49, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Andrew Josephson, S.; Kamel, H. The acute stroke care revolution enhancing access to therapeutic advances. JAMA J. Am. Med. Assoc. 2018, 320, 1239–1240. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Shibata, K.; Ohata, H.; Hasumi, K.; Honda, K. Altered gene expression in an embolic stroke model after thrombolysis with tissue plasminogen activator and Stachybotrys microspora triprenyl phenol-7. J. Pharmacol. Sci. 2014, 125. [Google Scholar] [CrossRef]
- Miyazaki, T.; Kimura, Y.; Ohata, H.; Hashimoto, T.; Shibata, K.; Hasumi, K.; Honda, K. Distinct effects of tissue-type plasminogen activator and SMTP-7 on cerebrovascular inflammation following thrombolytic reperfusion. Stroke 2011, 42. [Google Scholar] [CrossRef]
- Huang, Y.; Ohta, Y.; Shang, J.; Morihara, R.; Nakano, Y.; Fukui, Y.; Liu, X.; Shi, X.; Feng, T.; Yamashita, T.; et al. Antineuroinflammatory Effect of SMTP-7 in Ischemic Mice. J. Stroke Cerebrovasc. Dis. 2018, 27. [Google Scholar] [CrossRef]
- Verstraete, M.; Bounameaux, H.; de Cock, F.; Van de Werf, F.; Collen, D. Pharmacokinetics and systemic fibrinogenolytic effects of recombinant human tissue-type plasminogen activator (rt-PA) in humans. J. Pharmacol. Exp. Ther. 1985, 235, 506–512. [Google Scholar]
- Verstraete, M.; Su, C.A.P.F.; Tanswell, P.; Feuerer, W.; Collen, D. Pharmacokinetics and effects on fibrinolytic and coagulation parameters of two doses of recombinant tissue-type plasminogen activator in healthy volunteers. Thromb. Haemost. 1986, 56, 1–5. [Google Scholar] [CrossRef]
- Tanswell, P.; Seifried, E.; Su, P.C.A.F.; Feuerer, W.; Rijken, D.C. Pharmacokinetics and systemic effects of tissue-type plasminogen activator in normal subjects. Clin. Pharmacol. Ther. 1989, 46, 155–162. [Google Scholar] [CrossRef]
- Jickling, G.C.; Liu, D.; Stamova, B.; Ander, B.P.; Zhan, X.; Lu, A.; Sharp, F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J. Cereb. Blood Flow Metab. 2014, 34, 185–199. [Google Scholar] [CrossRef]
- Inoue, S.; Kim, R.; Hoshino, Y.; Honda, K. Synthesis of Tricyclic Pyrano[2,3-e]isoindolin-3-ones as the Core Structure of Stachybotrin A, B, and C. ChemInform 2006, 37. [Google Scholar] [CrossRef]
- Tumma, N.; Jacolot, M.; Jean, M.; Chandrasekhar, S.; Van De Weghe, P. Synthetic studies towards stachybotrin C. Synlett 2012, 23, 2919–2922. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasumi, K.; Suzuki, E. Impact of SMTP Targeting Plasminogen and Soluble Epoxide Hydrolase on Thrombolysis, Inflammation, and Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 954. https://doi.org/10.3390/ijms22020954
Hasumi K, Suzuki E. Impact of SMTP Targeting Plasminogen and Soluble Epoxide Hydrolase on Thrombolysis, Inflammation, and Ischemic Stroke. International Journal of Molecular Sciences. 2021; 22(2):954. https://doi.org/10.3390/ijms22020954
Chicago/Turabian StyleHasumi, Keiji, and Eriko Suzuki. 2021. "Impact of SMTP Targeting Plasminogen and Soluble Epoxide Hydrolase on Thrombolysis, Inflammation, and Ischemic Stroke" International Journal of Molecular Sciences 22, no. 2: 954. https://doi.org/10.3390/ijms22020954
APA StyleHasumi, K., & Suzuki, E. (2021). Impact of SMTP Targeting Plasminogen and Soluble Epoxide Hydrolase on Thrombolysis, Inflammation, and Ischemic Stroke. International Journal of Molecular Sciences, 22(2), 954. https://doi.org/10.3390/ijms22020954