Advances in Research of Adult Gliomas

 , ,
, ,
Abstract
1. Introduction
2. Epidemiology and Classification of Adult Gliomas
3. Experimental Models of Adult Glioma
3.1. Cell Lines
3.2. Cerebral Organoids
3.3. Organotypic Brain Slice Cultures
3.4. In Vivo Models of Human Gliomas
3.5. Genetically Engineering Mouse Models of Glioma
3.6. Xenograft Models
3.7. Xenograft in ‘Humanized’ Mice
4. Intratumour Heterogeneity and Tumour Evolution of Adult Gliomas
4.1. Intra-Tumour Heterogeneity Using Novel Tissue Sampling
4.2. Complex Environmental Factors Shaping Dynamic Intratumour Gene Expressions
5. Tumour Microenvironment Variations in Tumour Type, Subtypes, and Type of Treatment
6. Computational and Mathematical Modelling of Glioma
6.1. Modelling Parameters of Glioma Invasion and Progression
6.2. Modelling Parameters of Blood–Brain Barrier Permeability
6.3. Discovering Evolutionary Patterns and Understanding the Emergence of Drug Resistance
6.4. Virtual Screening for New Drug Molecules and Virtual Clinical Trials
7. Epigenetic Alterations Found within Glioma, from Identification of Novel Biomarkers to Predictors of Clinical Outcomes and Treatment Options
7.1. Methylation Patterns Differ between IDHWT and IDHmut Gliomas
7.2. Methylation Shifts in Recurrent Glioma Represents a Switch to a More Aggressive Phenotype
7.3. Identification of a New DNA Modification in Glioblastoma
8. Liquid Biopsies Could Revolutionise Detection and Diagnosis of Glioma
9. Current Treatments and Future Therapy
9.1. Surgery for Diffuse Gliomas
9.2. Radiotherapy and Chemotherapy for Diffuse Gliomas
9.3. Immunotherapy and Targeted Therapy
9.4. Future Trials and Therapies
10. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jessen, K.R.; Mirsky, R. Glial cells in the enteric nervous system contain glial fibrillary acidic protein. Nature 1980, 286, 736–737. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Yang, R.; Li, K.; Yi, G.; Li, Z.; Guo, J.; Zhang, Z.; Junxiang, P.; Liu, Y.; Qi, S.; et al. Establishment of age group classification for risk stratification in glioma patients. BMC Neurol. 2020, 20, 310. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Brain Tumours: Rise in Glioblastoma Multiforme Incidence in England 1995-2015 Suggests an Adverse Environmental or Lifestyle Factor. J. Environ. Public Health 2018, 2018, 7910754. [Google Scholar] [CrossRef]
- Johnson, D.R. Rising incidence of glioblastoma and meningioma in the United States: Projections through 2050. J. Clin. Oncol. 2012, 30, 2065-2065. [Google Scholar] [CrossRef]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neuro-Oncology 2012, 107, 207–212. [Google Scholar] [CrossRef]
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus 2015, 38, E6. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chan, A.K.; Yao, Y.; Zhang, Z.; Chung, N.Y.; Liu, J.S.; Li, K.K.; Shi, Z.; Chan, D.T.; Poon, W.S.; Zhou, L.; et al. TERT promoter mutations contribute to subset prognostication of lower-grade gliomas. Mod. Pathol 2015, 28, 177–186. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Brandes, A.A.; Taphoorn, M.J.; Kros, J.M.; Kouwenhoven, M.C.; Delattre, J.Y.; Bernsen, H.J.; Frenay, M.; Tijssen, C.C.; Grisold, W.; et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 344–350. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Ohta, T.; Oh, J.E.; Kim, Y.H.; Kleihues, P.; Ohgaki, H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013, 126, 931–937. [Google Scholar] [CrossRef]
- Simon, M.; Hosen, I.; Gousias, K.; Rachakonda, S.; Heidenreich, B.; Gessi, M.; Schramm, J.; Hemminki, K.; Waha, A.; Kumar, R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro-Oncology 2015, 17, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network, Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef] [PubMed]
- Wick, W.; Hartmann, C.; Engel, C.; Stoffels, M.; Felsberg, J.; Stockhammer, F.; Sabel, M.C.; Koeppen, S.; Ketter, R.; Meyermann, R.; et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5874–5880. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Stupp, R.; Hegi, M.E.; van den Bent, M.; Tonn, J.C.; Sanson, M.; Wick, W.; Reifenberger, G. Personalized care in neuro-oncology coming of age: Why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro-Oncology 2012, 14 (Suppl. 4), iv100-108. [Google Scholar] [CrossRef]
- Jenkins, R.B.; Blair, H.; Ballman, K.V.; Giannini, C.; Arusell, R.M.; Law, M.; Flynn, H.; Passe, S.; Felten, S.; Brown, P.D.; et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006, 66, 9852–9861. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Gleize, V.; Alentorn, A.; Connen de Kerillis, L.; Labussiere, M.; Nadaradjane, A.A.; Mundwiller, E.; Ottolenghi, C.; Mangesius, S.; Rahimian, A.; Ducray, F.; et al. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann. Neurol. 2015, 78, 355–374. [Google Scholar] [CrossRef]
- Wesseling, P.; van den Bent, M.; Perry, A. Oligodendroglioma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 809–827. [Google Scholar] [CrossRef]
- Burnet, N.G.; Jefferies, S.J.; Benson, R.J.; Hunt, D.P.; Treasure, F.P. Years of life lost (YLL) from cancer is an important measure of population burden--and should be considered when allocating research funds. Br. J. Cancer 2005, 92, 241–245. [Google Scholar] [CrossRef]
- Nandakumar, P.; Mansouri, A.; Das, S. The Role of ATRX in Glioma Biology. Front. Oncol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Reuss, D.; Koelsche, C.; Capper, D.; Schittenhelm, J.; Heim, S.; Jones, D.T.; Pfister, S.M.; Herold-Mende, C.; Wick, W.; et al. Farewell to oligoastrocytoma: In situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol. 2014, 128, 551–559. [Google Scholar] [CrossRef]
- Wiestler, B.; Capper, D.; Sill, M.; Jones, D.T.; Hovestadt, V.; Sturm, D.; Koelsche, C.; Bertoni, A.; Schweizer, L.; Korshunov, A.; et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol. 2014, 128, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Diamond, E.L.; Wang, L.; Rosenblum, M.K. Mixed glioma with molecular features of composite oligodendroglioma and astrocytoma: A true “oligoastrocytoma”? Acta Neuropathol. 2015, 129, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, P.; Li, C.C.; Lee, M.; Shivalingam, B.; Brennan, J.; Suter, C.M.; Kaufman, K.; Lum, T.; Buckland, M.E. Oligoastrocytomas: Throwing the baby out with the bathwater? Acta Neuropathol. 2015, 129, 147–149. [Google Scholar] [CrossRef]
- Lenting, K.; Verhaak, R.; Ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef]
- Li, N.; Zhang, P.; Kiang, K.M.Y.; Cheng, Y.S.; Leung, G.K.K. Caffeine Sensitizes U87-MG Human Glioblastoma Cells to Temozolomide through Mitotic Catastrophe by Impeding G2 Arrest. BioMed Res. Int. 2018, 2018, 5364973. [Google Scholar] [CrossRef]
- Schrader, J.; Deuster, O.; Rinn, B.; Schulz, M.; Kautz, A.; Dodel, R.; Meyer, B.; Al-Abed, Y.; Balakrishnan, K.; Reese, J.P.; et al. Restoration of contact inhibition in human glioblastoma cell lines after MIF knockdown. BMC Cancer 2009, 9, 464. [Google Scholar] [CrossRef]
- Ernst, A.; Hofmann, S.; Ahmadi, R.; Becker, N.; Korshunov, A.; Engel, F.; Hartmann, C.; Felsberg, J.; Sabel, M.; Peterziel, H.; et al. Genomic and expression profiling of glioblastoma stem cell-like spheroid cultures identifies novel tumor-relevant genes associated with survival. Clin. Cancer Res. 2009, 15, 6541–6550. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re353. [Google Scholar] [CrossRef] [PubMed]
- Huszthy, P.C.; Daphu, I.; Niclou, S.P.; Stieber, D.; Nigro, J.M.; Sakariassen, P.O.; Miletic, H.; Thorsen, F.; Bjerkvig, R. In vivo models of primary brain tumors: Pitfalls and perspectives. Neuro-Oncology 2012, 14, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, A.; Stieber, D.; Enger, P.O.; Golebiewska, A.; Molven, A.; Svendsen, A.; Westermark, B.; Niclou, S.P.; Olsen, T.K.; Chekenya Enger, M.; et al. U-251 revisited: Genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. Cancer Med. 2014, 3, 812–824. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.A.; Tetzlaff, W.; Weiss, S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci 1992, 12, 4565–4574. [Google Scholar] [CrossRef]
- Vescovi, A.L.; Reynolds, B.A.; Fraser, D.D.; Weiss, S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron 1993, 11, 951–966. [Google Scholar] [CrossRef]
- Balvers, R.K.; Kleijn, A.; Kloezeman, J.J.; French, P.J.; Kremer, A.; van den Bent, M.J.; Dirven, C.M.; Leenstra, S.; Lamfers, M.L. Serum-free culture success of glial tumors is related to specific molecular profiles and expression of extracellular matrix-associated gene modules. Neuro-Oncology 2013, 15, 1684–1695. [Google Scholar] [CrossRef]
- Conti, L.; Pollard, S.M.; Gorba, T.; Reitano, E.; Toselli, M.; Biella, G.; Sun, Y.; Sanzone, S.; Ying, Q.L.; Cattaneo, E.; et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005, 3, e283. [Google Scholar] [CrossRef]
- Brewer, G.J.; Torricelli, J.R.; Evege, E.K.; Price, P.J. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 1993, 35, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, R.; Padmashri, R.; Dunaevsky, A.; Liu, Y.; Dreyfus, C.F.; Jiang, P. Pluripotent Stem Cell-Derived Cerebral Organoids Reveal Human Oligodendrogenesis with Dorsal and Ventral Origins. Stem Cell Rep. 2019, 12, 890–905. [Google Scholar] [CrossRef]
- Jin, M.Z.; Han, R.R.; Qiu, G.Z.; Ju, X.C.; Lou, G.; Jin, W.L. Organoids: An intermediate modeling platform in precision oncology. Cancer Lett. 2018, 414, 174–180. [Google Scholar] [CrossRef]
- Qian, X.; Song, H.; Ming, G.L. Brain organoids: Advances, applications and challenges. Development 2019, 146. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e3205. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid technology and applications in cancer research. J. Hematol. Oncol. 2018, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro-Oncology 2020, 22, 1138–1149. [Google Scholar] [CrossRef]
- Matsumura, H.; Ohnishi, T.; Kanemura, Y.; Maruno, M.; Yoshimine, T. Quantitative analysis of glioma cell invasion by confocal laser scanning microscopy in a novel brain slice model. Biochem. Biophys. Res. Commun. 2000, 269, 513–520. [Google Scholar] [CrossRef]
- Jensen, S.S.; Meyer, M.; Petterson, S.A.; Halle, B.; Rosager, A.M.; Aaberg-Jessen, C.; Thomassen, M.; Burton, M.; Kruse, T.A.; Kristensen, B.W. Establishment and Characterization of a Tumor Stem Cell-Based Glioblastoma Invasion Model. PLoS ONE 2016, 11, e0159746. [Google Scholar] [CrossRef] [PubMed]
- Minami, N.; Maeda, Y.; Shibao, S.; Arima, Y.; Ohka, F.; Kondo, Y.; Maruyama, K.; Kusuhara, M.; Sasayama, T.; Kohmura, E.; et al. Organotypic brain explant culture as a drug evaluation system for malignant brain tumors. Cancer Med. 2017, 6, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Marques-Torrejon, M.A.; Gangoso, E.; Pollard, S.M. Modelling glioblastoma tumour-host cell interactions using adult brain organotypic slice co-culture. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Costa, B.; Strelau, J.; Mittelbronn, M.; Angel, P.; Peterziel, H. An advanced glioma cell invasion assay based on organotypic brain slice cultures. BMC Cancer 2018, 18, 103. [Google Scholar] [CrossRef]
- Fomchenko, E.I.; Holland, E.C. Mouse models of brain tumors and their applications in preclinical trials. Clin. Cancer Res. 2006, 12, 5288–5297. [Google Scholar] [CrossRef]
- Kijima, N.; Kanemura, Y. Mouse Models of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane (AU), QLD, Australia, 2017. [Google Scholar] [CrossRef]
- Simeonova, I.; Huillard, E. In vivo models of brain tumors: Roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell Mol. Life Sci. 2014, 71, 4007–4026. [Google Scholar] [CrossRef]
- Reilly, K.M.; Loisel, D.A.; Bronson, R.T.; McLaughlin, M.E.; Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 2000, 26, 109–113. [Google Scholar] [CrossRef]
- Ding, H.; Roncari, L.; Shannon, P.; Wu, X.; Lau, N.; Karaskova, J.; Gutmann, D.H.; Squire, J.A.; Nagy, A.; Guha, A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001, 61, 3826–3836. [Google Scholar]
- Marumoto, T.; Tashiro, A.; Friedmann-Morvinski, D.; Scadeng, M.; Soda, Y.; Gage, F.H.; Verma, I.M. Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 2009, 15, 110–116. [Google Scholar] [CrossRef]
- Ding, H.; Shannon, P.; Lau, N.; Wu, X.; Roncari, L.; Baldwin, R.L.; Takebayashi, H.; Nagy, A.; Gutmann, D.H.; Guha, A. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res. 2003, 63, 1106–1113. [Google Scholar]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef]
- Brandenburg, S.; Muller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Maas, S.L.N.; Nieland, L.; Wei, Z.; Cheah, P.S.; Tai, E.; Kolsteeg, C.J.; Dusoswa, S.A.; Ting, D.T.; Hickman, S.; et al. Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep. 2019, 28, 3105–3119.e3107. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.; Su, Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis. Model. Mech. 2008, 1, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.M.; Keating, A.K. Creating anatomically accurate and reproducible intracranial xenografts of human brain tumors. J. Vis. Exp. 2014, e52017. [Google Scholar] [CrossRef] [PubMed]
- Irtenkauf, S.M.; Sobiechowski, S.; Hasselbach, L.A.; Nelson, K.K.; Transou, A.D.; Carlton, E.T.; Mikkelsen, T.; deCarvalho, A.C. Optimization of Glioblastoma Mouse Orthotopic Xenograft Models for Translational Research. Comp. Med. 2017, 67, 300–314. [Google Scholar] [PubMed]
- Lee, J.; Jo, D.H.; Kim, J.H.; Cho, C.S.; Han, J.E.; Kim, Y.; Park, H.; Yoo, S.H.; Yu, Y.S.; Moon, H.E.; et al. Development of a patient-derived xenograft model of glioblastoma via intravitreal injection in mice. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, J.; Liu, W.N.; Zhao, Y. Cancer Immunotherapies and Humanized Mouse Drug Testing Platforms. Transl Oncol 2019, 12, 987–995. [Google Scholar] [CrossRef]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef]
- Bernard, D.; Peakman, M.; Hayday, A.C. Establishing humanized mice using stem cells: Maximizing the potential. Clin. Exp. Immunol. 2008, 152, 406–414. [Google Scholar] [CrossRef]
- Nassereddine, S.; Rafei, H.; Elbahesh, E.; Tabbara, I. Acute Graft Versus Host Disease: A Comprehensive Review. Anticancer Res. 2017, 37, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 67, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Semenkow, S.; Li, S.; Kahlert, U.D.; Raabe, E.H.; Xu, J.; Arnold, A.; Janowski, M.; Oh, B.C.; Brandacher, G.; Bulte, J.W.M.; et al. An immunocompetent mouse model of human glioblastoma. Oncotarget 2017, 8, 61072–61082. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cao, Y.; Sundgren, P.C.; Tsien, C.I.; Chenevert, T.T.; Junck, L. Physiologic and metabolic magnetic resonance imaging in gliomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 1228–1235. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Spiteri, I.; Sottoriva, A.; Touloumis, A.; Ber, S.; Price, S.J.; Heywood, R.; Francis, N.J.; Howarth, K.D.; Collins, V.P.; et al. Contributions to drug resistance in glioblastoma derived from malignant cells in the sub-ependymal zone. Cancer Res. 2015, 75, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef]
- Mandel, J.J.; Yust-Katz, S.; Patel, A.J.; Cachia, D.; Liu, D.; Park, M.; Yuan, Y.; Kent, T.A.; de Groot, J.F. Inability of positive phase II clinical trials of investigational treatments to subsequently predict positive phase III clinical trials in glioblastoma. Neuro-Oncology 2018, 20, 113–122. [Google Scholar] [CrossRef]
- Mandel, J.J.; Youssef, M.; Ludmir, E.; Yust-Katz, S.; Patel, A.J.; De Groot, J.F. Highlighting the need for reliable clinical trials in glioblastoma. Expert Rev. Anticancer 2018, 18, 1031–1040. [Google Scholar] [CrossRef]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol 2014, 232, 165–177. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Huse, J.T.; Phillips, H.S.; Brennan, C.W. Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 2011, 59, 1190–1199. [Google Scholar] [CrossRef]
- Zheng, S.; Chheda, M.G.; Verhaak, R.G. Studying a complex tumor: Potential and pitfalls. Cancer J. 2012, 18, 107–114. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Ozawa, T.; Riester, M.; Cheng, Y.K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef]
- Wolf, D.A.; Hesterman, J.Y.; Sullivan, J.M.; Orcutt, K.D.; Silva, M.D.; Lobo, M.; Wellman, T.; Hoppin, J.; Verma, A. Dynamic dual-isotope molecular imaging elucidates principles for optimizing intrathecal drug delivery. JCI Insight 2016, 1, e85311. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Yan, J.; Kong, L.Y.; Hu, J.; Gabrusiewicz, K.; Dibra, D.; Xia, X.; Heimberger, A.B.; Li, S. FGL2 as a Multimodality Regulator of Tumor-Mediated Immune Suppression and Therapeutic Target in Gliomas. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Huettner, C.; Czub, S.; Kerkau, S.; Roggendorf, W.; Tonn, J.C. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997, 17, 3217–3224. [Google Scholar] [PubMed]
- Wagner, S.; Czub, S.; Greif, M.; Vince, G.H.; Suss, N.; Kerkau, S.; Rieckmann, P.; Roggendorf, W.; Roosen, K.; Tonn, J.C. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int. J. Cancer 1999, 82, 12–16. [Google Scholar] [CrossRef]
- Doucette, T.; Rao, G.; Rao, A.; Shen, L.; Aldape, K.; Wei, J.; Dziurzynski, K.; Gilbert, M.; Heimberger, A.B. Immune heterogeneity of glioblastoma subtypes: Extrapolation from the cancer genome atlas. Cancer Immunol Res. 2013, 1, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [PubMed]
- Le, L.Q.; Shipman, T.; Burns, D.K.; Parada, L.F. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell 2009, 4, 453–463. [Google Scholar] [CrossRef]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Lun, M.; Lok, E.; Gautam, S.; Wu, E.; Wong, E.T. The natural history of extracranial metastasis from glioblastoma multiforme. J. Neuro-Oncology 2011, 105, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Noroxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. Esmo Open 2016, 1, e000144. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, J.C.L.; Talkenberger, K.; Seifert, M.; Klink, B.; Hawkins-Daarud, A.; Swanson, K.R.; Hatzikirou, H.; Deutsch, A. The biology and mathematical modelling of glioma invasion: A review. J. R Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neuro-Oncology 2004, 70, 217–228. [Google Scholar] [CrossRef]
- Aubert, M.; Badoual, M.; Christov, C.; Grammaticos, B. A model for glioma cell migration on collagen and astrocytes. J. R Soc. Interface 2008, 5, 75–83. [Google Scholar] [CrossRef]
- Oliveira, R.; Christov, C.; Guillamo, J.S.; de Bouard, S.; Palfi, S.; Venance, L.; Tardy, M.; Peschanski, M. Contribution of gap junctional communication between tumor cells and astroglia to the invasion of the brain parenchyma by human glioblastomas. BMC Cell Biol. 2005, 6, 7. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6. [Google Scholar] [CrossRef]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ’Go or grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gallagher, E.; Jorgensen, C.; Troendle, E.P.; Hu, D.; Searson, P.C.; Ulmschneider, M.B. An experimentally validated approach to calculate the blood-brain barrier permeability of small molecules. Sci. Rep. 2019, 9, 6117. [Google Scholar] [CrossRef]
- Goliaei, A.; Adhikari, U.; Berkowitz, M.L. Opening of the blood-brain barrier tight junction due to shock wave induced bubble collapse: A molecular dynamics simulation study. ACS Chem. Neurosci. 2015, 6, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, A.; Banerjee, R. Nanobubble Liposome Complexes for Diagnostic Imaging and Ultrasound-Triggered Drug Delivery in Cancers: A Theranostic Approach. ACS Omega 2019, 4, 15567–15580. [Google Scholar] [CrossRef]
- Caravagna, G.; Giarratano, Y.; Ramazzotti, D.; Tomlinson, I.; Graham, T.A.; Sanguinetti, G.; Sottoriva, A. Detecting repeated cancer evolution from multi-region tumor sequencing data. Nat. Methods 2018, 15, 707–714. [Google Scholar] [CrossRef]
- Brady, S.W.; McQuerry, J.A.; Qiao, Y.; Piccolo, S.R.; Shrestha, G.; Jenkins, D.F.; Layer, R.M.; Pedersen, B.S.; Miller, R.H.; Esch, A.; et al. Combating subclonal evolution of resistant cancer phenotypes. Nat. Commun. 2017, 8, 1231. [Google Scholar] [CrossRef]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef]
- Zhao, B.; Hemann, M.T.; Lauffenburger, D.A. Modeling Tumor Clonal Evolution for Drug Combinations Design. Trends Cancer 2016, 2, 144–158. [Google Scholar] [CrossRef]
- Acar, A.; Nichol, D.; Fernandez-Mateos, J.; Cresswell, G.D.; Barozzi, I.; Hong, S.P.; Spiteri, I.; Stubbs, M.; Burke, R.; Stewart, A.; et al. Exploiting evolutionary herding to control drug resistance in cancer. BioRxiv 2019, 566950. [Google Scholar] [CrossRef]
- Grier, J.T.; Batchelor, T. Low-grade gliomas in adults. Oncologist 2006, 11, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, V.M.; Ayala-Hernandez, L.E.; Belmonte-Beitia, J.; Schucht, P.; Murek, M.; Raabe, A.; Sepulveda, J. Computational design of improved standardized chemotherapy protocols for grade II oligodendrogliomas. PLoS Comput Biol. 2019, 15, e1006778. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Cova, T.; Sousa, J.; Vitorino, C.; Pais, A. Computational modeling in glioblastoma: From the prediction of blood-brain barrier permeability to the simulation of tumor behavior. Future Med. Chem. 2018, 10, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, L.; Zheng, L.; Zhang, M.; Qiu, C.; Zhang, Y.; Du, D.; Niu, B. 2D-QSAR and 3D-QSAR Analyses for EGFR Inhibitors. BioMed Res. Int. 2017, 2017, 4649191. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D., Jr. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Li, X.; He, L.; Zhu, Y. Computer-aided design of temozolomide derivatives based on alkylglycerone phosphate synthase structure with isothiocyanate and their pharmacokinetic/toxicity prediction and anti-tumor activity in vitro. Biomed. Rep. 2018, 8, 235–240. [Google Scholar] [CrossRef]
- Shen, L.; Song, C.X.; He, C.; Zhang, Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu. Rev. Biochem 2014, 83, 585–614. [Google Scholar] [CrossRef]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin 2015, 8, 24. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, M.; Ficz, G.; Oxley, D.; Raiber, E.A.; Bachman, M.; Booth, M.J.; Andrews, S.; Balasubramanian, S.; Reik, W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013, 14, R119. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Turcan, S. Epigenetic Reprogramming for Targeting IDH-Mutant Malignant Gliomas. Cancers 2019, 11, 1616. [Google Scholar] [CrossRef] [PubMed]
- Unruh, D.; Zewde, M.; Buss, A.; Drumm, M.R.; Tran, A.N.; Scholtens, D.M.; Horbinski, C. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci. Rep. 2019, 9, 8946. [Google Scholar] [CrossRef]
- Gusyatiner, O.; Hegi, M.E. Glioma epigenetics: From subclassification to novel treatment options. Semin Cancer Biol. 2018, 51, 50–58. [Google Scholar] [CrossRef]
- de Souza, C.F.; Sabedot, T.S.; Malta, T.M.; Stetson, L.; Morozova, O.; Sokolov, A.; Laird, P.W.; Wiznerowicz, M.; Iavarone, A.; Snyder, J.; et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep. 2018, 23, 637–651. [Google Scholar] [CrossRef]
- Xiao, C.L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.Q.; Luo, F.; et al. N(6)-Methyladenine DNA Modification in the Human Genome. Mol. Cell 2018, 71, 306–318.e307. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, T.P.; Gimple, R.C.; Li, Z.; Prager, B.C.; Wu, Q.; Yu, Y.; Wang, P.; Wang, Y.; Gorkin, D.U.; et al. N(6)-methyladenine DNA Modification in Glioblastoma. Cell 2018, 175, 1228–1243.e1220. [Google Scholar] [CrossRef]
- Saenz-Antonanzas, A.; Auzmendi-Iriarte, J.; Carrasco-Garcia, E.; Moreno-Cugnon, L.; Ruiz, I.; Villanua, J.; Egana, L.; Otaegui, D.; Sampron, N.; Matheu, A. Liquid Biopsy in Glioblastoma: Opportunities, Applications and Challenges. Cancers 2019, 11, 950. [Google Scholar] [CrossRef]
- Shankar, G.M.; Balaj, L.; Stott, S.L.; Nahed, B.; Carter, B.S. Liquid biopsy for brain tumors. Expert Rev. Mol. Diagn 2017, 17, 943–947. [Google Scholar] [CrossRef]
- Pantel, K. Blood-Based Analysis of Circulating Cell-Free DNA and Tumor Cells for Early Cancer Detection. PLoS Med. 2016, 13, e1002205. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Sabedot, T.; Malta, T.; Nelson, K.; Snyder, J.; Wells, M.; deCarvalho, A.; Mukherjee, A.; Chitale, D.; Mosella, M.; et al. Detection of glioma and prognostic subtypes by non-invasive circulating cell-free DNA methylation markers. bioRxiv 2019, 601245. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Petzold, A.; Bennett, J.L.; Berven, F.S.; Brundin, L.; Comabella, M.; Franciotta, D.; Frederiksen, J.L.; Fleming, J.O.; Furlan, R.; et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009, 73, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Berger, M.S. Surgical oncology for gliomas: The state of the art. Nat. Rev. Clin. Oncol. 2018, 15, 112–125. [Google Scholar] [CrossRef]
- National Guideline, A. National Guideline, A. National Institute for Health and Care Excellence: Clinical Guidelines. In Brain Tumours (Primary) and Brain Metastases in Adults; National Institute for Health and Care Excellence (UK) Copyright © NICE 2018.: London, UK, 2018. [Google Scholar]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef]
- Gulati, S.; Jakola, A.S.; Nerland, U.S.; Weber, C.; Solheim, O. The risk of getting worse: Surgically acquired deficits, perioperative complications, and functional outcomes after primary resection of glioblastoma. World Neurosurg. 2011, 76, 572–579. [Google Scholar] [CrossRef]
- Rahman, M.; Abbatematteo, J.; De Leo, E.K.; Kubilis, P.S.; Vaziri, S.; Bova, F.; Sayour, E.; Mitchell, D.; Quinones-Hinojosa, A. The effects of new or worsened postoperative neurological deficits on survival of patients with glioblastoma. J. Neurosurg. 2017, 127, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Pichlmeier, U.; Bink, A.; Schackert, G.; Stummer, W. Resection and survival in glioblastoma multiforme: An RTOG recursive partitioning analysis of ALA study patients. Neuro-Oncology 2008, 10, 1025–1034. [Google Scholar] [CrossRef]
- Szelényi, A.; Bello, L.; Duffau, H.; Fava, E.; Feigl, G.C.; Galanda, M.; Neuloh, G.; Signorelli, F.; Sala, F. Intraoperative electrical stimulation in awake craniotomy: Methodological aspects of current practice. Neurosurg. Focus 2010, 28, E7. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C.; Robles, S.G.; Zwinderman, A.H.; Duffau, H.; Berger, M.S. Impact of intraoperative stimulation brain mapping on glioma surgery outcome: A meta-analysis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2559–2565. [Google Scholar] [CrossRef] [PubMed]
- Schucht, P.; Beck, J.; Abu-Isa, J.; Andereggen, L.; Murek, M.; Seidel, K.; Stieglitz, L.; Raabe, A. Gross total resection rates in contemporary glioblastoma surgery: Results of an institutional protocol combining 5-aminolevulinic acid intraoperative fluorescence imaging and brain mapping. Neurosurgery 2012, 71, 927–935; discussion 926–935. [Google Scholar] [CrossRef]
- Schucht, P.; Seidel, K.; Beck, J.; Murek, M.; Jilch, A.; Wiest, R.; Fung, C.; Raabe, A. Intraoperative monopolar mapping during 5-ALA-guided resections of glioblastomas adjacent to motor eloquent areas: Evaluation of resection rates and neurological outcome. Neurosurg. Focus 2014, 37, E16. [Google Scholar] [CrossRef]
- Senft, C.; Bink, A.; Franz, K.; Vatter, H.; Gasser, T.; Seifert, V. Intraoperative MRI guidance and extent of resection in glioma surgery: A randomised, controlled trial. Lancet Oncol. 2011, 12, 997–1003. [Google Scholar] [CrossRef]
- Mahboob, S.; McPhillips, R.; Qiu, Z.; Jiang, Y.; Meggs, C.; Schiavone, G.; Button, T.; Desmulliez, M.; Demore, C.; Cochran, S.; et al. Intraoperative Ultrasound-Guided Resection of Gliomas: A Meta-Analysis and Review of the Literature. World Neurosurg. 2016, 92, 255–263. [Google Scholar] [CrossRef]
- Jenkinson, M.D.; Barone, D.G.; Bryant, A.; Vale, L.; Bulbeck, H.; Lawrie, T.A.; Hart, M.G.; Watts, C. Intraoperative imaging technology to maximise extent of resection for glioma. Cochrane Database Syst. Rev. 2018, 1, Cd012788. [Google Scholar] [CrossRef] [PubMed]
- Gorlia, T.; Wu, W.; Wang, M.; Baumert, B.G.; Mehta, M.; Buckner, J.C.; Shaw, E.; Brown, P.; Stupp, R.; Galanis, E.; et al. New validated prognostic models and prognostic calculators in patients with low-grade gliomas diagnosed by central pathology review: A pooled analysis of EORTC/RTOG/NCCTG phase III clinical trials. Neuro-Oncology 2013, 15, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Buckner, J.; Giannini, C.; Eckel-Passow, J.; Lachance, D.; Parney, I.; Laack, N.; Jenkins, R. Management of diffuse low-grade gliomas in adults - use of molecular diagnostics. Nat. Rev. Neurol. 2017, 13, 340–351. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Afra, D.; de Witte, O.; Ben Hassel, M.; Schraub, S.; Hoang-Xuan, K.; Malmstrom, P.O.; Collette, L.; Pierart, M.; Mirimanoff, R.; et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: The EORTC 22845 randomised trial. Lancet 2005, 366, 985–990. [Google Scholar] [CrossRef]
- Baumert, B.G.; Hegi, M.E.; van den Bent, M.J.; von Deimling, A.; Gorlia, T.; Hoang-Xuan, K.; Brandes, A.A.; Kantor, G.; Taphoorn, M.J.B.; Hassel, M.B.; et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): A randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016, 17, 1521–1532. [Google Scholar] [CrossRef]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.B.; Brown, P.D.; Felten, S.J.; Wu, W.; Buckner, J.C.; Arusell, R.M.; Curran, W.J.; Abrams, R.A.; Schiff, D.; Shaw, E.G. Validation of EORTC prognostic factors for adults with low-grade glioma: A report using intergroup 86-72-51. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.B.; Maat, B.; Hatlevoll, R.; Menten, J.; Rutten, E.H.; Thomas, D.G.; Mascarenhas, F.; Horiot, J.C.; Parvinen, L.M.; van Reijn, M.; et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 549–556. [Google Scholar] [CrossRef]
- Shaw, E.; Arusell, R.; Scheithauer, B.; O’Fallon, J.; O’Neill, B.; Dinapoli, R.; Nelson, D.; Earle, J.; Jones, C.; Cascino, T.; et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: Initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 2267–2276. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Malmstrom, A.; Gronberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sabel, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715. [Google Scholar] [CrossRef]
- Bush, N.A.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Piantadosi, S.; Burger, P.C.; Walker, M.; Selker, R.; Vick, N.A.; Black, K.; Sisti, M.; Brem, S.; Mohr, G.; et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet 1995, 345, 1008–1012. [Google Scholar] [CrossRef]
- Harrington, S.E.; Smith, T.J. The role of chemotherapy at the end of life:”when is enough, enough?”. JAMA 2008, 299, 2667–2678. [Google Scholar] [CrossRef] [PubMed]
- McGovern, P.C.; Lautenbach, E.; Brennan, P.J.; Lustig, R.A.; Fishman, N.O. Risk factors for postcraniotomy surgical site infection after 1,3-bis (2-chloroethyl)-1-nitrosourea (Gliadel) wafer placement. Clin. Infect. Dis 2003, 36, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Meinel, T.; Ewelt, C.; Martus, P.; Jakobs, O.; Felsberg, J.; Reifenberger, G. Prospective cohort study of radiotherapy with concomitant and adjuvant temozolomide chemotherapy for glioblastoma patients with no or minimal residual enhancing tumor load after surgery. J. Neuro-Oncology 2012, 108, 89–97. [Google Scholar] [CrossRef]
- McGirt, M.J.; Than, K.D.; Weingart, J.D.; Chaichana, K.L.; Attenello, F.J.; Olivi, A.; Laterra, J.; Kleinberg, L.R.; Grossman, S.A.; Brem, H.; et al. Gliadel (BCNU) wafer plus concomitant temozolomide therapy after primary resection of glioblastoma multiforme. J. Neurosurg. 2009, 110, 583–588. [Google Scholar] [CrossRef]
- Filley, A.C.; Dey, M. Dendritic cell based vaccination strategy: An evolving paradigm. J. Neuro-Oncology 2017, 133, 223–235. [Google Scholar] [CrossRef]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharm. 2017, 8, 242. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Xiao, Q.; Yang, S.; Ding, G.; Luo, M. Anti-vascular endothelial growth factor in glioblastoma: A systematic review and meta-analysis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2018, 39, 2021–2031. [Google Scholar] [CrossRef]
- Wick, W.; Weller, M.; Weiler, M.; Batchelor, T.; Yung, A.W.; Platten, M. Pathway inhibition: Emerging molecular targets for treating glioblastoma. Neuro-Oncology 2011, 13, 566–579. [Google Scholar] [CrossRef]
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2020, 22, 684–693. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. Acs Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol. Cancer Res. Mcr. 2016, 14, 976–983. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]


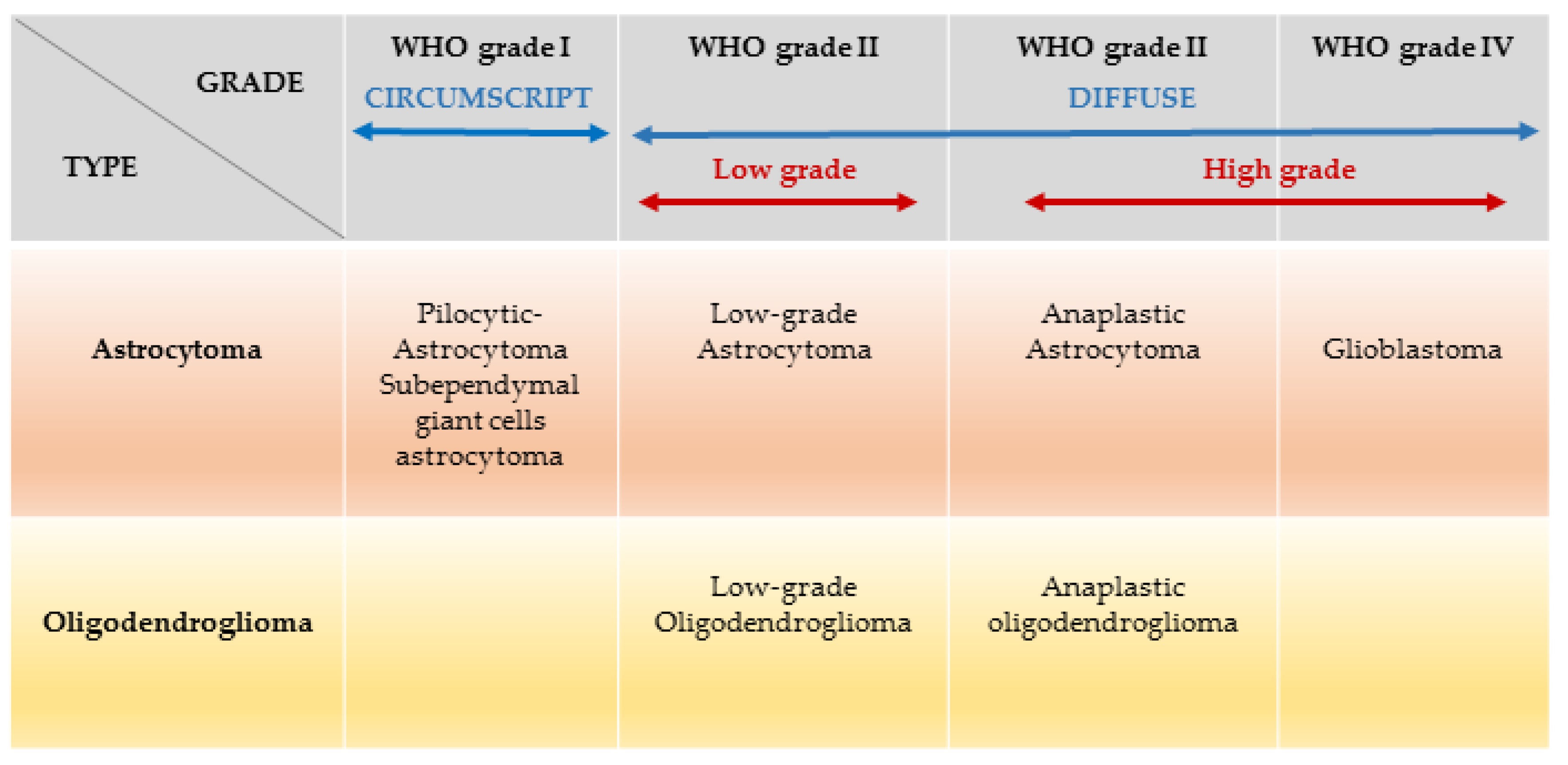{kind=link}
| Cell Type | Classification | Histological Grading | Molecular Classification |
|---|---|---|---|
| ‘Diffuse’ astrocytomas | Diffuse astrocytoma (A) | II | IDH wildtype (30%) IDH mutant (70%) |
| Anaplastic astrocytoma (AA) | III | ||
| Glioblastoma (GB) | IV | IDH wildtype (90%) IDH mutant (10%) | |
| Oligodendrogliomas | Oligodendroglioma (O) | II | IDH mutant (100%) 1p19q codeleted (100%) |
| Anaplastic oligodendroglioma (AO) | III |
| Summary | Advantages | Disadvantages | |
|---|---|---|---|
Monolayer cell culture |
|
|
|
Neuro/tumoursphere culture |
|
|
|
Cerebral organoids |
|
|
|
Organotypic brain slice culture |
|
|
|
Genetically engineered mouse models |
|
|
|
Xenograft mouse models |
|
|
|
“Humanised” xenograft mouse models |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finch, A.; Solomou, G.; Wykes, V.; Pohl, U.; Bardella, C.; Watts, C. Advances in Research of Adult Gliomas. Int. J. Mol. Sci. 2021, 22, 924. https://doi.org/10.3390/ijms22020924
Finch A, Solomou G, Wykes V, Pohl U, Bardella C, Watts C. Advances in Research of Adult Gliomas. International Journal of Molecular Sciences. 2021; 22(2):924. https://doi.org/10.3390/ijms22020924
Chicago/Turabian StyleFinch, Alina, Georgios Solomou, Victoria Wykes, Ute Pohl, Chiara Bardella, and Colin Watts. 2021. "Advances in Research of Adult Gliomas" International Journal of Molecular Sciences 22, no. 2: 924. https://doi.org/10.3390/ijms22020924
APA StyleFinch, A., Solomou, G., Wykes, V., Pohl, U., Bardella, C., & Watts, C. (2021). Advances in Research of Adult Gliomas. International Journal of Molecular Sciences, 22(2), 924. https://doi.org/10.3390/ijms22020924