Molecular Changes Underlying Hypertrophic Scarring Following Burns Involve Specific Deregulations at All Wound Healing Stages (Inflammation, Proliferation and Maturation)

 ,
,  and
and 
Abstract
:1. Introduction
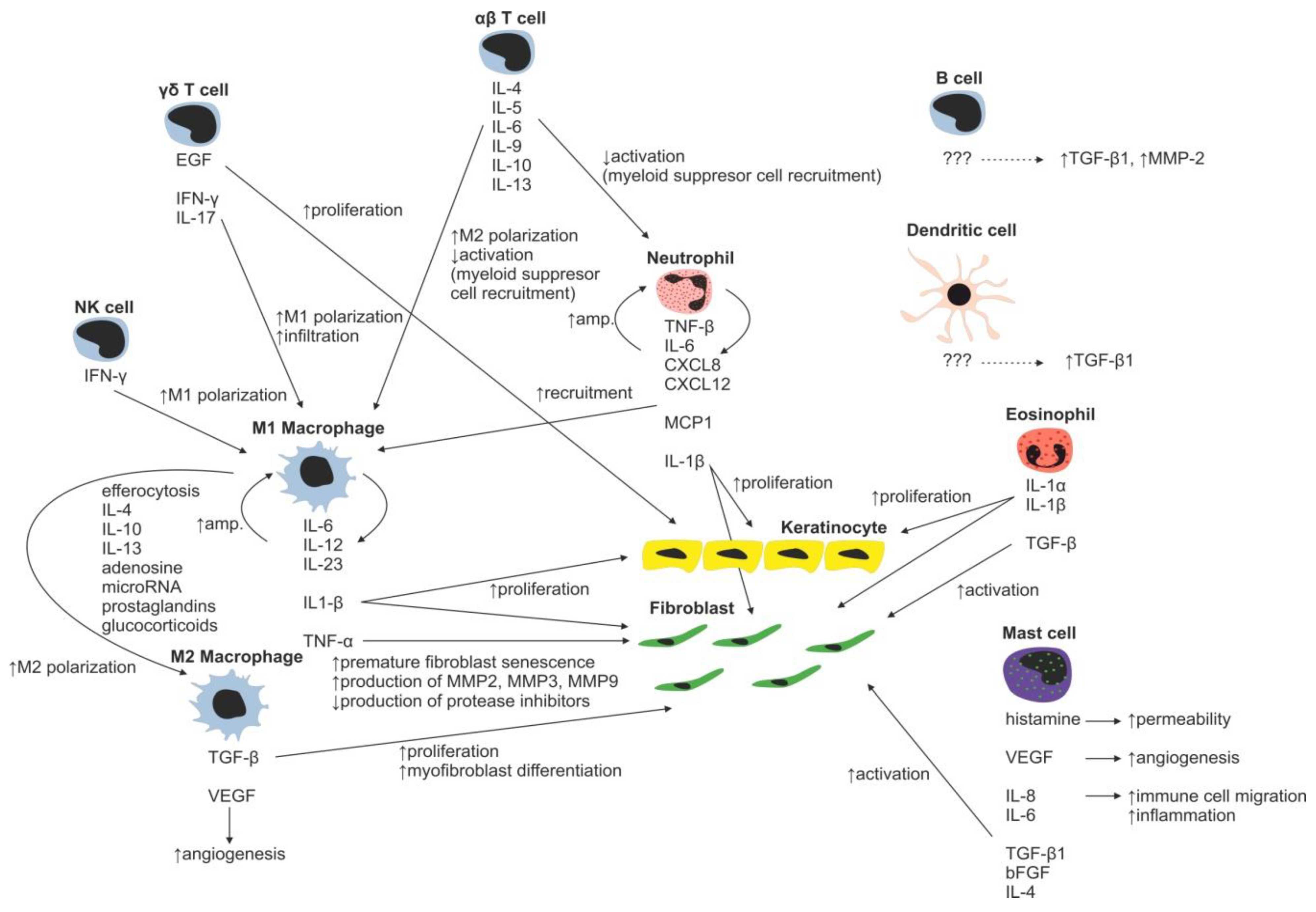
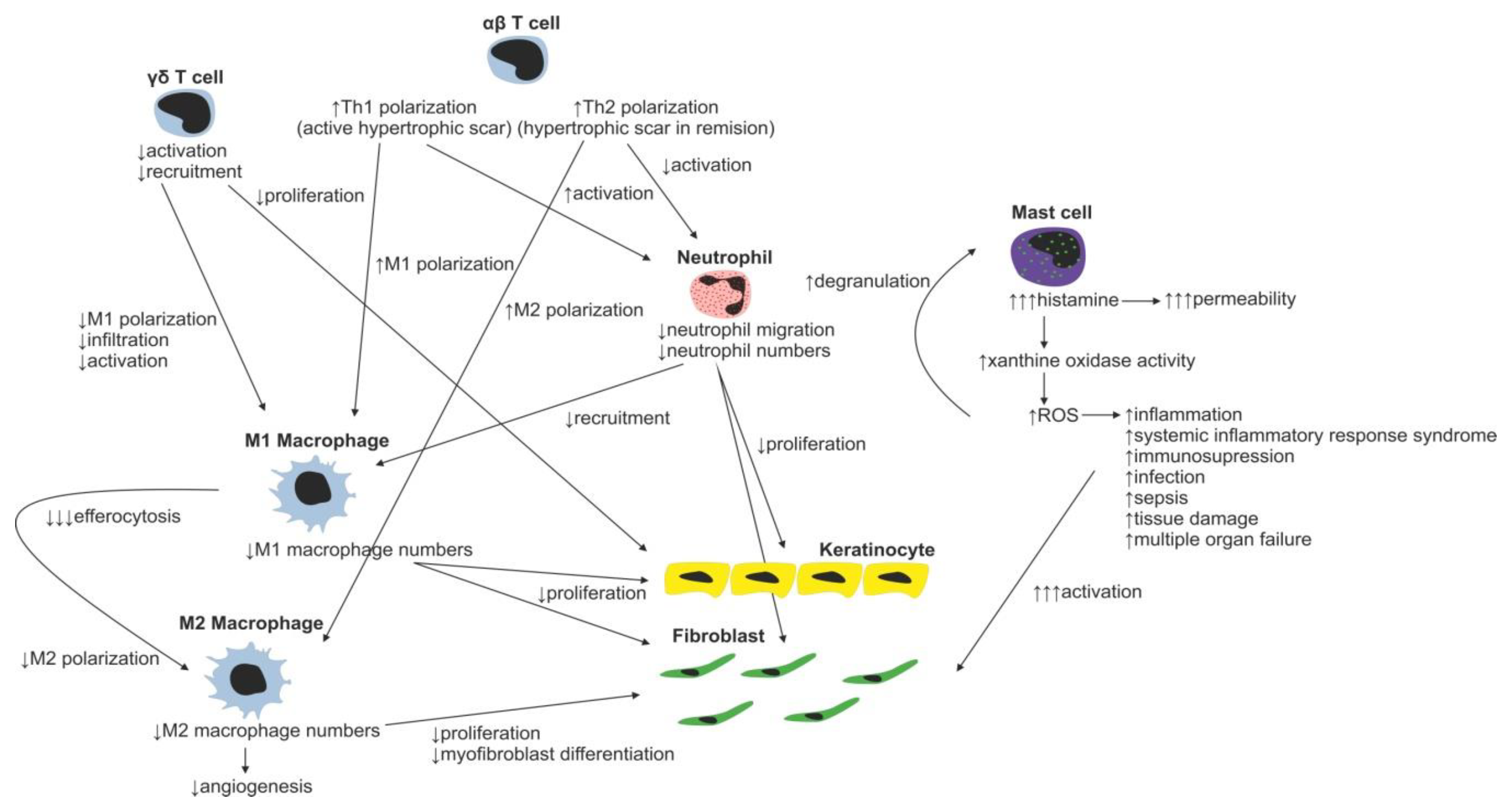
2. Inflammatory Phase of Wound Healing
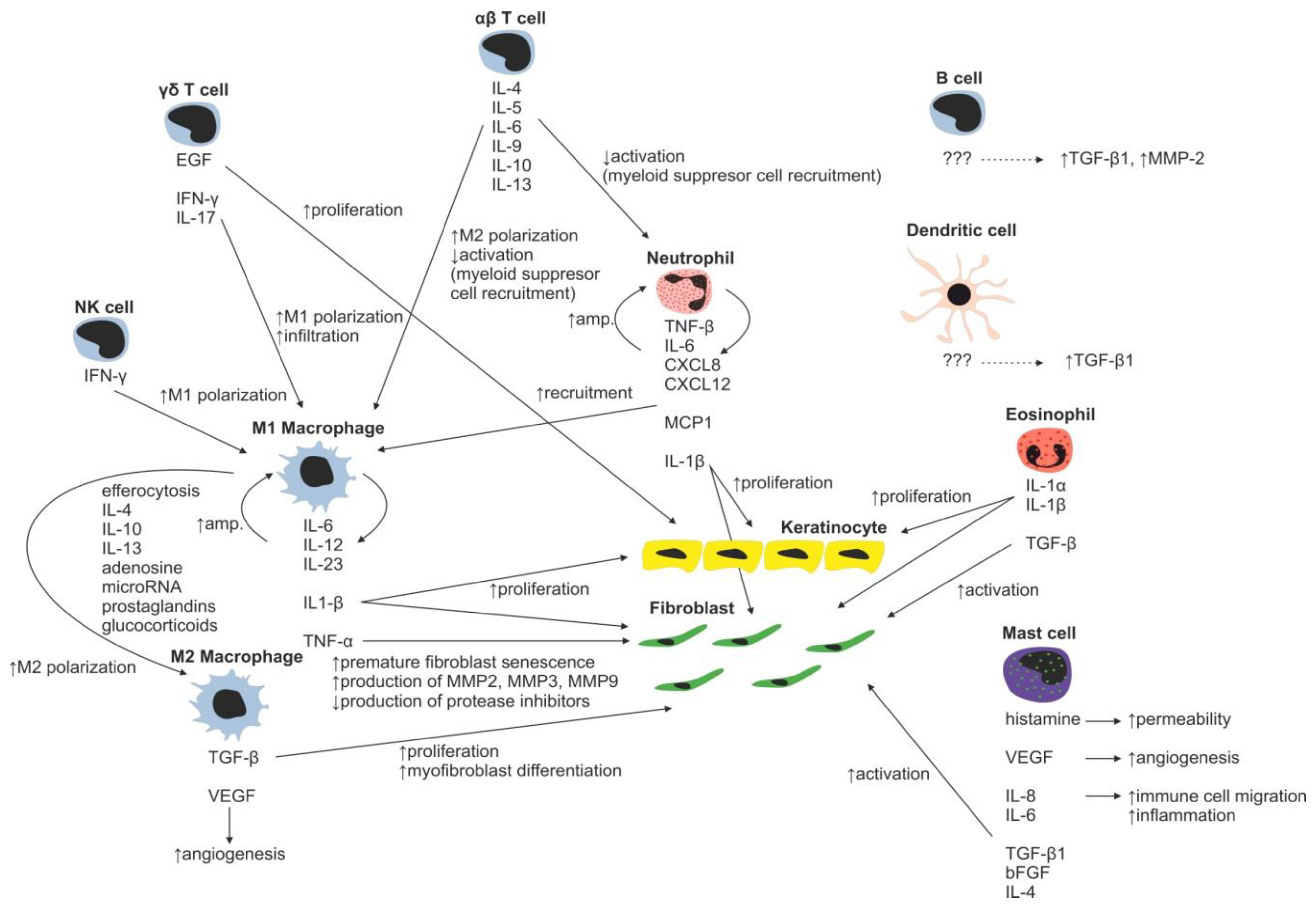
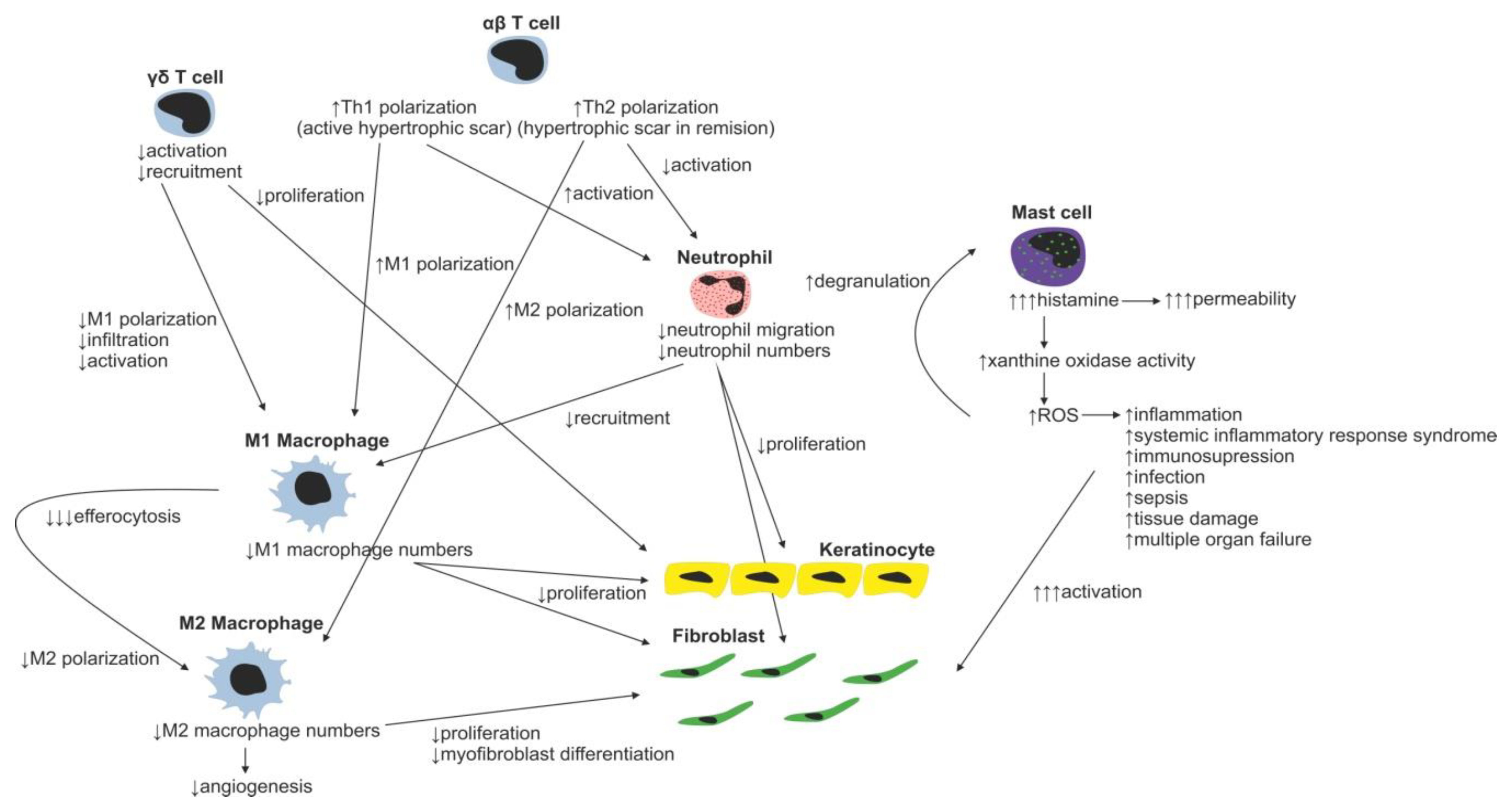
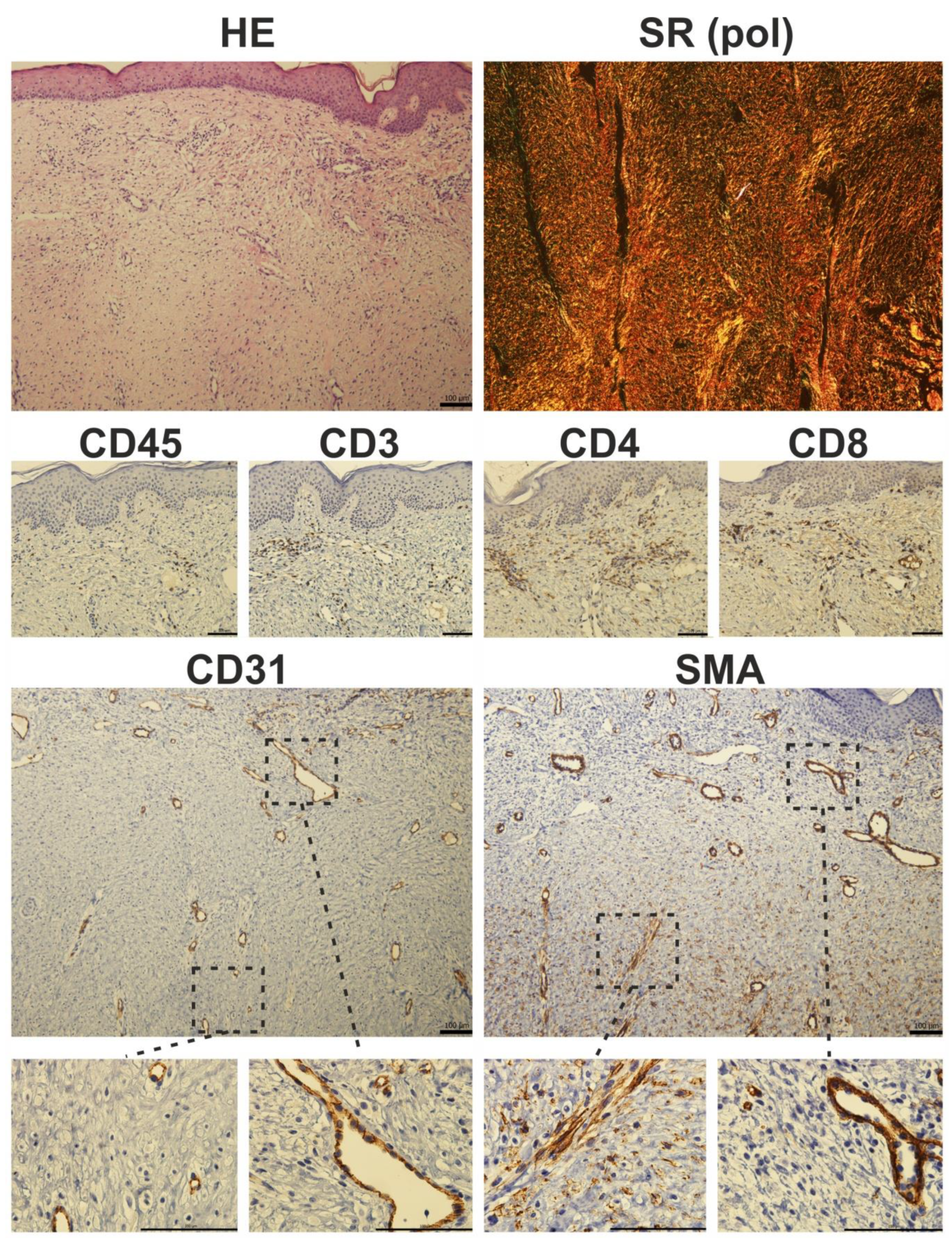
Immune Cells
3. Proliferation Phase of Wound Healing
3.1. Epidermis Regeneration
Epithelial-to-Mesenchymal/Mesenchymal-to-Epithelial Transition
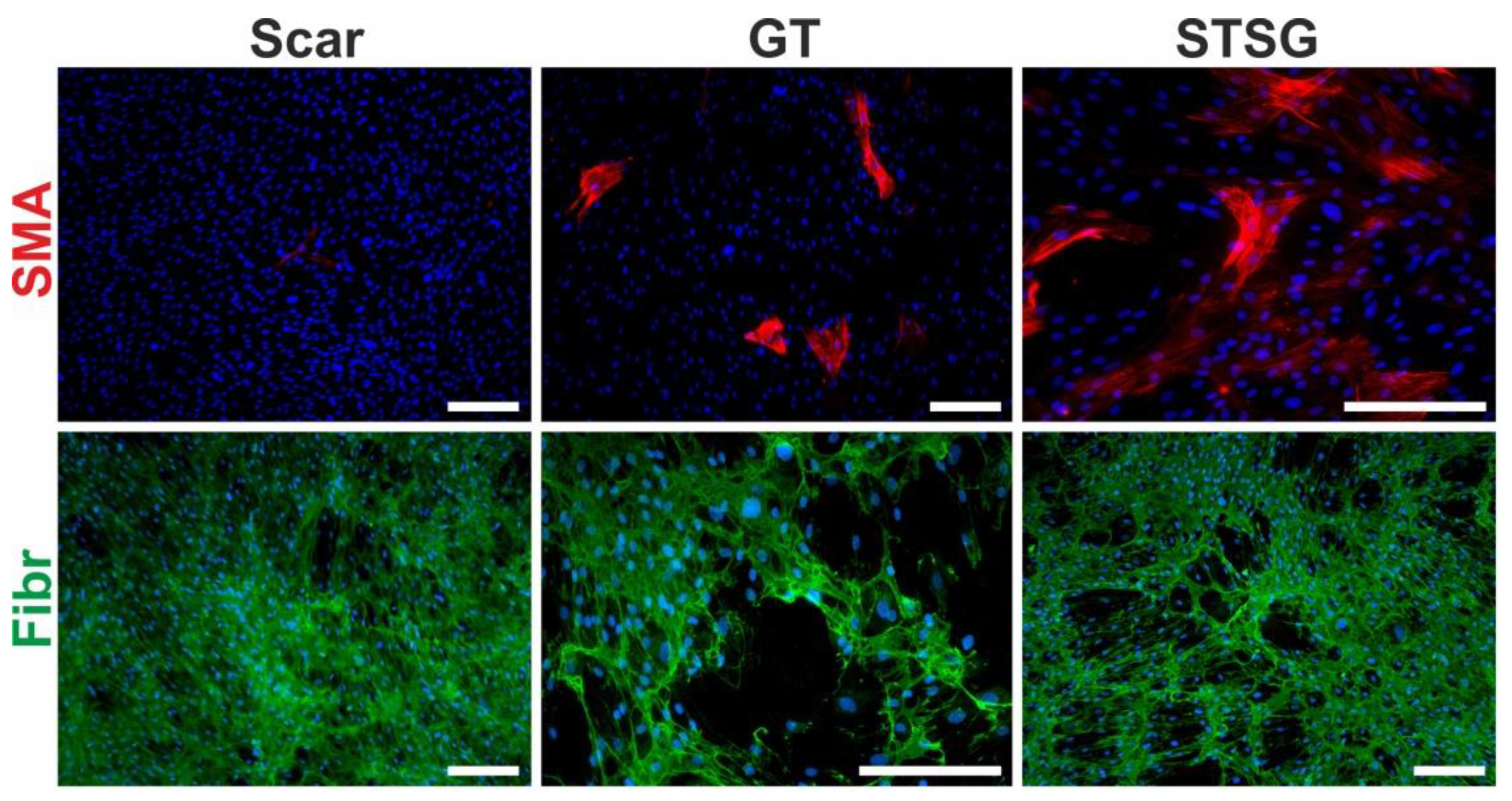
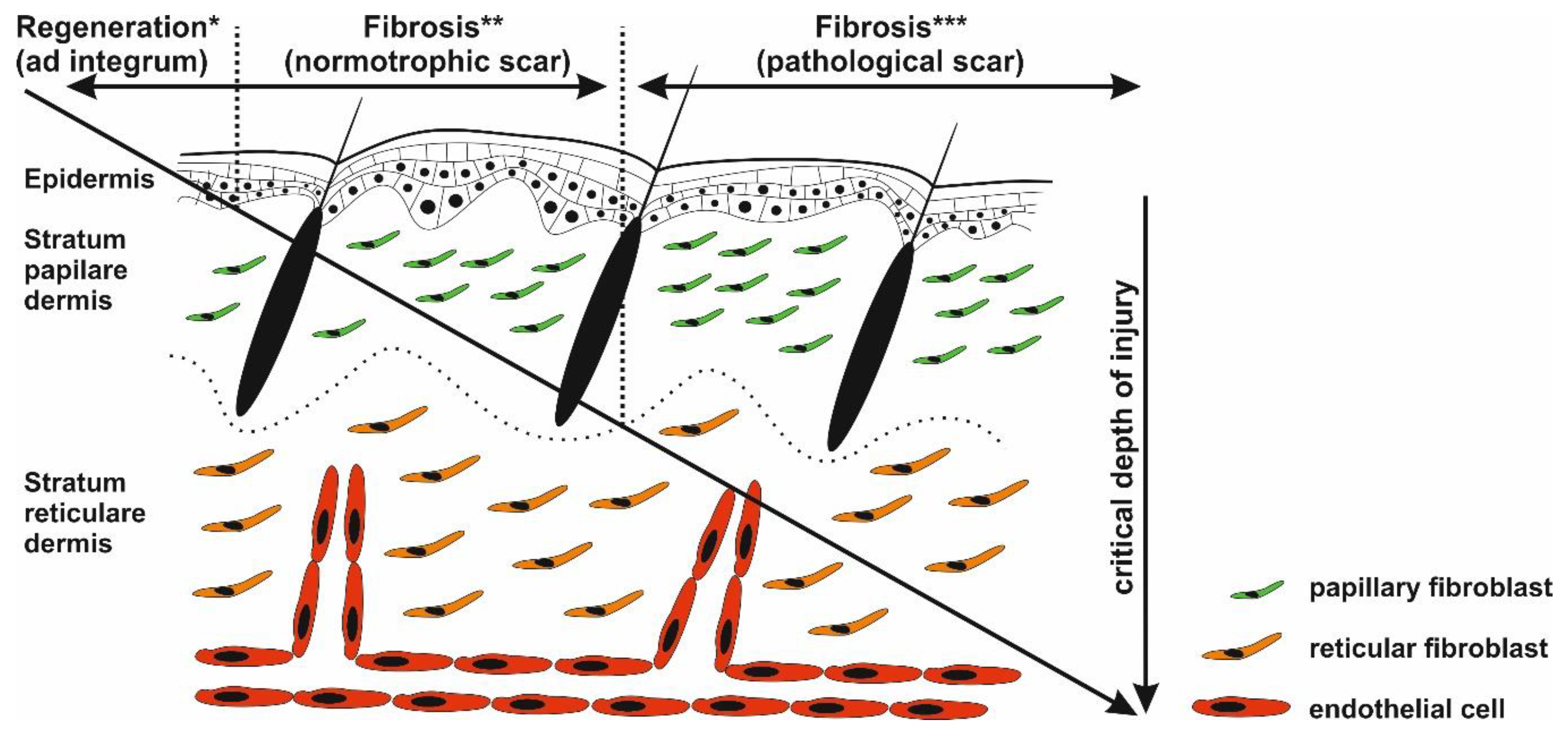
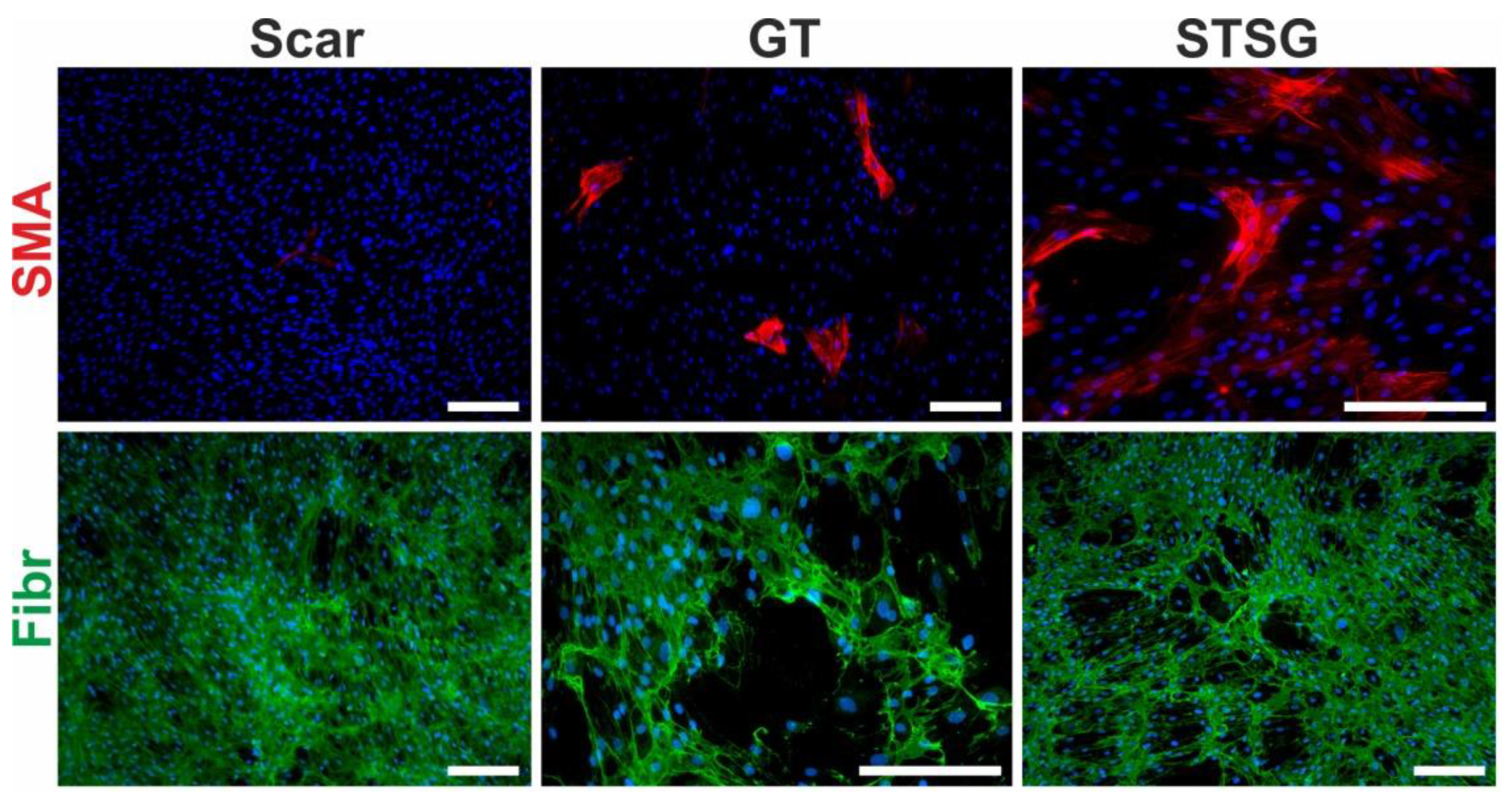
3.2. Fibroblast, the Key Cell
ECM Deposition
3.3. Angiogenesis
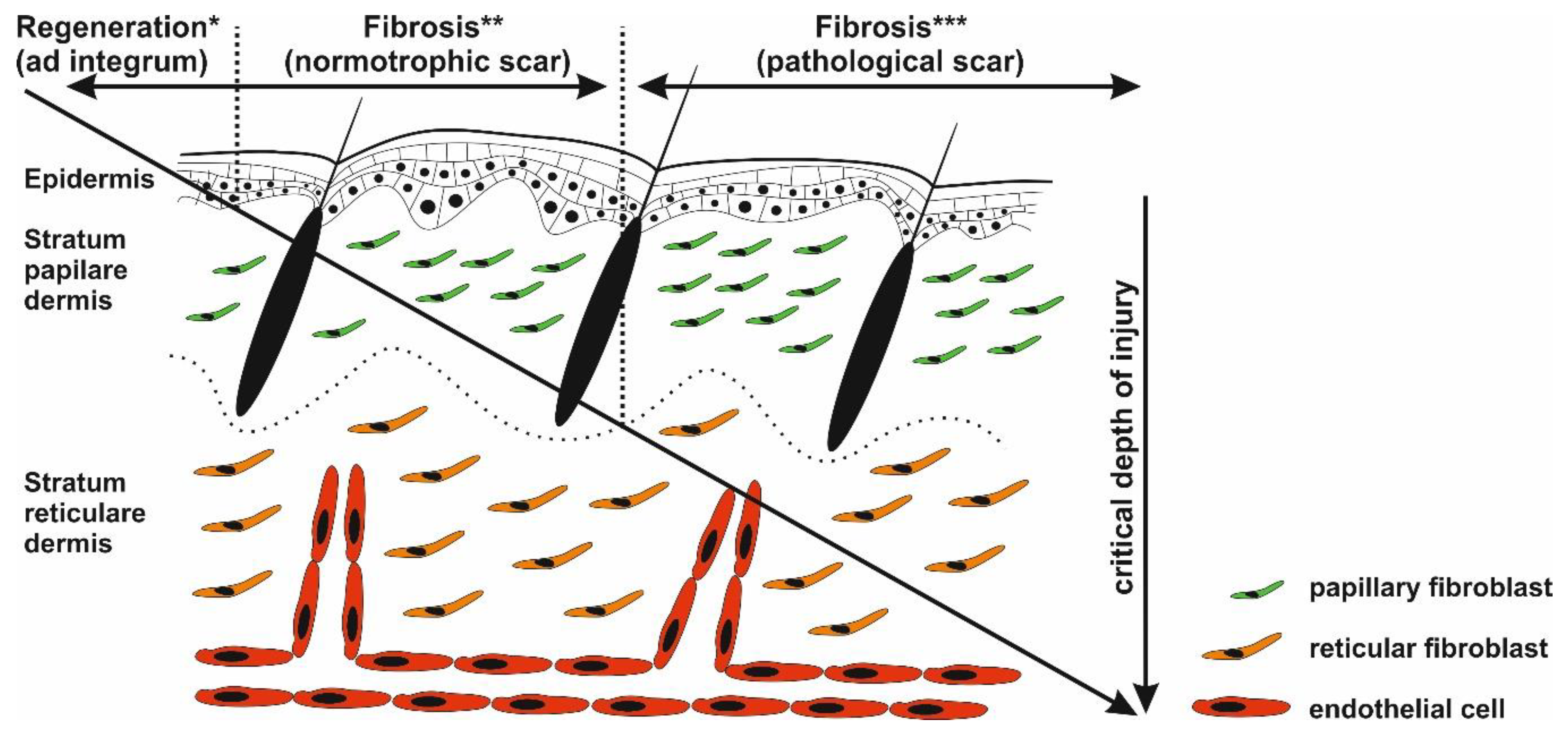
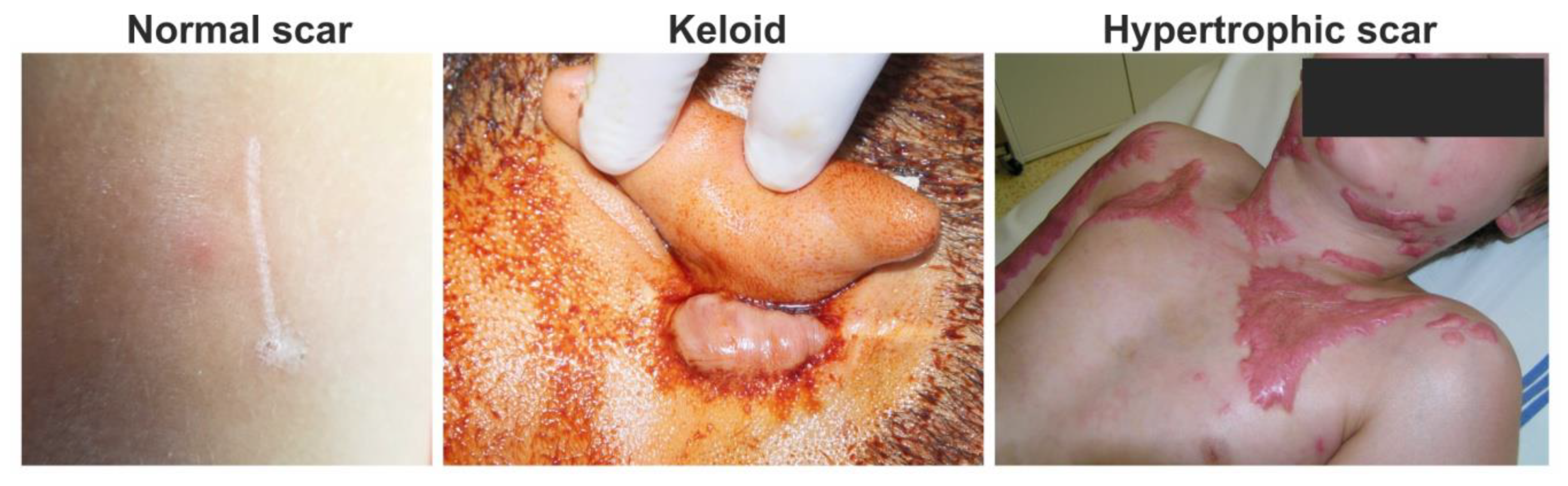
4. Maturation and Remodeling Phase of Wound Healing
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aFGF | Acidic fibroblast growth factor |
| bFGF | Basic fibroblast growth factor |
| CAF | Cancer-associated fibroblast |
| CD | Cluster of differentiation |
| CTGF | Connective tissue growth factor |
| CXCL | Chemokine ligand |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EMT | Epithelial-mesenchymal transition |
| FAK | Focal adhesion kinase |
| Fibr | Fibronectin |
| FOXA | Forkhead Box A |
| Gal | Galectin |
| GRHL2 | Grainyhead-like protein 2 homolog |
| GATA | guanine, adenine, thymine, adenine |
| GT | Granulation tissue |
| HGF | Hepatocyte growth factor |
| HIF | Hypoxia inducible factor |
| HNF1A | HNF1 homeobox A |
| IGF | Insulin growth factor |
| IL | Interleukin |
| IFN | Interferon |
| KLF4 | Kruppel-like factor 4 |
| MAPK | Mitogen-activated protein kinase |
| MET | Mesenchymal-to-epithelial transition |
| MMP | Matrix metalloproteinase |
| OVOL2 | Ovo-like 2 |
| PDGF | Platelet-derived growth factor |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphoinositide 3-kinase |
| ROS | Reactive oxygen species |
| SMA | Smooth muscle actin |
| STSG | Split thickness skin graft |
| TIMP | Tissue inhibitor of matrix metalloproteinase |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| TP63 | Tumor protein p63 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| WME | Wound microenvironment |
References
- Bayat, A.; McGrouther, D.A.; Ferguson, M.W. Skin scarring. BMJ 2003, 326, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.S.; Dover, J.S.; Arndt, K.A.; Kaminer, M.S. Scar management: Keloid, hypertrophic, atrophic, and acne scars. Semin. Cutan. Med. Surg 2002, 21, 46–75. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.; McAdams, T.; Morgan, R.; Parshley, P.F.; Pike, R.C.; Riggs, P.; Carpenter, J.E. Pruritus in burns: A descriptive study. J. Burn Care Rehabil. 1988, 9, 305–308. [Google Scholar] [PubMed]
- Robert, R.; Blakeney, P.; Villarreal, C.; Meyer, W.J., 3rd. Anxiety: Current practices in assessment and treatment of anxiety of burn patients. Burns 2000, 26, 549–552. [Google Scholar] [CrossRef]
- Taal, L.A.; Faber, A.W. Posttraumatic stress and maladjustment among adult burn survivors 1-2 years postburn. Burns 1998, 24, 285–292. [Google Scholar] [CrossRef]
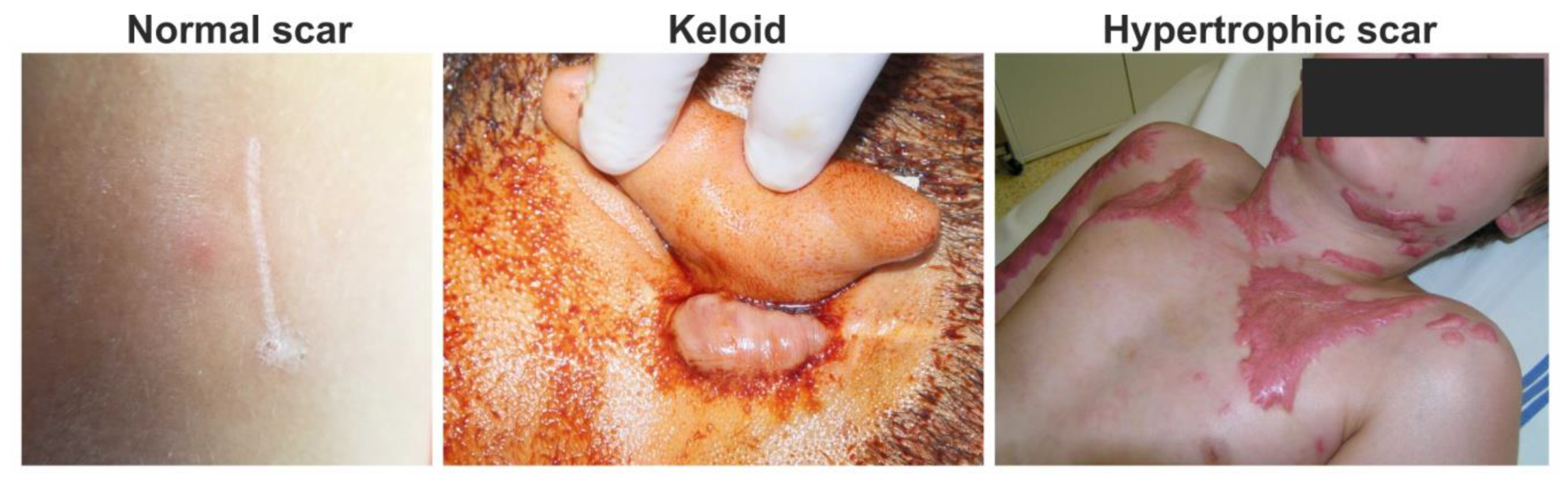
- Slemp, A.E.; Kirschner, R.E. Keloids and scars: A review of keloids and scars, their pathogenesis, risk factors, and management. Curr. Opin. Pediatr. 2006, 18, 396–402. [Google Scholar] [CrossRef]
- Atiyeh, B.S. Nonsurgical management of hypertrophic scars: Evidence-based therapies, standard practices, and emerging methods. Aesthetic. Plast. Surg. 2007, 31, 468–492; discussion 493–464. [Google Scholar] [CrossRef]
- Lindley, L.E.; Stojadinovic, O.; Pastar, I.; Tomic-Canic, M. Biology and biomarkers for wound healing. Plast. Reconstr. Surg. 2016, 138, 18S–28S. [Google Scholar] [CrossRef]
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol. Med. 2011, 17, 113–125. [Google Scholar] [CrossRef]
- Berman, B.; Maderal, A.; Raphael, B. Keloids and hypertrophic scars: Pathophysiology, classification, and treatment. Dermatol. Surg. 2017, 43 (Suppl 1), S3–S18. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessen, F.B.; Schalkwijk, J.; Vos, H.; Timens, W. Hypertrophic scar formation is associated with an increased number of epidermal langerhans cells. J. Pathol. 2004, 202, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurtner, G.C.; Dauskardt, R.H.; Wong, V.W.; Bhatt, K.A.; Wu, K.; Vial, I.N.; Padois, K.; Korman, J.M.; Longaker, M.T. Improving cutaneous scar formation by controlling the mechanical environment: Large animal and phase i studies. Ann. Surg. 2011, 254, 217–225. [Google Scholar] [CrossRef]
- Meyer, M.; McGrouther, D.A. A study relating wound tension to scar morphology in the pre-sternal scar using langers technique. Br. J. Plast. Surg. 1991, 44, 291–294. [Google Scholar] [CrossRef]
- Wray, R.C. Force required for wound closure and scar appearance. Plast. Reconstr. Surg. 1983, 72, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Farina, J.A., Jr.; Rosique, M.J.; Rosique, R.G. Curbing inflammation in burn patients. Int. J. Inflam. 2013, 2013, 715645. [Google Scholar] [CrossRef] [Green Version]
- Schultz, G.S.; Chin, G.A.; Moldawer, L.; Diegelmann, R.F. Principles of wound healing. In Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists; Fitridge, R., Thompson, M., Eds.; University of Adelaide Press: Adelaide, SA, Australia, 2011. [Google Scholar]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How it all starts: Initiation of the clotting cascade. Crit. Rev. Biochem Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.H.; Huang, B.S.; Horng, H.C.; Yeh, C.C.; Chen, Y.J. Wound healing. J. Chin. Med. Assoc. 2018, 81, 94–101. [Google Scholar] [CrossRef]
- Niessen, F.B.; Spauwen, P.H.; Schalkwijk, J.; Kon, M. On the nature of hypertrophic scars and keloids: A review. Plast. Reconstr. Surg. 1999, 104, 1435–1458. [Google Scholar] [CrossRef]
- Balaji, S.; Watson, C.L.; Ranjan, R.; King, A.; Bollyky, P.L.; Keswani, S.G. Chemokine involvement in fetal and adult wound healing. Adv. Wound Care 2015, 4, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Martins-Green, M.; Petreaca, M.; Wang, L. Chemokines and their receptors are key players in the orchestra that regulates wound healing. Adv. Wound Care 2013, 2, 327–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fivenson, D.P.; Faria, D.T.; Nickoloff, B.J.; Poverini, P.J.; Kunkel, S.; Burdick, M.; Strieter, R.M. Chemokine and inflammatory cytokine changes during chronic wound healing. Wound Repair. Regen. 1997, 5, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Ind. J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef]
- Ozgok Kangal, M.K.; Regan, J.P. Wound healing. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Lin, F.; Nguyen, C.M.; Wang, S.J.; Saadi, W.; Gross, S.P.; Jeon, N.L. Effective neutrophil chemotaxis is strongly influenced by mean il-8 concentration. Biochem. Biophys. Res. Commun. 2004, 319, 576–581. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mocsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in chronic wounds. Int. J. Mol. Sci 2016, 17, 2085. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Widgerow, A.D. Cellular resolution of inflammation—Catabasis. Wound Repair Regen. 2012, 20, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.L.; Ambravaneswaran, V.; Agrawal, N.; Bilodeau, M.; Toner, M.; Tompkins, R.G.; Fagan, S.; Irimia, D. Burn injury reduces neutrophil directional migration speed in microfluidic devices. PLoS ONE 2010, 5, e11921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egners, A.; Erdem, M.; Cramer, T. The response of macrophages and neutrophils to hypoxia in the context of cancer and other inflammatory diseases. Mediat. Inflamm. 2016, 2016, 2053646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodero, M.P.; Legrand, J.M.; Bou-Gharios, G.; Khosrotehrani, K. Wound-associated macrophages control collagen 1alpha2 transcription during the early stages of skin wound healing. Exp. Dermatol. 2013, 22, 143–145. [Google Scholar] [CrossRef] [Green Version]
- Rodero, M.P.; Licata, F.; Poupel, L.; Hamon, P.; Khosrotehrani, K.; Combadiere, C.; Boissonnas, A. In vivo imaging reveals a pioneer wave of monocyte recruitment into mouse skin wounds. PLoS ONE 2014, 9, e108212. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, K.P.; Palmer, J.S.; Cronau, S.; Seppanen, E.; Olver, S.; Raffelt, N.C.; Kuns, R.; Pettit, A.R.; Clouston, A.; Wainwright, B.; et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 2010, 116, 3955–3963. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.O. Molecular interactions in macrophage activation. Immunol. Today 1989, 10, 33–35. [Google Scholar] [CrossRef]
- Adams, D.O.; Koerner, T.J. Gene regulation in macrophage development and activation. Year Immunol. 1989, 4, 159–180. [Google Scholar]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Verreck, F.A.; de Boer, T.; Langenberg, D.M.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human il-23-producing type 1 macrophages promote but il-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Porta, C.; Morlacchi, S.; Banfi, S.; Strauss, L.; Rimoldi, M.; Totaro, M.G.; Riboldi, E. Origin and functions of tumor-associated myeloid cells (tamcs). Cancer Microenviron. 2012, 5, 133–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaja-Milatovic, S.; Richmond, A. Cxc chemokines and their receptors: A case for a significant biological role in cutaneous wound healing. Histol. Histopathol. 2008, 23, 1399–1407. [Google Scholar] [PubMed]
- Mendez, M.V.; Stanley, A.; Park, H.Y.; Shon, K.; Phillips, T.; Menzoian, J.O. Fibroblasts cultured from venous ulcers display cellular characteristics of senescence. J. Vasc. Surg. 1998, 28, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving tgf-beta, pge2, and paf. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Shahid, M.Q.; Bai, L.; Lu, Z.Z.; Chen, Y.H.; Jiang, L.; Diao, M.Y.; Liu, X.D.; Lu, Y.G. Evaluation of genetic diversity and development of a core collection of wild rice (oryza rufipogon griff.) populations in china. PLoS ONE 2015, 10, e0145990. [Google Scholar] [CrossRef] [Green Version]
- Nosbaum, A.; Prevel, N.; Truong, H.A.; Mehta, P.; Ettinger, M.; Scharschmidt, T.C.; Ali, N.H.; Pauli, M.L.; Abbas, A.K.; Rosenblum, M.D. Cutting edge: Regulatory t cells facilitate cutaneous wound healing. J. Immunol. 2016, 196, 2010–2014. [Google Scholar] [CrossRef]
- Brancato, S.K.; Albina, J.E. Wound macrophages as key regulators of repair: Origin, phenotype, and function. Am. J. Pathol. 2011, 178, 19–25. [Google Scholar] [CrossRef]
- Xiu, F.; Jeschke, M.G. Perturbed mononuclear phagocyte system in severely burned and septic patients. Shock 2013, 40, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage phenotypes regulate scar formation and chronic wound healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory m1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, S.; Lin, E.J.; Tartar, D. Immunology of wound healing. Curr. Dermatol. Rep. 2018, 7, 350–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, T.; Kabashima, K.; Miyachi, Y. The panoply of alphabetat cells in the skin. J. Dermatol. Sci. 2014, 76, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havran, W.L.; Jameson, J.M. Epidermal t cells and wound healing. J. Immunol. 2010, 184, 5423–5428. [Google Scholar] [CrossRef]
- Jensen, K.D.; Su, X.; Shin, S.; Li, L.; Youssef, S.; Yamasaki, S.; Steinman, L.; Saito, T.; Locksley, R.M.; Davis, M.M.; et al. Thymic selection determines gammadelta t cell effector fate: Antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity 2008, 29, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Schmolka, N.; Wencker, M.; Hayday, A.C.; Silva-Santos, B. Epigenetic and transcriptional regulation of gammadelta t cell differentiation: Programming cells for responses in time and space. Semin. Immunol. 2015, 27, 19–25. [Google Scholar] [CrossRef]
- Larouche, J.; Sheoran, S.; Maruyama, K.; Martino, M.M. Immune regulation of skin wound healing: Mechanisms and novel therapeutic targets. Adv. Wound Care 2018, 7, 209–231. [Google Scholar] [CrossRef]
- Schwacha, M.G. Gammadelta t-cells: Potential regulators of the post-burn inflammatory response. Burns 2009, 35, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Rani, M.; Schwacha, M.G. The composition of t-cell subsets are altered in the burn wound early after injury. PLoS ONE 2017, 12, e0179015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyce, D.E.; Jones, W.D.; Ruge, F.; Harding, K.G.; Moore, K. The role of lymphocytes in human dermal wound healing. Br. J. Dermatol. 2000, 143, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Bernabei, P.; Rigamonti, L.; Ariotti, S.; Stella, M.; Castagnoli, C.; Novelli, F. Functional analysis of t lymphocytes infiltrating the dermis and epidermis of post-burn hypertrophic scar tissues. Burns 1999, 25, 43–48. [Google Scholar] [CrossRef]
- Pileri, D.; Accardo Palombo, A.; D’Amelio, L.; D’Arpa, N.; Amato, G.; Masellis, A.; Cataldo, V.; Mogavero, R.; Napoli, B.; Lombardo, C.; et al. Concentrations of cytokines il-6 and il-10 in plasma of burn patients: Their relationship to sepsis and outcome. Ann. Burns Fire Disast. 2008, 21, 182–185. [Google Scholar]
- Diehl, S.; Rincon, M. The two faces of il-6 on th1/th2 differentiation. Mol. Immunol. 2002, 39, 531–536. [Google Scholar] [CrossRef]
- Hager, S.; Foldenauer, A.C.; Rennekampff, H.O.; Deisz, R.; Kopp, R.; Tenenhaus, M.; Gernot, M.; Pallua, N. Interleukin-6 serum levels correlate with severity of burn injury but not with gender. J. Burn Care Res. 2018, 39, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Entezami, K.Z.; Mosavi, T. Determination of lymphocytes surface markers in patients with thermal burns and the influence of burn size on mononuclear cell subsets. Med. J. Islam Repub. Iran 2017, 31, 38. [Google Scholar] [CrossRef]
- Rose, L.F.; Chan, R.K. The burn wound microenvironment. Adv. Wound Care 2016, 5, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Wilgus, T.A.; Wulff, B.C. The importance of mast cells in dermal scarring. Adv. Wound Care 2014, 3, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Komi, D.E.A.; Khomtchouk, K.; Santa Maria, P.L. A review of the contribution of mast cells in wound healing: Involved molecular and cellular mechanisms. Clin. Rev. Allergy Immunol. 2020, 58, 298–312. [Google Scholar] [CrossRef]
- Au, S.R.; Au, K.; Saggers, G.C.; Karne, N.; Ehrlich, H.P. Rat mast cells communicate with fibroblasts via gap junction intercellular communications. J. Cell Biochem. 2007, 100, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.T.; Saggers, G.C.; Moyer, K.E.; Ehrlich, H.P. Rat mast cells enhance fibroblast proliferation and fibroblast-populated collagen lattice contraction through gap junctional intercellular communications. Plast. Reconstr. Surg. 2011, 127, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.T.; Ehrlich, H.P. Through gap junction communications, co-cultured mast cells and fibroblasts generate fibroblast activities allied with hypertrophic scarring. Plast. Reconstr. Surg. 2013, 131, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Schrementi, M.E.; Ranzer, M.J.; Wilgus, T.A.; DiPietro, L.A. Blockade of mast cell activation reduces cutaneous scar formation. PLoS ONE 2014, 9, e85226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilgus, T.A. Immune cells in the healing skin wound: Influential players at each stage of repair. Pharmacol. Res. 2008, 58, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.X.; Arroyo, C.; Garcia, I.; Blasco, R.; Obispo, J.M.; Hamann, C.; Espejo, L. Role of mast cells in the pathogenesis of postburn inflammatory response: Reactive oxygen species as mast cell stimulators. Burns 2000, 26, 145–147. [Google Scholar] [CrossRef]
- Parihar, A.; Parihar, M.S.; Milner, S.; Bhat, S. Oxidative stress and anti-oxidative mobilization in burn injury. Burns 2008, 34, 6–17. [Google Scholar] [CrossRef]
- Sirbulescu, R.F.; Boehm, C.K.; Soon, E.; Wilks, M.Q.; Ilies, I.; Yuan, H.; Maxner, B.; Chronos, N.; Kaittanis, C.; Normandin, M.D.; et al. Mature b cells accelerate wound healing after acute and chronic diabetic skin lesions. Wound Repair. Regen. 2017, 25, 774–791. [Google Scholar] [CrossRef]
- Iwata, Y.; Yoshizaki, A.; Komura, K.; Shimizu, K.; Ogawa, F.; Hara, T.; Muroi, E.; Bae, S.; Takenaka, M.; Yukami, T.; et al. Cd19, a response regulator of b lymphocytes, regulates wound healing through hyaluronan-induced tlr4 signaling. Am. J. Pathol. 2009, 175, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Vinish, M.; Cui, W.; Stafford, E.; Bae, L.; Hawkins, H.; Cox, R.; Toliver-Kinsky, T. Dendritic cells modulate burn wound healing by enhancing early proliferation. Wound Repair. Regen. 2016, 24, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Gomes, I.; Mathur, S.K.; Espenshade, B.M.; Mori, Y.; Varga, J.; Ackerman, S.J. Eosinophil-fibroblast interactions induce fibroblast il-6 secretion and extracellular matrix gene expression: Implications in fibrogenesis. J. Allergy Clin. Immunol. 2005, 116, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Childs, D.R.; Murthy, A.S. Overview of wound healing and management. Surg. Clin. North. Am. 2017, 97, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.A. Role of fibronectin in normal wound healing. Int. Wound J. 2015, 12, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.P.; Krummel, T.M. Regulation of wound healing from a connective tissue perspective. Wound Repair. Regen. 1996, 4, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, J.H. Fibroblasts and myofibroblasts in wound healing: Force generation and measurement. J. Tissue Viabil. 2011, 20, 108–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in wound healing: A comprehensive review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, E.; Raghavan, S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet. 2002, 3, 199–209. [Google Scholar] [CrossRef]
- Rousselle, P.; Braye, F.; Dayan, G. Re-epithelialization of adult skin wounds: Cellular mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 344–365. [Google Scholar] [CrossRef]
- Lavker, R.M.; Sun, T.T. Epidermal stem cells: Properties, markers, and location. Proc. Natl. Acad. Sci. USA 2000, 97, 13473–13475. [Google Scholar] [CrossRef] [Green Version]
- Watt, S.M.; Pleat, J.M. Stem cells, niches and scaffolds: Applications to burns and wound care. Adv. Drug Deliv. Rev. 2018, 123, 82–106. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Zhang, L.J. Keratin 6, 16 and 17-critical barrier alarmin molecules in skin wounds and psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigal, C.; Pieraggi, M.T.; Vincent, C.; Prost, C.; Bouisou, H.; Serre, G. Healing of full-thickness cutaneous wounds in the pig. I. Immunohistochemical study of epidermo-dermal junction regeneration. J. Investig. Dermatol. 1991, 96, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darby, I.A.; Laverdet, B.; Bonte, F.; Desmouliere, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Parvani, J.G.; Schiemann, W.P. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J. Mamm. Gland Biol. Neoplas. 2010, 15, 169–190. [Google Scholar] [CrossRef] [Green Version]
- Bolos, V.; Peinado, H.; Perez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor slug represses e-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with snail and e47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Xiong, M.; Jiang, L.; Zhou, Y.; Qiu, W.; Fang, L.; Tan, R.; Wen, P.; Yang, J. The mir-200 family regulates tgf-beta1-induced renal tubular epithelial to mesenchymal transition through smad pathway by targeting zeb1 and zeb2 expression. Am. J. Physiol. Renal Physiol. 2012, 302, F369–F379. [Google Scholar] [CrossRef] [Green Version]
- Radisky, D.C.; Kenny, P.A.; Bissell, M.J. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via emt? J. Cell. Biochem. 2007, 101, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safferling, K.; Sutterlin, T.; Westphal, K.; Ernst, C.; Breuhahn, K.; James, M.; Jager, D.; Halama, N.; Grabe, N. Wound healing revised: A novel reepithelialization mechanism revealed by in vitro and in silico models. J. Cell. Biol. 2013, 203, 691–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragona, M.; Dekoninck, S.; Rulands, S.; Lenglez, S.; Mascre, G.; Simons, B.D.; Blanpain, C. Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat. Commun. 2017, 8, 14684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Wang, Y.; Haensel, D.; Lee, B.; Dai, X.; Nie, Q. Multiscale modeling of layer formation in epidermis. PLoS Comput. Biol. 2018, 14, e1006006. [Google Scholar] [CrossRef] [PubMed]
- Gal, P.; Varinska, L.; Faber, L.; Novak, S.; Szabo, P.; Mitrengova, P.; Mirossay, A.; Mucaji, P.; Smetana, K. How signaling molecules regulate tumor microenvironment: Parallels to wound repair. Molecules 2017, 22, 1818. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.Z.; Xiao, S.C.; Luo, P.F.; Huang, G.F.; Wang, G.Y.; Zhu, S.H.; Wu, M.J.; Xia, Z.F. An epidermal stem cells niche microenvironment created by engineered human amniotic membrane. Biomaterials 2011, 32, 7801–7811. [Google Scholar] [CrossRef]
- Freedberg, I.M.; Tomic-Canic, M.; Komine, M.; Blumenberg, M. Keratins and the keratinocyte activation cycle. J. Invest. Dermatol. 2001, 116, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, J.; Kirsner, R. Pathophysiology of acute wound healing. Clin. Dermatol. 2007, 25, 9–18. [Google Scholar] [CrossRef]
- Fisher, G.; Rittie, L. Restoration of the basement membrane after wounding: A hallmark of young human skin altered with aging. J. Cell Commun Signal. 2018, 12, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Chimenti, I.; Rosa, P.; Angelini, F.; Pagano, F.; Calogero, A.; Giacomello, A.; Messina, E. Emt/met at the crossroad of stemness, regeneration and oncogenesis: The ying-yang equilibrium recapitulated in cell spheroids. Cancers 2017, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting epithelial-mesenchymal transition (emt) to overcome drug resistance in cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, V.; Brabletz, T.; Ceppi, P. Targeting emt in cancer with repurposed metabolic inhibitors. Trends Cancer 2020, 6, 942–950. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of pka leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.F.; Liu, A.J.; Krishnakumar, R.; Freimer, J.W.; DeVeale, B.; Blelloch, R. Grhl2-dependent enhancer switching maintains a pluripotent stem cell transcriptional subnetwork after exit from naive pluripotency. Cell Stem Cell 2018, 23, 226–238 e224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D.; Sud, S.; Schumann, T.; Wilkinson, J.E.; Zaslavsky, A.; Li, H.; et al. Transcription factors ovol1 and ovol2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 2013, 8, e76773. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Villarreal-Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary morphogenesis and regeneration require the inhibition of emt at terminal end buds by ovol2 transcriptional repressor. Dev. Cell 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaku, M.; Grimm, S.A.; Shimbo, T.; Perera, L.; Menafra, R.; Stunnenberg, H.G.; Archer, T.K.; Machida, S.; Kurumizaka, H.; Wade, P.A. Gata3-dependent cellular reprogramming requires activation-domain dependent recruitment of a chromatin remodeler. Genome Biol. 2016, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Jagle, S.; Busch, H.; Freihen, V.; Beyes, S.; Schrempp, M.; Boerries, M.; Hecht, A. Snail1-mediated downregulation of foxa proteins facilitates the inactivation of transcriptional enhancer elements at key epithelial genes in colorectal cancer cells. PLoS Genet. 2017, 13, e1007109. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Liu, J.; Yang, J.; Chen, Y.; Chen, J.; Ni, S.; Song, H.; Zeng, L.; Ding, K.; Pei, D. Bmps functionally replace klf4 and support efficient reprogramming of mouse fibroblasts by oct4 alone. Cell Res. 2011, 21, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, L.; Mao, S.Q.; Li, Z.; Chen, J.; Zhang, R.R.; Wu, H.P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and tdg mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, K.; Talukdar, I.; Patil, V.S.; Dang, J.; Li, Z.; Chang, K.Y.; Lu, C.C.; Delorme-Walker, V.; Dermardirossian, C.; Anderson, K.; et al. Kinome-wide functional analysis highlights the role of cytoskeletal remodeling in somatic cell reprogramming. Cell Stem Cell 2014, 14, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, S.; Brabletz, T. The zeb/mir-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Liu, Y.; Noguchi, S.; Murray, M.; Chang, J.C.; Kishima, M.; Nishimura, H.; Hashimoto, K.; Minoda, A.; Suzuki, H. Ovol2 induces mesenchymal-to-epithelial transition in fibroblasts and enhances cell-state reprogramming towards epithelial lineages. Sci. Rep. 2019, 9, 6490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an nf-kappab-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [Green Version]
- desJardins-Park, H.E.; Chinta, M.S.; Foster, D.S.; Borrelli, M.R.; Shen, A.H.; Wan, D.C.; Longaker, M.T. Fibroblast heterogeneity in and its implications for plastic and reconstructive surgery: A basic science review. Plast. Reconstr. Surg. Glob. Open 2020, 8, e2927. [Google Scholar]
- Vorstandlechner, V.; Laggner, M.; Kalinina, P.; Haslik, W.; Radtke, C.; Shaw, L.; Lichtenberger, B.M.; Tschachler, E.; Ankersmit, H.J.; Mildner, M. Deciphering the functional heterogeneity of skin fibroblasts using single-cell rna sequencing. FASEB J. 2020, 34, 3677–3692. [Google Scholar] [CrossRef]
- Jiang, D.; Rinkevich, Y. Scars or regeneration?-dermal fibroblasts as drivers of diverse skin wound responses. Int. J. Mol. Sci 2020, 21, 617. [Google Scholar] [CrossRef] [Green Version]
- Reilkoff, R.A.; Bucala, R.; Herzog, E.L. Fibrocytes: Emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 2011, 11, 427–435. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, Z.; He, W.; Gao, H.; Bai, Y.; Yang, S.; Zhang, L.; Zhan, R.; Tan, J.; Zhou, J.; et al. P311 induces the transdifferentiation of epidermal stem cells to myofibroblast-like cells by stimulating transforming growth factor beta1 expression. Stem Cell Res. Ther. 2016, 7, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saikia, P.; Crabb, J.S.; Dibbin, L.L.; Juszczak, M.J.; Willard, B.; Jang, G.F.; Shiju, T.M.; Crabb, J.W.; Wilson, S.E. Quantitative proteomic comparison of myofibroblasts derived from bone marrow and cornea. Sci Rep. 2020, 10, 16717. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (endomt) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, L.D.; Shupp, J.W.; Smith, R.D.; Alkhalil, A.; Moffatt, L.T.; Carney, B.C. Galectin-1 production is elevated in hypertrophic scar. Wound Repair. Regen. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Chen, J.S.; Wu, M.H.; Hsieh, I.S.; Liang, C.H.; Hsu, C.L.; Hong, T.M.; Chen, Y.L. Galectin-1 accelerates wound healing by regulating the neuropilin-1/smad3/nox4 pathway and ros production in myofibroblasts. J. Investig. Dermatol. 2015, 135, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotendorst, G.R.; Duncan, M.R. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 2005, 19, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Glim, J.E.; Niessen, F.B.; Everts, V.; van Egmond, M.; Beelen, R.H. Platelet derived growth factor-cc secreted by m2 macrophages induces alpha-smooth muscle actin expression by dermal and gingival fibroblasts. Immunobiology 2013, 218, 924–929. [Google Scholar] [CrossRef]
- Lian, N.; Li, T. Growth factor pathways in hypertrophic scars: Molecular pathogenesis and therapeutic implications. Biomed. Pharmacother. 2016, 84, 42–50. [Google Scholar] [CrossRef]
- Limandjaja, G.C.; Niessen, F.B.; Scheper, R.J.; Gibbs, S. Hypertrophic scars and keloids: Overview of the evidence and practical guide for differentiating between these abnormal scars. Exp. Dermatol. 2020, 30, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Ghazawi, F.M.; Zargham, R.; Gilardino, M.S.; Sasseville, D.; Jafarian, F. Insights into the pathophysiology of hypertrophic scars and keloids: How do they differ? Adv. Skin Wound Care 2018, 31, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Honnegowda, T.; Kumar, P.; Udupa, E.; Kumar, S.; Kumar, U.; Rao, P. Role of angiogenesis and angiogenic factors in acute and chronic wound healing. Plast. Aesthet. Res. 2015, 2, 243. [Google Scholar]
- Breier, G.; Blum, S.; Peli, J.; Groot, M.; Wild, C.; Risau, W.; Reichmann, E. Transforming growth factor-beta and ras regulate the vegf/vegf-receptor system during tumor angiogenesis. Int. J. Cancer 2002, 97, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Benjamin, L.; Zeng, H.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008, 11, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F.; Nagy, J.A.; Feng, D.; Brown, L.F.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr. Top. Microbiol. Immunol. 1999, 237, 97–132. [Google Scholar]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, 3005. [Google Scholar] [CrossRef] [Green Version]
- Majima, M.; Hayashi, I.; Muramatsu, M.; Katada, J.; Yamashina, S.; Katori, M. Cyclo-oxygenase-2 enhances basic fibroblast growth factor-induced angiogenesis through induction of vascular endothelial growth factor in rat sponge implants. Br. J. Pharmacol. 2000, 130, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Pintucci, G.; Froum, S.; Pinnell, J.; Mignatti, P.; Rafii, S.; Green, D. Trophic effects of platelets on cultured endothelial cells are mediated by platelet-associated fibroblast growth factor-2 (fgf-2) and vascular endothelial growth factor (vegf). Thromb. Haemost. 2002, 88, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.G.; Raveendran, R. An insight into the possibilities of fibroblast growth factor in periodontal regeneration. J. Ind. Soc. Periodontol. 2014, 18, 289–292. [Google Scholar] [CrossRef]
- Yoshida, S.; Yoshida, A.; Matsui, H.; Takada, Y.; Ishibashi, T. Involvement of macrophage chemotactic protein-1 and interleukin-1beta during inflammatory but not basic fibroblast growth factor-dependent neovascularization in the mouse cornea. Lab. Invest. J. Tech. Methods Pathol. 2003, 83, 927–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Mans, S.; Banz, Y.; Mueller, B.U.; Pabst, T. The angiogenesis inhibitor vasostatin is regulated by neutrophil elastase-dependent cleavage of calreticulin in aml patients. Blood 2012, 120, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Inoki, I.; Shiomi, T.; Hashimoto, G.; Enomoto, H.; Nakamura, H.; Makino, K.; Ikeda, E.; Takata, S.; Kobayashi, K.; Okada, Y. Connective tissue growth factor binds vascular endothelial growth factor (vegf) and inhibits vegf-induced angiogenesis. FASEB J. 2002, 16, 219–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, D.; Bauer, J.; Schoenberger, J. Blockade of neoangiogenesis, a new and promising technique to control the growth of malignant tumors and their metastases. Curr. Vasc. Pharmacol. 2009, 7, 347–357. [Google Scholar] [CrossRef]
- Bootle-Wilbraham, C.A.; Tazzyman, S.; Thompson, W.D.; Stirk, C.M.; Lewis, C.E. Fibrin fragment e stimulates the proliferation, migration and differentiation of human microvascular endothelial cells in vitro. Angiogenesis 2001, 4, 269–275. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, L.; Wei, X.; Tan, Y.S.; Tong, L.; Chang, R.; Ghanamah, M.S.; Reinblatt, M.; Marti, G.P.; Harmon, J.W.; et al. Impaired angiogenesis and mobilization of circulating angiogenic cells in hif-1alpha heterozygous-null mice after burn wounding. Wound Repair Regen. 2010, 18, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Fox, A.; Smythe, J.; Fisher, N.; Tyler, M.P.; McGrouther, D.A.; Watt, S.M.; Harris, A.L. Mobilization of endothelial progenitor cells into the circulation in burned patients. Br. J. Surg. 2008, 95, 244–251. [Google Scholar] [CrossRef]
- Foresta, C.; Schipilliti, M.; De Toni, L.; Magagna, S.; Lancerotto, L.; Azzena, B.; Vindigni, V.; Mazzoleni, F. Blood levels, apoptosis, and homing of the endothelial progenitor cells after skin burns and escharectomy. J. Trauma 2011, 70, 459–465. [Google Scholar] [CrossRef]
- Bates, D.O.; Heald, R.I.; Curry, F.E.; Williams, B. Vascular endothelial growth factor increases rana vascular permeability and compliance by different signalling pathways. J. Physiol. 2001, 533, 263–272. [Google Scholar] [CrossRef]
- Zittermann, S.I.; Issekutz, A.C. Endothelial growth factors vegf and bfgf differentially enhance monocyte and neutrophil recruitment to inflammation. J. Leukoc. Biol. 2006, 80, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Detmar, M.; Brown, L.F.; Schon, M.P.; Elicker, B.M.; Velasco, P.; Richard, L.; Fukumura, D.; Monsky, W.; Claffey, K.P.; Jain, R.K. Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of vegf transgenic mice. J. Investig. Dermatol. 1998, 111, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulff, B.C.; Parent, A.E.; Meleski, M.A.; DiPietro, L.A.; Schrementi, M.E.; Wilgus, T.A. Mast cells contribute to scar formation during fetal wound healing. J. Investig. Dermatol. 2012, 132, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Reinders, M.E.; Sho, M.; Izawa, A.; Wang, P.; Mukhopadhyay, D.; Koss, K.E.; Geehan, C.S.; Luster, A.D.; Sayegh, M.H.; Briscoe, D.M. Proinflammatory functions of vascular endothelial growth factor in alloimmunity. J. Clin. Investig. 2003, 112, 1655–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagabir, R.; Byers, R.J.; Chaudhry, I.H.; Muller, W.; Paus, R.; Bayat, A. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br. J. Dermatol. 2012, 167, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.A.; Mantovani, A.; Marme, D. Migration of human monocytes in response to vascular endothelial growth factor (vegf) is mediated via the vegf receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockmann, C.; Kirmse, S.; Helfrich, I.; Weidemann, A.; Takeda, N.; Doedens, A.; Johnson, R.S. A wound size-dependent effect of myeloid cell-derived vascular endothelial growth factor on wound healing. J. Investig. Dermatol. 2011, 131, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, J.; Tam, B.Y.; Sundram, U.; von Degenfeld, G.; Blau, H.M.; Kuo, C.J.; Cooke, J.P. Discordant effects of a soluble vegf receptor on wound healing and angiogenesis. Gene Ther. 2004, 11, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.; Hubner, G.; Breier, G.; Longaker, M.T.; Greenhalgh, D.G.; Werner, S. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J. Biol. Chem. 1995, 270, 12607–12613. [Google Scholar] [CrossRef] [Green Version]
- Wilgus, T.A.; Ferreira, A.M.; Oberyszyn, T.M.; Bergdall, V.K.; Dipietro, L.A. Regulation of scar formation by vascular endothelial growth factor. Lab. Invest. J. Tech. Methods Pathol. 2008, 88, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.F.; Xu, Y.B.; Tang, J.M.; Yang, R.H.; Liu, X.S. Hoxa9 regulates angiogenesis in human hypertrophic scars: Induction of vegf secretion by epidermal stem cells. Int. J. Clin. Exp. Pathol. 2014, 7, 2998–3007. [Google Scholar] [PubMed]
- Wang, J.; Chen, H.; Shankowsky, H.A.; Scott, P.G.; Tredget, E.E. Improved scar in postburn patients following interferon-alpha2b treatment is associated with decreased angiogenesis mediated by vascular endothelial cell growth factor. J. Interf. Cytok. Res. 2008, 28, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Mejia, I.; Bodapati, S.; Chen, K.T.; Diaz, B. Pancreatic adenocarcinoma invasiveness and the tumor microenvironment: From biology to clinical trials. Biomedicines 2020, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Jobe, N.P.; Zivicova, V.; Mifkova, A.; Rosel, D.; Dvorankova, B.; Kodet, O.; Strnad, H.; Kolar, M.; Sedo, A.; Smetana, K., Jr.; et al. Fibroblasts potentiate melanoma cells in vitro invasiveness induced by uv-irradiated keratinocytes. Histochem. Cell Biol. 2018, 149, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Boyuk, E.; Saracoglu, Z.N.; Arik, D. Cutaneous leiomyoma mimicking a keloid. Acta Dermatovenerol. Croat. 2020, 28, 116. [Google Scholar]
- Zhou, B.Y.; Wang, W.B.; Wu, X.L.; Zhang, W.J.; Zhou, G.D.; Gao, Z.; Liu, W. Nintedanib inhibits keloid fibroblast functions by blocking the phosphorylation of multiple kinases and enhancing receptor internalization. Acta Pharmacol. Sin. 2020, 41, 1234–1245. [Google Scholar] [CrossRef]
- Schulz, J.N.; Plomann, M.; Sengle, G.; Gullberg, D.; Krieg, T.; Eckes, B. New developments on skin fibrosis—Essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 2018, 68, 522–532. [Google Scholar] [CrossRef]
- Barnes, L.A.; Marshall, C.D.; Leavitt, T.; Hu, M.S.; Moore, A.L.; Gonzalez, J.G.; Longaker, M.T.; Gurtner, G.C. Mechanical forces in cutaneous wound healing: Emerging therapies to minimize scar formation. Adv. Wound Care 2018, 7, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Foreman, D.M.; Ferguson, M.W. Neutralisation of tgf-beta 1 and tgf-beta 2 or exogenous addition of tgf-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995, 108, 985–1002. [Google Scholar]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef] [Green Version]
- Januszyk, M.; Kwon, S.H.; Wong, V.W.; Padmanabhan, J.; Maan, Z.N.; Whittam, A.J.; Major, M.R.; Gurtner, G.C. The role of focal adhesion kinase in keratinocyte fibrogenic gene expression. Int. J. Mol. Sci. 2017, 18, 1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Eng. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladin, D.A.; Hou, Z.; Patel, D.; McPhail, M.; Olson, J.C.; Saed, G.M.; Fivenson, D.P. P53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Repair Regen. 1998, 6, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.; Khumalo, N.; Bayat, A. Understanding keloid pathobiology from a quasi-neoplastic perspective: Less of a scar and more of a chronic inflammatory disease with cancer-like tendencies. Front. Immunol 2019, 10, 1810. [Google Scholar] [CrossRef]
- Rees, P.A.; Greaves, N.S.; Baguneid, M.; Bayat, A. Chemokines in wound healing and as potential therapeutic targets for reducing cutaneous scarring. Adv. Wound Care 2015, 4, 687–703. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Budd, D.C.; Shih, B.; Seifert, O.; Beaton, A.; Wright, T.; Dempsey, M.; Kelly, F.; Egerton, J.; Marshall, R.P.; et al. Transforming growth factor beta gene signatures are spatially enriched in keloid tissue biopsies and ex vivo-cultured keloid fibroblasts. Acta Derm. Venereol. 2017, 97, 10–16. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter/Cell Type | Papillary Fibroblasts | Reticular Fibroblasts | Hypertrophic Fibroblasts | CAFs 1 |
| cell size | + | ++ | + | + |
| proliferation | ++ | + | ++ | +/− |
| collagen | ++ | ++ | ++ | ++++ |
| collagenase activity | ++++ | + | + | ? |
| α-SMA | + | +++ | +++ | +++ |
| collagen contraction | + | +++ | +++ | +++ |
| TGF-β | + | + | + | + |
| TGF-β receptor 2 | + | +++ | +++ | +/+++ |
| CTGF | + | +++ | +++ | ++++ |
| osteopontin | + | +++ | +++ | +++ |
| decorin | ++++ | + | + | +++ |
| versican | + | +++ | +++ | ++++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čoma, M.; Fröhlichová, L.; Urban, L.; Zajíček, R.; Urban, T.; Szabo, P.; Novák, Š.; Fetissov, V.; Dvořánková, B.; Smetana, K., Jr.; et al. Molecular Changes Underlying Hypertrophic Scarring Following Burns Involve Specific Deregulations at All Wound Healing Stages (Inflammation, Proliferation and Maturation). Int. J. Mol. Sci. 2021, 22, 897. https://doi.org/10.3390/ijms22020897
Čoma M, Fröhlichová L, Urban L, Zajíček R, Urban T, Szabo P, Novák Š, Fetissov V, Dvořánková B, Smetana K Jr., et al. Molecular Changes Underlying Hypertrophic Scarring Following Burns Involve Specific Deregulations at All Wound Healing Stages (Inflammation, Proliferation and Maturation). International Journal of Molecular Sciences. 2021; 22(2):897. https://doi.org/10.3390/ijms22020897
Chicago/Turabian StyleČoma, Matúš, Lucia Fröhlichová, Lukáš Urban, Robert Zajíček, Tomáš Urban, Pavol Szabo, Štěpán Novák, Vitaly Fetissov, Barbora Dvořánková, Karel Smetana, Jr., and et al. 2021. "Molecular Changes Underlying Hypertrophic Scarring Following Burns Involve Specific Deregulations at All Wound Healing Stages (Inflammation, Proliferation and Maturation)" International Journal of Molecular Sciences 22, no. 2: 897. https://doi.org/10.3390/ijms22020897
APA StyleČoma, M., Fröhlichová, L., Urban, L., Zajíček, R., Urban, T., Szabo, P., Novák, Š., Fetissov, V., Dvořánková, B., Smetana, K., Jr., & Gál, P. (2021). Molecular Changes Underlying Hypertrophic Scarring Following Burns Involve Specific Deregulations at All Wound Healing Stages (Inflammation, Proliferation and Maturation). International Journal of Molecular Sciences, 22(2), 897. https://doi.org/10.3390/ijms22020897