A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake?



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanisms to Control the Ca2+ Conductance of VDAC
2.1. Structure and Electrophysiological Properties of VDACs
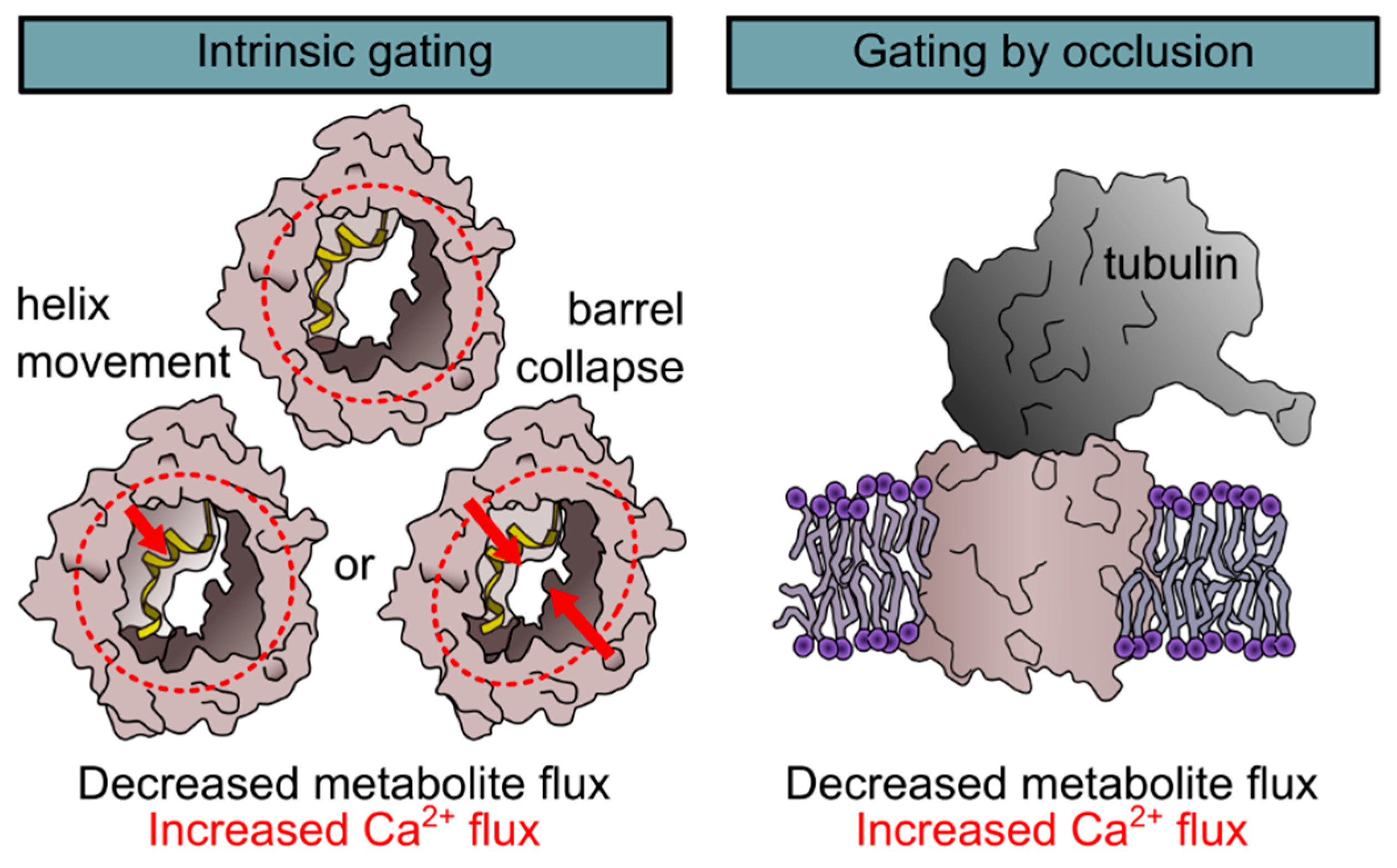
2.1.1. Regulation of Ca2+ Conductance by Gating
2.1.2. Regulation of Ca2+ Conductance by Occlusion
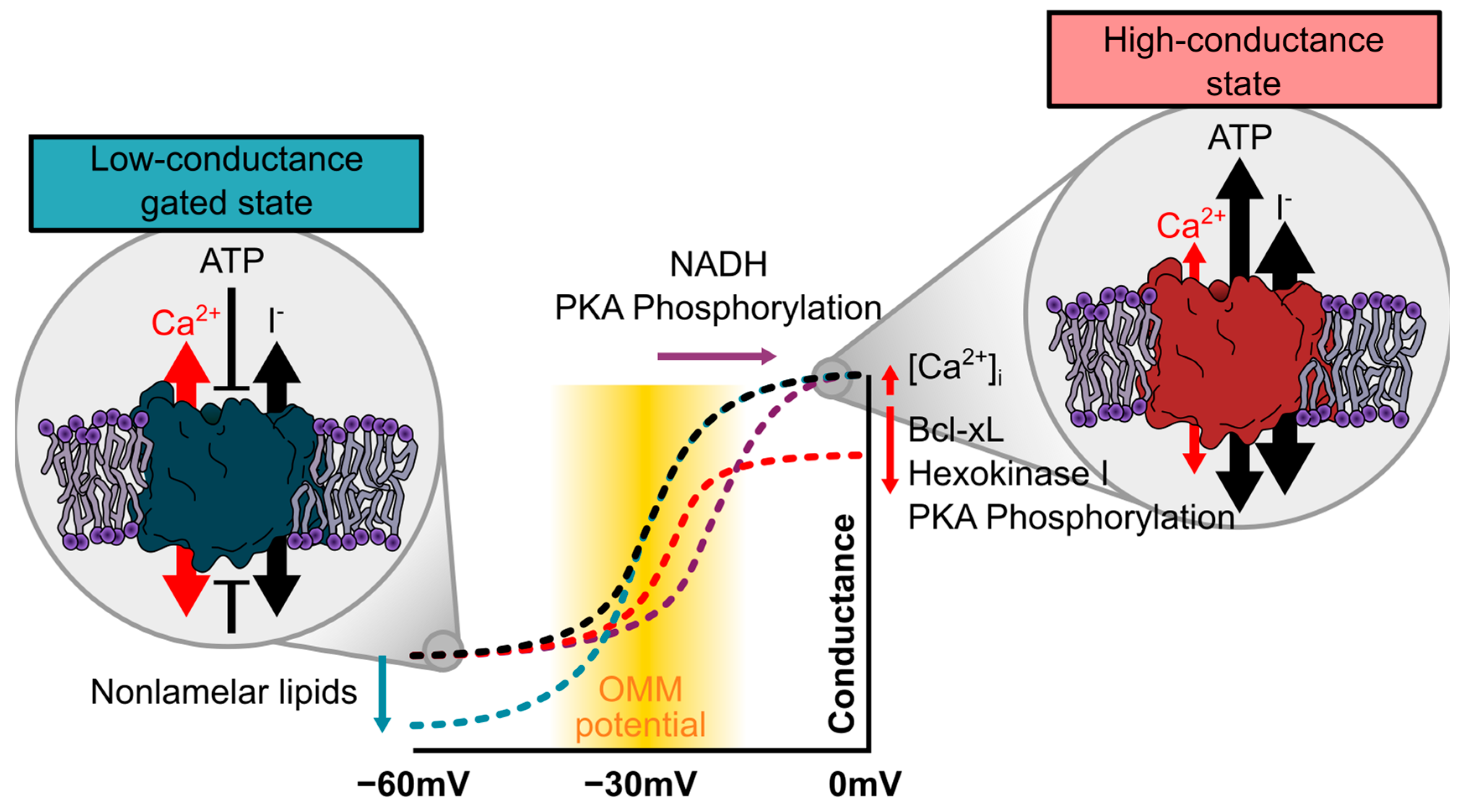
2.2. Modification of the Gating Profile of VDAC
2.2.1. An OMM Potential as a Trigger for VDAC Gating
2.2.2. Effects of Ca2+ on VDAC Gating
2.2.3. Modulation of VDAC Gating by Posttranslational Modification
2.2.4. Modulation of VDAC Gating through the Lipidic Environment
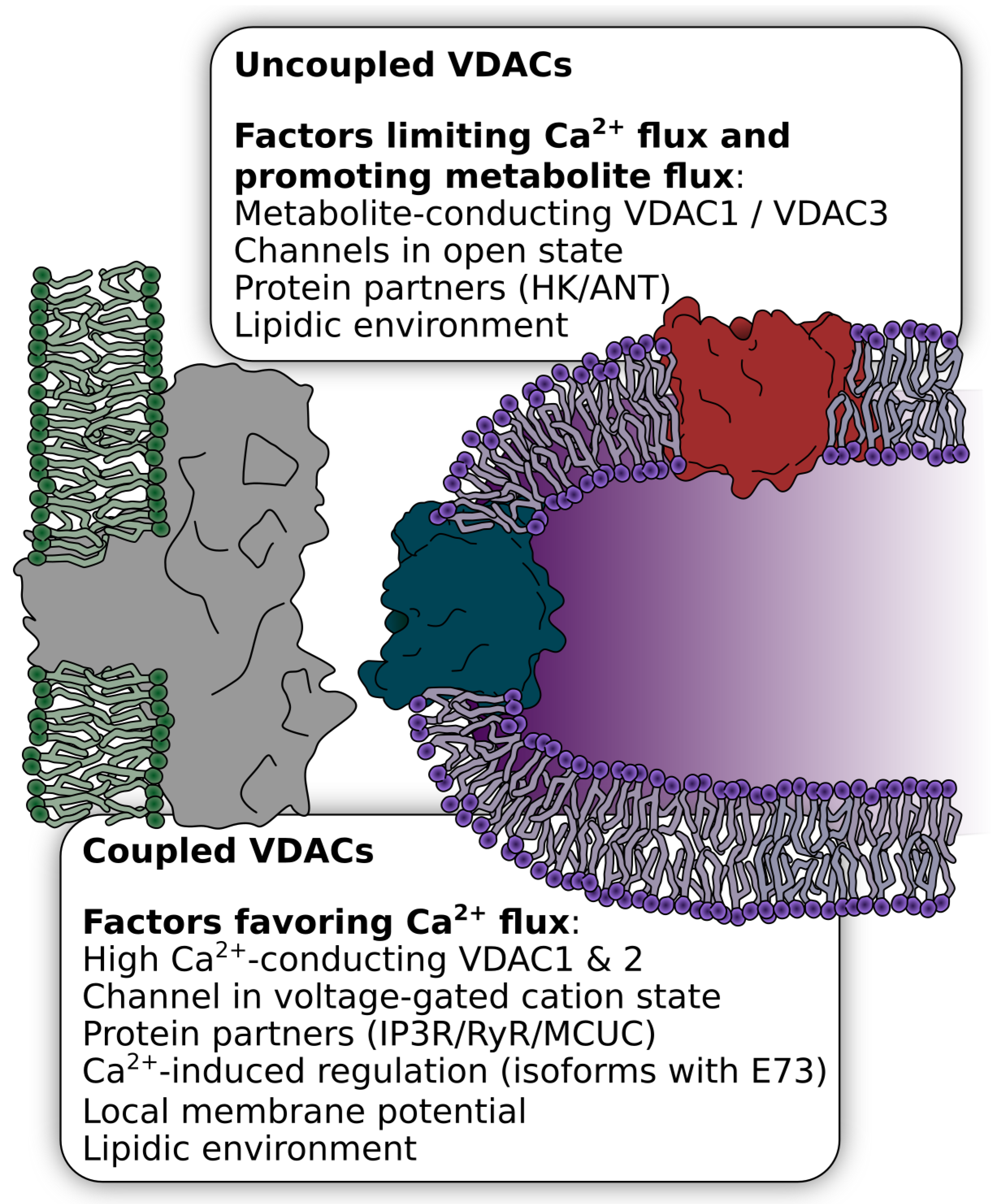
2.3. Regulation of VDAC Ca2+ Flux by Protein Partners
2.4. Regulation by Subcellular Localization in Ca2+ Microdomains
2.4.1. Regulation of the OMM Ca2+ Flux by Different VDAC Isoforms
2.4.2. Regulation of OMM Ca2+ Flux by VDAC Expression Levels
3. Physiological Relevance of a Ca2+ Flux Regulation at the OMM
4. Regulation of VDAC-Mediated Ca2+ Flux under Pathophysiological Conditions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slater, E.C.; Cleland, K.W. The calcium content of isolated heart-muscle sarcosomes. Biochem. J. 1953, 54, xxii. [Google Scholar] [PubMed]
- DeLuca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef]
- Ham, S.J.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, Q.; Tian, X.; Qian, N.; Chai, P.; Liu, B.; Hu, J.; Blackstone, C.; Zhu, D.; Teng, J.; et al. DNA damage triggers tubular endoplasmic reticulum extension to promote apoptosis by facilitating ER-mitochondria signaling. Cell Res. 2018, 28, 833–854. [Google Scholar] [CrossRef]
- Schweitzer, M.K.; Wilting, F.; Sedej, S.; Dreizehnter, L.; Dupper, N.J.; Tian, Q.; Moretti, A.; My, I.; Kwon, O.; Priori, S.G.; et al. Suppression of Arrhythmia by Enhancing Mitochondrial Ca2+ Uptake in Catecholaminergic Ventricular Tachycardia Models. JACC Basic Transl. Sci. 2017, 2, 737–746. [Google Scholar] [CrossRef]
- Wilting, F.; Kopp, R.; Gurnev, P.A.; Schedel, A.; Dupper, N.J.; Kwon, O.; Nicke, A.; Gudermann, T.; Schredelseker, J. The antiarrhythmic compound efsevin directly modulates voltage-dependent anion channel 2 by binding to its inner wall and enhancing mitochondrial Ca2+ uptake. Br. J. Pharmacol. 2020, 177, 2947–2958. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.-P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef] [PubMed]
- Schredelseker, J.; Paz, A.; López, C.J.; Altenbach, C.; Leung, C.S.; Drexler, M.K.; Chen, J.-N.; Hubbell, W.L.; Abramson, J. High-Resolution Structure and Double Electron-Electron Resonance of the Zebrafish Voltage Dependent Anion Channel 2 Reveal an Oligomeric Population. J. Biol. Chem. 2014, 289, 12566–12577. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.R.; Mahalakshmi, R. Evolutionary selection of a 19-stranded mitochondrial β-barrel scaffold bears structural and functional significance. J. Biol. Chem. 2020, 295, 14653–14665. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, O.P.; Paz, A.; Adelman, J.; Colletier, J.-P.; Abramson, J.; Grabe, M. Characterizing ATP Permeation through the Voltage-Dependent Anion Channel VDAC. Biophys. J. 2014, 106, 147a–148a. [Google Scholar] [CrossRef]
- Colombini, M. Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 1989, 111, 103–111. [Google Scholar] [CrossRef]
- Menzel, V.A.; Cassará, M.C.; Benz, R.; De Pinto, V.; Messina, A.; Cunsolo, V.; Saletti, R.; Hinsch, K.; Hinsch, E. Molecular and functional characterization of VDAC2 purified from mammal spermatozoa. Biosci. Rep. 2009, 29, 351–362. [Google Scholar] [CrossRef]
- Mertins, B.; Psakis, G.; Grosse, W.; Back, K.C.; Salisowski, A.; Reiss, P.; Koert, U.; Essen, L.-O. Flexibility of the N-Terminal mVDAC1 Segment Controls the Channel’s Gating Behavior. PLoS ONE 2012, 7, e47938. [Google Scholar] [CrossRef]
- Guardiani, C.; Magrì, A.; Karachitos, A.; Di Rosa, M.C.; Reina, S.; Bodrenko, I.; Messina, A.; Kmita, H.; Ceccarelli, M.; De Pinto, V. yVDAC2, the second mitochondrial porin isoform of Saccharomyces cerevisiae. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 270–279. [Google Scholar] [CrossRef]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta 2007, 1768, 2510–2515. [Google Scholar] [CrossRef]
- Zachariae, U.; Schneider, R.; Briones, R.; Gattin, Z.; Demers, J.-P.; Giller, K.; Maier, E.; Zweckstetter, M.; Griesinger, C.; Becker, S.; et al. β-Barrel Mobility Underlies Closure of the Voltage-Dependent Anion Channel. Structure 2012, 20, 1540–1549. [Google Scholar] [CrossRef]
- Pavlov, E.; Grigoriev, S.M.; Dejean, L.M.; Zweihorn, C.L.; Mannella, C.A.; Kinnally, K.W. The mitochondrial channel VDAC has a cation-selective open state. Biochim. Biophys. Acta Bioenerg. 2005, 1710, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Checchetto, V.; Reina, S.; Magrì, A.; Szabo, I.; De Pinto, V. Recombinant human voltage dependent anion selective channel isoform 3 (hVDAC3) forms pores with a very small conductance. Cell. Physiol. Biochem. 2014, 34, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Queralt-Martín, M.; Bergdoll, L.; Teijido, O.; Munshi, N.; Jacobs, D.; Kuszak, A.J.; Protchenko, O.; Reina, S.; Magrì, A.; De Pinto, V.; et al. A lower affinity to cytosolic proteins reveals VDAC3 isoform-specific role in mitochondrial biology. J. Gen. Physiol. 2020, 152. [Google Scholar] [CrossRef]
- Lemeshko, V.V. Model of the Outer Membrane Potential Generation by the Inner Membrane of Mitochondria. Biophys. J. 2002, 82, 684–692. [Google Scholar] [CrossRef][Green Version]
- Adams, V.; Bosch, W.; Schlegel, J.; Wallimann, T.; Brdiczka, D. Further characterization of contact sites from mitochondria of different tissues: Topology of peripheral kinases. Biochim. Biophys. Acta Biomembr. 1989, 981, 213–225. [Google Scholar] [CrossRef]
- Porcelli, A.M.; Ghelli, A.; Zanna, C.; Pinton, P.; Rizzuto, R.; Rugolo, M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem. Biophys. Res. Commun. 2005, 326, 799–804. [Google Scholar] [CrossRef]
- Báthori, G.; Csordás, G.; Garcia-Perez, C.; Davies, E.; Hajnóczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 2006, 281, 17347–17358. [Google Scholar] [CrossRef]
- Lee, A.C.; Xu, X.; Colombini, M. The role of pyridine dinucleotides in regulating the permeability of the mitochondrial outer membrane. J. Biol. Chem. 1996, 271, 26724–26731. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Kazemi, N.; Weinrich, M.; Bezrukov, S.M. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J. Biol. Chem. 2006, 281. [Google Scholar] [CrossRef]
- Van Liefferinge, F.; Krammer, E.-M.; Sengupta, D.; Prévost, M. Lipid composition and salt concentration as regulatory factors of the anion selectivity of VDAC studied by coarse-grained molecular dynamics simulations. Chem. Phys. Lipids 2019, 220, 66–76. [Google Scholar] [CrossRef]
- Budelier, M.M.; Cheng, W.W.L.; Bergdoll, L.; Chen, Z.-W.; Janetka, J.W.; Abramson, J.; Krishnan, K.; Mydock-McGrane, L.; Covey, D.F.; Whitelegge, J.P.; et al. Photoaffinity labeling with cholesterol analogues precisely maps a cholesterol-binding site in voltage-dependent anion channel-1. J. Biol. Chem. 2017, 292, 9294–9304. [Google Scholar] [CrossRef] [PubMed]
- Briones, R.; Weichbrodt, C.; Paltrinieri, L.; Mey, I.; Villinger, S.; Giller, K.; Lange, A.; Zweckstetter, M.; Griesinger, C.; Becker, S.; et al. Voltage Dependence of Conformational Dynamics and Subconducting States of VDAC-1. Biophys. J. 2016, 111, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Queralt-Martín, M.; Hoogerheide, D.P.; Noskov, S.Y.; Berezhkovskii, A.M.; Rostovtseva, T.K.; Bezrukov, S.M. VDAC gating thermodynamics, but not gating kinetics, are virtually temperature-independent. Biophys. J. 2020, 119, 2584–2592. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Ahmed, T.; Chirasani, V.R.; Singh, P.K.; Maji, S.K.; Senapati, S.; Bera, A.K. Modulation of the mitochondrial voltage dependent anion channel (VDAC) by curcumin. Biochim. Biophys. Acta 2015, 1848, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Majumdar, D.; Vallabhaneni, S.; Bera, A.K. Aspirin induces cell death by directly modulating mitochondrial voltage-dependent anion channel (VDAC). Sci. Rep. 2017, 7, 45184. [Google Scholar] [CrossRef]
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J. Biol. Chem. 2013, 288, 19870–19881. [Google Scholar] [CrossRef]
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007, 26, 2606–2620. [Google Scholar] [CrossRef]
- Okazaki, M.; Kurabayashi, K.; Asanuma, M.; Saito, Y.; Dodo, K.; Sodeoka, M. VDAC3 gating is activated by suppression of disulfide-bond formation between the N-terminal region and the bottom of the pore. Biochim. Biophys. Acta Biomembr. 2015, 1848, 3188–3196. [Google Scholar] [CrossRef]
- Maurya, S.R.; Mahalakshmi, R. N-helix and Cysteines Inter-regulate Human Mitochondrial VDAC-2 Function and Biochemistry. J. Biol. Chem. 2015, 290, 30240–30250. [Google Scholar] [CrossRef]
- Shuvo, S.R.; Ferens, F.G.; Court, D.A. The N-terminus of VDAC: Structure, mutational analysis, and a potential role in regulating barrel shape. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1350–1361. [Google Scholar] [CrossRef]
- Choudhary, O.P.; Ujwal, R.; Kowallis, W.; Coalson, R.; Abramson, J.; Grabe, M. The Electrostatics of VDAC: Implications for Selectivity and Gating. J. Mol. Biol. 2010, 396, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Teijido, O.; Ujwal, R.; Hillerdal, C.-O.; Kullman, L.; Rostovtseva, T.K.; Abramson, J. Affixing N-terminal α-helix to the wall of the voltage-dependent anion channel does not prevent its voltage gating. J. Biol. Chem. 2012, 287, 11437–11445. [Google Scholar] [CrossRef] [PubMed]
- Zizi, M.; Forte, M.; Blachly-Dyson, E.; Colombini, M. NADH regulates the gating of VDAC, the mitochondrial outer membrane channel. J. Biol. Chem. 1994, 269, 1614–1616. [Google Scholar] [CrossRef]
- Böhm, R.; Amodeo, G.F.; Murlidaran, S.; Chavali, S.; Wagner, G.; Winterhalter, M.; Brannigan, G.; Hiller, S. The Structural Basis for Low Conductance in the Membrane Protein VDAC upon β-NADH Binding and Voltage Gating. Structure 2020, 28, 206–214.e4. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Hoogerheide, D.P.; Rovini, A.; Sirajuddin, M.; Bezrukov, S.M. Sequence diversity of tubulin isotypes in regulation of the mitochondrial voltage-dependent anion channel. J. Biol. Chem. 2018, 293, 10949–10962. [Google Scholar] [CrossRef]
- Puurand, M.; Tepp, K.; Timohhina, N.; Aid, J.; Shevchuk, I.; Chekulayev, V.; Kaambre, T. Tubulin βII and βIII Isoforms as the Regulators of VDAC Channel Permeability in Health and Disease. Cells 2019, 8, 239. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef]
- Hoogerheide, D.P.; Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Mechanism of α-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale 2017, 9, 183–192. [Google Scholar] [CrossRef]
- Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett. 2011, 585, 2363–2366. [Google Scholar] [CrossRef]
- Simson, P.; Jepihhina, N.; Laasmaa, M.; Peterson, P.; Birkedal, R.; Vendelin, M. Restricted ADP movement in cardiomyocytes: Cytosolic diffusion obstacles are complemented with a small number of open mitochondrial voltage-dependent anion channels. J. Mol. Cell. Cardiol. 2016, 97, 197–203. [Google Scholar] [CrossRef]
- Lemeshko, V.V. VDAC electronics: 1. VDAC-hexo(gluco)kinase generator of the mitochondrial outer membrane potential. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Lemeshko, V.V. VDAC electronics: 5. Mechanism and computational model of hexokinase-dependent generation of the outer membrane potential in brain mitochondria. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2599–2607. [Google Scholar] [CrossRef]
- Lemeshko, V.V. Electrical control of the cell energy metabolism at the level of mitochondrial outer membrane. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183493. [Google Scholar] [CrossRef] [PubMed]
- García-Pérez, C.; Schneider, T.G.; Hajnóczky, G.; Csordás, G. Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1907-15. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hao, Y.; Chen, H.; He, Q.; Yuan, Z.; Cheng, J. Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 2015, 6, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Abu-Hamad, S.; Zaid, H.; Nahon, E.; Shoshan-Barmatz, V. Localization of the voltage-dependent anion channel-1 Ca2+-binding sites. Cell Calcium 2007, 41, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Villinger, S.; Mari, S.A.; Giller, K.; Griesinger, C.; Becker, S.; Müller, D.J.; Zweckstetter, M. Molecular Plasticity of the Human Voltage-Dependent Anion Channel Embedded Into a Membrane. Structure 2016, 24, 585–594. [Google Scholar] [CrossRef]
- Villinger, S.; Briones, R.; Giller, K.; Zachariae, U.; Lange, A.; de Groot, B.L.; Griesinger, C.; Becker, S.; Zweckstetter, M. Functional dynamics in the voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2010, 107, 22546–22551. [Google Scholar] [CrossRef]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef]
- Schredelseker, J.; Shimizu, H.; Huang, J.; Lin, B.; Chen, J.-N. Regulation of Voltage-Dependent Anion Channel 2 at Glutamate 73 is Critical for its Role in Cardiac Calcium Handling. Biophys. J. 2012, 102, 312a. [Google Scholar] [CrossRef]
- Bera, A.K.; Ghosh, S. Dual mode of gating of voltage-dependent anion channel as revealed by phosphorylation. J. Struct. Biol. 2001, 135, 67–72. [Google Scholar] [CrossRef]
- Banerjee, J.; Ghosh, S. Phosphorylation of rat brain mitochondrial voltage-dependent anion as a potential tool to control leakage of cytochrome c. J. Neurochem. 2006, 98, 670–676. [Google Scholar] [CrossRef]
- Gupta, R.; Ghosh, S. Phosphorylation of purified mitochondrial Voltage-Dependent Anion Channel by c-Jun N-terminal Kinase-3 modifies channel voltage-dependence. Biochim. Open 2017, 4, 78–87. [Google Scholar] [CrossRef]
- Gatliff, J.; East, D.A.; Singh, A.; Alvarez, M.S.; Frison, M.; Matic, I.; Ferraina, C.; Sampson, N.; Turkheimer, F.; Campanella, M. A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis. 2017, 8, e2896. [Google Scholar] [CrossRef]
- Baines, C.P.; Song, C.-X.; Zheng, Y.-T.; Wang, G.-W.; Zhang, J.; Wang, O.-L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, K.L.; Maldonado, E.N.; Lemasters, J.J.; Rostovtseva, T.K.; Bezrukov, S.M. Phosphorylation of Voltage-Dependent Anion Channel by Serine/Threonine Kinases Governs Its Interaction with Tubulin. PLoS ONE 2011, 6, e25539. [Google Scholar] [CrossRef] [PubMed]
- Rovini, A.; Gurnev, P.A.; Beilina, A.; Queralt-Martín, M.; Rosencrans, W.; Cookson, M.R.; Bezrukov, S.M.; Rostovtseva, T.K. Molecular mechanism of olesoxime-mediated neuroprotection through targeting α-synuclein interaction with mitochondrial VDAC. Cell. Mol. Life Sci. 2020, 77, 3611–3626. [Google Scholar] [CrossRef] [PubMed]
- Crimi, M.; Esposti, M.D. Apoptosis-induced changes in mitochondrial lipids. Biochim. Biophys. Acta 2011, 1813, 551–557. [Google Scholar] [CrossRef]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent Anion Channel 1-based Peptides Interact with Bcl-2 to Prevent Antiapoptotic Activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shah, K.; Bradbury, N.A.; Li, C.; White, C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014, 5, e1482. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 2015, 290, 9150–9161. [Google Scholar] [CrossRef] [PubMed]
- Tornero, D.; Posadas, I.; Ceña, V. Bcl-xL Blocks a Mitochondrial Inner Membrane Channel and Prevents Ca2+ Overload-Mediated Cell Death. PLoS ONE 2011, 6, e20423. [Google Scholar] [CrossRef] [PubMed]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef]
- Tamse, C.; Lu, X.; Mortel, E.; Cabrales, E.; Feng, W.; Schaefer, S. The Peripheral Benzodiazepine Receptor Modulates Ca2+ Transport through the VDAC in Rat Heart Mitochondria. J. Clin. Basic Cardiol. 2008, 11, 24–29. [Google Scholar]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef]
- Viola, H.M.; Adams, A.M.; Davies, S.M.K.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, 111, E2905–E2914. [Google Scholar] [CrossRef]
- Min, C.K.; Yeom, D.R.; Lee, K.-E.; Kwon, H.-K.; Kang, M.; Kim, Y.-S.; Park, Z.Y.; Jeon, H.; Kim, D.H. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 2012, 447, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581.e4. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Ramesh, V.; Franzini-Armstrong, C.; Sheu, S.-S. Transport of Ca2+ from Sarcoplasmic Reticulum to Mitochondria in Rat Ventricular Myocytes. J. Bioenerg. Biomembr. 2000, 32, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Brdiczka, D.G.; Zorov, D.B. Mitochondrial contact sites: Their role in energy metabolism and apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Guan, N.; Ren, Y.-L.; Wei, Q.-J.; Tao, Y.-H.; Yang, G.-S.; Liu, X.-Y.; Bu, D.-F.; Zhang, Y.; Zhu, S.-N. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yücel, Y.Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M.; et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa2+ overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020, 27, 1907–1923. [Google Scholar] [CrossRef]
- Caterino, M.; Ruoppolo, M.; Mandola, A.; Costanzo, M.; Orrù, S.; Imperlini, E. Protein-protein interaction networks as a new perspective to evaluate distinct functional roles of voltage-dependent anion channel isoforms. Mol. Biosyst. 2017, 13, 2466–2476. [Google Scholar] [CrossRef]
- Anflous, K.; Armstrong, D.D.; Craigen, W.J. Altered mitochondrial sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J. Biol. Chem. 2001, 276, 1954–1960. [Google Scholar] [CrossRef]
- Raghavan, A.; Sheiko, T.V.; Graham, B.H.; Craigen, W.J. Voltage-dependant anion channels: Novel insights into isoform function through genetic models. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1477–1485. [Google Scholar] [CrossRef]
- Amodeo, G.F.; Scorciapino, M.A.; Messina, A.; De Pinto, V.; Ceccarelli, M. Charged Residues Distribution Modulates Selectivity of the Open State of Human Isoforms of the Voltage Dependent Anion-Selective Channel. PLoS ONE 2014, 9, e103879. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.Y.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef]
- Sasaki, K.; Donthamsetty, R.; Heldak, M.; Cho, Y.-E.; Scott, B.T.; Makino, A. VDAC: Old protein with new roles in diabetes. Am. J. Physiol. Physiol. 2012, 303, C1055–C1060. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.-C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Sheng, Y.; Fu, X.; Cheng, H.; Zhou, R. MYBL2 guides autophagy suppressor VDAC2 in the developing ovary to inhibit autophagy through a complex of VDAC2-BECN1-BCL2L1 in mammals. Autophagy 2015, 11, 1081–1098. [Google Scholar] [CrossRef]
- Weisthal, S.; Keinan, N.; Ben-Hail, D.; Arif, T.; Shoshan-Barmatz, V. Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2270–2281. [Google Scholar] [CrossRef]
- Allouche, M.; Pertuiset, C.; Robert, J.L.; Martel, C.; Veneziano, R.; Henry, C.; el dein, O.S.; Saint, N.; Brenner, C.; Chopineau, J. ANT-VDAC1 interaction is direct and depends on ANT isoform conformation in vitro. Biochem. Biophys. Res. Commun. 2012, 429, 12–17. [Google Scholar] [CrossRef]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; La Piana, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Glucose-6-phosphate tips the balance in modulating apoptosis in cerebellar granule cells. FEBS Lett. 2015, 589, 651–658. [Google Scholar] [CrossRef][Green Version]
- Dubey, A.K.; Godbole, A.; Mathew, M.K. Regulation of VDAC trafficking modulates cell death. Cell Death Discov. 2016, 2, 16085. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Müller, M.; McNeal, A.; Lovy, A.; Jaňa, F.; Bustos, G.; Urra, F.; Smith, N.; Molgó, J.; Diehl, J.A.; et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca2+ Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 2016, 14, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Yan, D.; Deng, H.; Chen, W.; An, G. Novel Molecular Regulators of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL)-Induced Apoptosis in NSCLC Cells. Clin. Lab. 2015, 61. [Google Scholar] [CrossRef] [PubMed]
- Simamura, E.; Shimada, H.; Ishigaki, Y.; Hatta, T.; Higashi, N.; Hirai, K.-I. Bioreductive activation of quinone antitumor drugs by mitochondrial voltage-dependent anion channel 1. Anat. Sci. Int. 2008, 83, 261–266. [Google Scholar] [CrossRef]
- Grills, C.; Jithesh, P.V.; Blayney, J.; Zhang, S.-D.; Fennell, D.A. Gene Expression Meta-Analysis Identifies VDAC1 as a Predictor of Poor Outcome in Early Stage Non-Small Cell Lung Cancer. PLoS ONE 2011, 6, e14635. [Google Scholar] [CrossRef]
- Rimessi, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Pinton, P. Mitochondrial Ca2+ Remodeling is a Prime Factor in Oncogenic Behavior. Front. Oncol. 2015, 5, 143. [Google Scholar] [CrossRef]
- Koren, I.; Raviv, Z.; Shoshan-Barmatz, V. Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol. Ther. 2010, 9, 1046–1052. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro. Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential Role of BNIP3 in Cardiac Remodeling, Myocardial Stiffness, and Endoplasmic Reticulum. Circ. Hear. Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef]
- Pan, M.; Han, Y.; Basu, A.; Dai, A.; Si, R.; Willson, C.; Balistrieri, A.; Scott, B.T.; Makino, A. Overexpression of hexokinase 2 reduces mitochondrial calcium overload in coronary endothelial cells of type 2 diabetic mice. Am. J. Physiol. Physiol. 2018, 314, C732–C740. [Google Scholar] [CrossRef]
- Miyamae, M.; Camacho, S.A.; Weiner, M.W.; Figueredo, V.M. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am. J. Physiol. 1996. [Google Scholar] [CrossRef] [PubMed]
- de J García-Rivas, G.; Carvajal, K.; Correa, F.; Zazueta, C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Liu, D.; Tang, L.; Yin, D.; Yin, S.; Lai, S.; Yao, J.; He, M. Long-term oral resveratrol intake provides nutritional preconditioning against myocardial ischemia/reperfusion injury: Involvement of VDAC1 downregulation. Mol. Nutr. Food Res. 2015, 59, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Thiebaut, P.-A.; Paillard, M.; Ducreux, S.; Abrial, M.; Crola Da Silva, C.; Durand, A.; Alam, M.R.; Van Coppenolle, F.; Sheu, S.-S.; et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2016, 23, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.-A.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B.; et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2016, 59, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Thivolet, C.; Vial, G.; Cassel, R.; Rieusset, J.; Madec, A.-M. Reduction of endoplasmic reticulum- mitochondria interactions in beta cells from patients with type 2 diabetes. PLoS ONE 2017, 12, e0182027. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Dia, M.; Gomez, L.; Thibault, H.; Tessier, N.; Leon, C.; Chouabe, C.; Ducreux, S.; Gallo-Bona, N.; Tubbs, E.; Bendridi, N.; et al. Reduced reticulum–mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res. Cardiol. 2020, 115, 74. [Google Scholar] [CrossRef]
- Joo, H.K.; Lee, Y.R.; Lim, S.Y.; Lee, E.J.; Choi, S.; Cho, E.J.; Park, M.S.; Ryoo, S.; Park, J.B.; Jeon, B.H. Peripheral benzodiazepine receptor regulates vascular endothelial activations via suppression of the voltage-dependent anion channel-1. FEBS Lett. 2012, 586, 1349–1355. [Google Scholar] [CrossRef]
- Thai, P.N.; Daugherty, D.J.; Frederich, B.J.; Lu, X.; Deng, W.; Bers, D.M.; Dedkova, E.N.; Schaefer, S. Cardiac-specific Conditional Knockout of the 18-kDa Mitochondrial Translocator Protein Protects from Pressure Overload Induced Heart Failure. Sci. Rep. 2018, 8, 16213. [Google Scholar] [CrossRef]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER–Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Ziviani, E. A trio has turned into a quartet: DJ-1 interacts with the IP3R-Grp75-VDAC complex to control ER-mitochondria interaction. Cell Calcium 2020, 87, 102186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Wang, W.; Hwang, J.; Namgung, U.; Min, K.-T. Increased ER–mitochondria tethering promotes axon regeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 16074–16079. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sander, P.; Gudermann, T.; Schredelseker, J. A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? Int. J. Mol. Sci. 2021, 22, 946. https://doi.org/10.3390/ijms22020946
Sander P, Gudermann T, Schredelseker J. A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? International Journal of Molecular Sciences. 2021; 22(2):946. https://doi.org/10.3390/ijms22020946
Chicago/Turabian StyleSander, Paulina, Thomas Gudermann, and Johann Schredelseker. 2021. "A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake?" International Journal of Molecular Sciences 22, no. 2: 946. https://doi.org/10.3390/ijms22020946
APA StyleSander, P., Gudermann, T., & Schredelseker, J. (2021). A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? International Journal of Molecular Sciences, 22(2), 946. https://doi.org/10.3390/ijms22020946