Molecular and Clinical Features of EGFR-TKI-Associated Lung Injury

 ,
, 
Abstract
1. Introduction
2. Development of EGFR-TKIs and Their AEs
2.1. Development of EGFR-TKIs in Clinical Studies
2.1.1. First-Generation EGFR-TKIs
2.1.2. Second-Generation EGFR-TKIs
2.1.3. Third-Generation EGFR-TKI
2.2. Adverse Events of EGFR-TKIs and Their Management
2.2.1. Rash, Paronychia, and Stomatitis
2.2.2. Diarrhea
2.2.3. Elevated Liver Transaminases
2.2.4. Interstitial Lung Disease
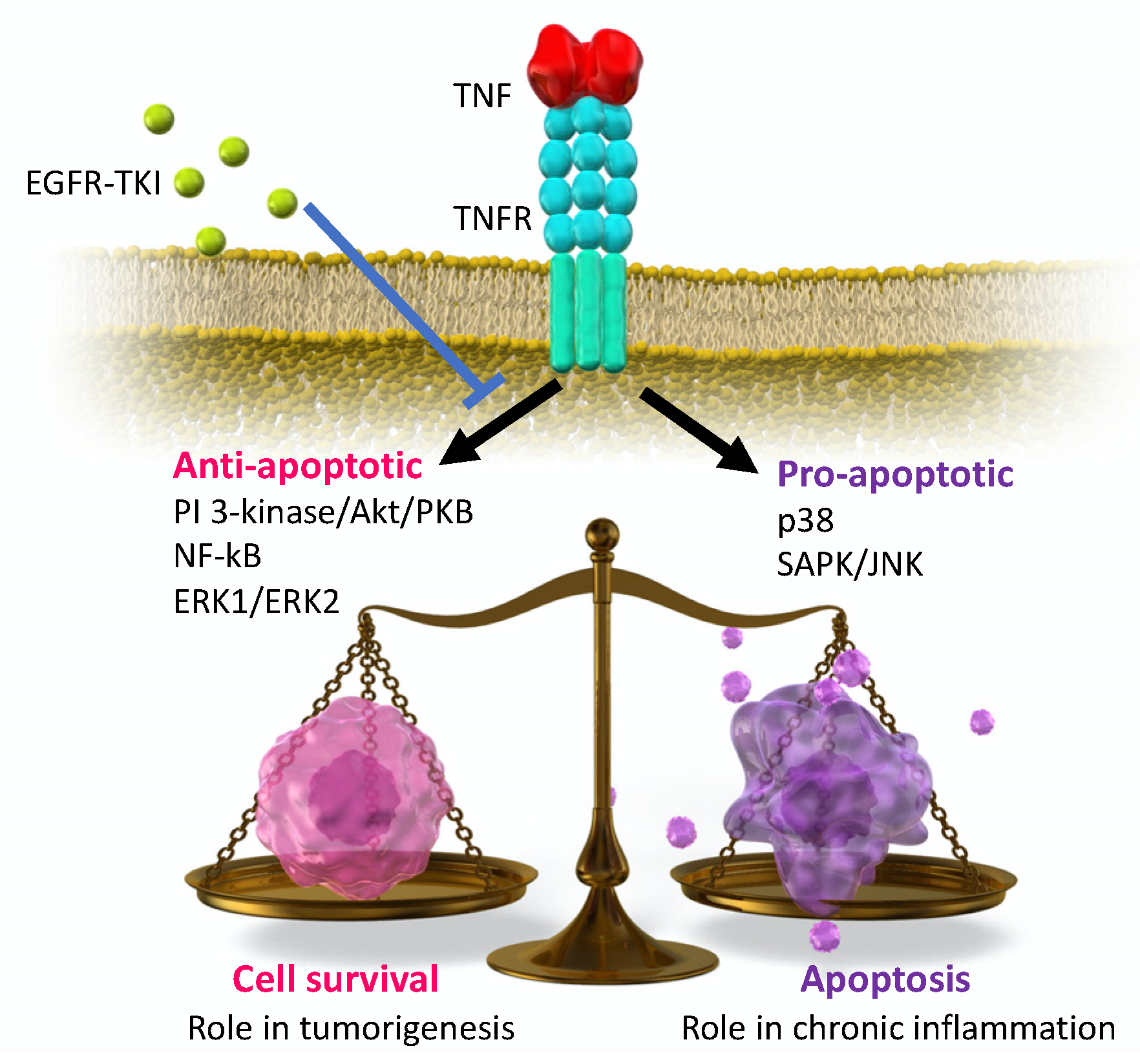
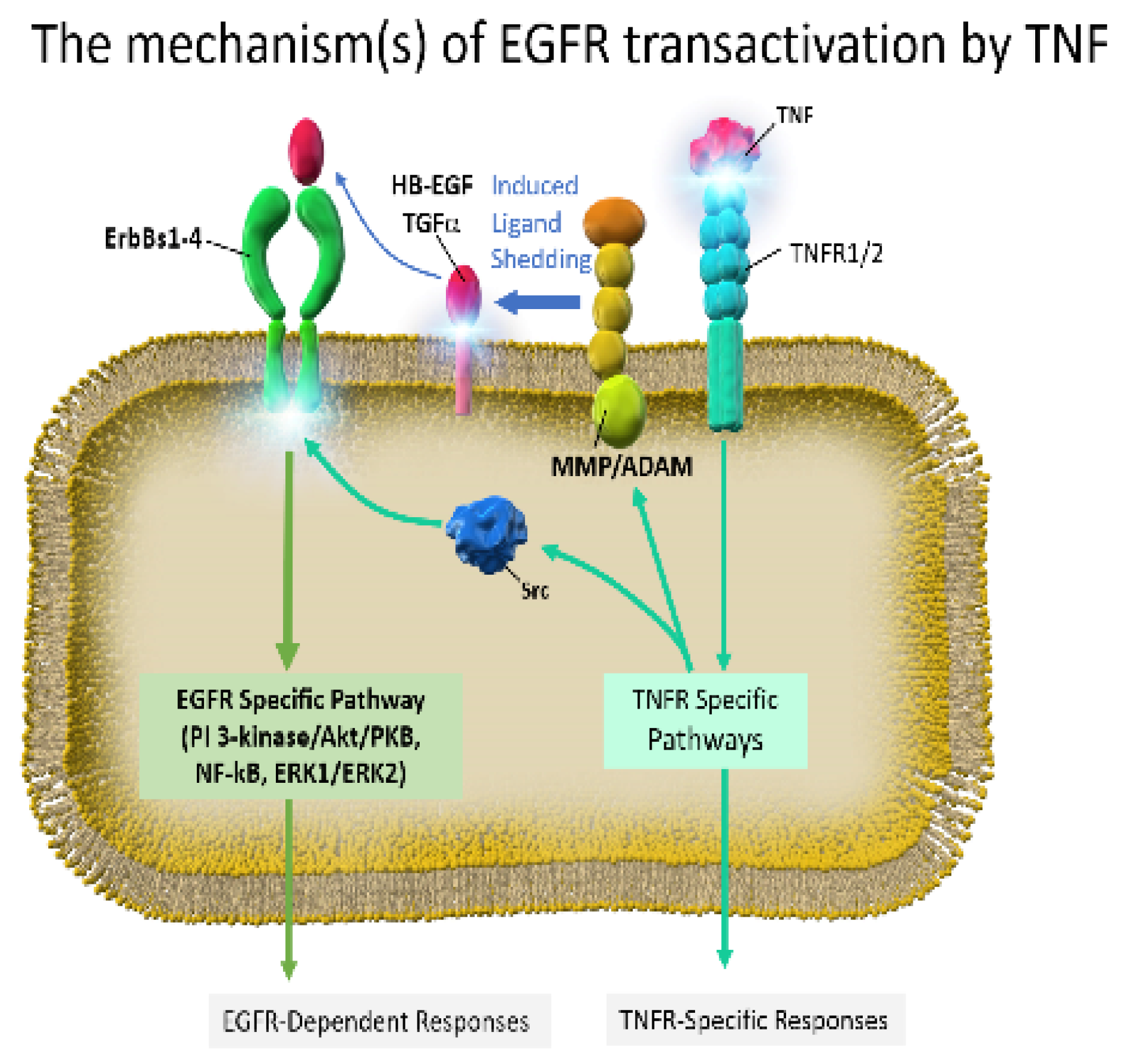
3. Preclinical Studies of EGFR-TKI Related Lung Injury
4. Inflammatory Cytokines on EGFR-TKI Resistance
4.1. Expression of Inflammatory Cytokines on EGFR-TKI Resistance
4.2. Possible EGFR-TKI Resistance Mechanisms in Inflammation
5. Clinical Studies of EGFR-TKI-Related Lung Injury
5.1. EGFR-TKI Monotherapy
5.2. Combination of EGFR-TKIs and Chemotherapeutic Agents or Angiogenesis Inhibitors
5.3. Combination of EGFR-TKIs and ICIs
6. Diagnosis and Therapeutics of EGFR-TKI-Related Lung Injury
6.1. Diagnosis
6.2. Therapeutics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Proto, C.; Ferrara, R.; Signorelli, D.; Lo Russo, G.; Galli, G.; Imbimbo, M.; Prelaj, A.; Zilembo, N.; Ganzinelli, M.; Pallavicini, L.M.; et al. Choosing wisely first line immunotherapy in non-small cell lung cancer (NSCLC): What to add and what to leave out. Cancer Treat. Rev. 2019, 75, 39–51. [Google Scholar] [CrossRef]
- Yatabe, Y.; Kerr, K.M.; Utomo, A.; Rajadurai, P.; Tran, V.K.; Du, X.; Chou, T.Y.; Enriquez, M.L.; Lee, G.K.; Iqbal, J.; et al. EGFR mutation testing practices within the Asia Pacific region: Results of a multicenter diagnostic survey. J. Thorac. Oncol. 2015, 10, 438–445. [Google Scholar] [CrossRef]
- Le, T.; Gerber, D.E. Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used? Cancers 2019, 11, 366. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Ren, S.; Li, Y.; Li, W.; Zhao, Z.; Jin, C.; Zhang, D. Fatal asymmetric interstitial lung disease after erlotinib for lung cancer. Respiration 2012, 84, 431–435. [Google Scholar] [CrossRef]
- Schacher-Kaufmann, S.; Pless, M. Acute Fatal Liver Toxicity under Erlotinib. Case Rep. Oncol. 2010, 3, 182–188. [Google Scholar] [CrossRef]
- Takeda, M.; Okamoto, I.; Tsurutani, J.; Oiso, N.; Kawada, A.; Nakagawa, K. Clinical impact of switching to a second EGFR-TKI after a severe AE related to a first EGFR-TKI in EGFR-mutated NSCLC. Jpn. J. Clin. Oncol. 2012, 42, 528–533. [Google Scholar] [CrossRef]
- Berge, E.M.; Doebele, R.C. Targeted therapies in non-small cell lung cancer: Emerging oncogene targets following the success of epidermal growth factor receptor. Semin. Oncol. 2014, 41, 110–125. [Google Scholar] [CrossRef]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Hirsh, V.; Boyer, M.; Yang, J.C.; Mok, T.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Overall survival data from the phase IIb LUX-Lung 7 trial. Ann. Oncol. 2017, 28, 270–277. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Okamoto, I.; Nakagawa, K. Pooled safety analysis of EGFR-TKI treatment for EGFR mutation-positive non-small cell lung cancer. Lung Cancer 2015, 88, 74–79. [Google Scholar] [CrossRef]
- Nakahara, T.; Moroi, Y.; Takayama, K.; Itoh, E.; Kido-Nakahara, M.; Nakanishi, Y.; Furue, M. Changes in sebum levels and the development of acneiform rash in patients with non-small cell lung cancer after treatment with EGFR inhibitors. Onco Targets Ther. 2015, 8, 259–263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Urata, Y.; Katakami, N.; Morita, S.; Kaji, R.; Yoshioka, H.; Seto, T.; Satouchi, M.; Iwamoto, Y.; Kanehara, M.; Fujimoto, D.; et al. Randomized Phase III Study Comparing Gefitinib With Erlotinib in Patients with Previously Treated Advanced Lung Adenocarcinoma: WJOG 5108L. J. Clin. Oncol. 2016, 34, 3248–3257. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, V. Managing treatment-related adverse events associated with egfr tyrosine kinase inhibitors in advanced non-small-cell lung cancer. Curr. Oncol. 2011, 18, 126–138. [Google Scholar] [CrossRef]
- Kudoh, S.; Kato, H.; Nishiwaki, Y.; Fukuoka, M.; Nakata, K.; Ichinose, Y.; Tsuboi, M.; Yokota, S.; Nakagawa, K.; Suga, M.; et al. Interstitial lung disease in Japanese patients with lung cancer: A cohort and nested case-control study. Am. J. Respir. Crit. Care Med. 2008, 177, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Gemma, A.; Kudoh, S.; Ando, M.; Ohe, Y.; Nakagawa, K.; Johkoh, T.; Yamazaki, N.; Arakawa, H.; Inoue, Y.; Ebina, M.; et al. Final safety and efficacy of erlotinib in the phase 4 POLARSTAR surveillance study of 10 708 Japanese patients with non-small-cell lung cancer. Cancer Sci. 2014, 105, 1584–1590. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nukiwa, T.; Gemma, A.; Yamamoto, N.; Mizushima, M.; Ochai, K.; Ikeda, R.; Azuma, H.; Nakanishi, Y. Real-world treatment of over 1600 Japanese patients with EGFR mutation-positive non-small cell lung cancer with daily afatinib. Int. J. Clin. Oncol. 2019, 24, 917–926. [Google Scholar] [CrossRef]
- Ohe, Y.; Kato, T.; Sakai, F.; Kusumoto, M.; Endo, M.; Saito, Y.; Baba, T.; Sata, M.; Yamaguchi, O.; Sakamoto, K.; et al. Real-world use of osimertinib for epidermal growth factor receptor T790M-positive non-small cell lung cancer in Japan. Jpn. J. Clin. Oncol. 2020, 50, 909–919. [Google Scholar] [CrossRef]
- Ando, M.; Okamoto, I.; Yamamoto, N.; Takeda, K.; Tamura, K.; Seto, T.; Ariyoshi, Y.; Fukuoka, M. Predictive factors for interstitial lung disease, antitumor response, and survival in non-small-cell lung cancer patients treated with gefitinib. J. Clin. Oncol. 2006, 24, 2549–2556. [Google Scholar] [CrossRef]
- Hotta, K.; Kiura, K.; Tabata, M.; Harita, S.; Gemba, K.; Yonei, T.; Bessho, A.; Maeda, T.; Moritaka, T.; Shibayama, T.; et al. Interstitial lung disease in Japanese patients with non-small cell lung cancer receiving gefitinib: An analysis of risk factors and treatment outcomes in Okayama Lung Cancer Study Group. Cancer J. 2005, 11, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoshiba, K.; Yokohori, N.; Nagai, A. Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosis. Cancer Res. 2003, 63, 5054–5059. [Google Scholar] [PubMed]
- Ishii, Y.; Fujimoto, S.; Fukuda, T. Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am. J. Respir. Crit. Care Med. 2006, 174, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, W.; Li, K.; Zhang, K.; Lin, C.; Han, R.; Lu, C.; Wang, Y.; Chen, H.; Sun, F.; et al. Metformin attenuates gefitinib-induced exacerbation of pulmonary fibrosis by inhibition of TGF-beta signaling pathway. Oncotarget 2015, 6, 43605–43619. [Google Scholar] [CrossRef]
- Del Barco, S.; Vazquez-Martin, A.; Cufi, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef]
- Moodley, Y.P.; Misso, N.L.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef]
- Saito, F.; Tasaka, S.; Inoue, K.; Miyamoto, K.; Nakano, Y.; Ogawa, Y.; Yamada, W.; Shiraishi, Y.; Hasegawa, N.; Fujishima, S.; et al. Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am. J. Respir. Cell Mol. Biol. 2008, 38, 566–571. [Google Scholar] [CrossRef]
- Ishiguro, Y.; Ishiguro, H.; Miyamoto, H. Epidermal growth factor receptor tyrosine kinase inhibition up-regulates interleukin-6 in cancer cells and induces subsequent development of interstitial pneumonia. Oncotarget 2013, 4, 550–559. [Google Scholar] [CrossRef]
- Wang, S.Z.; Rosenberger, C.L.; Bao, Y.X.; Stark, J.M.; Harrod, K.S. Clara cell secretory protein modulates lung inflammatory and immune responses to respiratory syncytial virus infection. J. Immunol. 2003, 171, 1051–1060. [Google Scholar] [CrossRef]
- Mango, G.W.; Johnston, C.J.; Reynolds, S.D.; Finkelstein, J.N.; Plopper, C.G.; Stripp, B.R. Clara cell secretory protein deficiency increases oxidant stress response in conducting airways. Am. J. Physiol. 1998, 275, L348–L356. [Google Scholar] [CrossRef]
- Jones, K.G.; Holland, J.F.; Foureman, G.L.; Bend, J.R.; Fouts, J.R. Xenobiotic metabolism in Clara cells and alveolar type II cells isolated from lungs of rats treated with beta-naphthoflavone. J. Pharmacol. Exp. Ther. 1983, 225, 316–319. [Google Scholar] [PubMed]
- Harada, C.; Kawaguchi, T.; Ogata-Suetsugu, S.; Yamada, M.; Hamada, N.; Maeyama, T.; Souzaki, R.; Tajiri, T.; Taguchi, T.; Kuwano, K.; et al. EGFR tyrosine kinase inhibition worsens acute lung injury in mice with repairing airway epithelium. Am. J. Respir. Crit. Care Med. 2011, 183, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Zwick, E.; Hackel, P.O.; Prenzel, N.; Ullrich, A. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol. Sci. 1999, 20, 408–412. [Google Scholar] [CrossRef]
- Shim, J.J.; Dabbagh, K.; Ueki, I.F.; Dao-Pick, T.; Burgel, P.R.; Takeyama, K.; Tam, D.C.; Nadel, J.A. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L134–L140. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Kuo, C.T.; Lin, C.C.; Hsieh, H.L.; Yang, C.M. IL-1beta induces expression of matrix metalloproteinase-9 and cell migration via a c-Src-dependent, growth factor receptor transactivation in A549 cells. Br. J. Pharmacol. 2010, 160, 1595–1610. [Google Scholar] [CrossRef]
- Yamaoka, T.; Yan, F.; Cao, H.; Hobbs, S.S.; Dise, R.S.; Tong, W.; Polk, D.B. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11772–11777. [Google Scholar] [CrossRef]
- Edelblum, K.L.; Yan, F.; Yamaoka, T.; Polk, D.B. Regulation of apoptosis during homeostasis and disease in the intestinal epithelium. Inflamm. Bowel. Dis. 2006, 12, 413–424. [Google Scholar] [CrossRef]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Yamaoka, T.; Arata, S.; Homma, M.; Homma, T.; Kusumoto, S.; Ando, K.; Manabe, R.; Kishino, Y.; Ohba, M.; Tsurutani, J.; et al. Blockade of EGFR Activation Promotes TNF-Induced Lung Epithelial Cell Apoptosis and Pulmonary Injury. Int. J. Mol. Sci. 2019, 20, 4021. [Google Scholar] [CrossRef]
- Liu, J.Y.; Brass, D.M.; Hoyle, G.W.; Brody, A.R. TNF-alpha receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am. J. Pathol. 1998, 153, 1839–1847. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Araki, K.; Vesin, C.; Garcia, I.; Kapanci, Y.; Whitsett, J.A.; Piguet, P.F.; Vassalli, P. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J. Clin. Investig. 1995, 96, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K. Transient Asymptomatic Pulmonary Opacities during Osimertinib Treatment: “Stop or Go” Decision. J. Thorac. Oncol. 2016, 11, 2051–2052. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Tanimoto, T.; Yuji, K.; Tojo, A. EGFR-TKI-Associated Interstitial Pneumonitis in Nivolumab-Treated Patients with Non-Small Cell Lung Cancer. JAMA Oncol. 2018, 4, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Azuma, A.; Kanazawa, M.; Kameda, H.; Kusumoto, M.; Genma, A.; Saijo, Y.; Sakai, F.; Sugiyama, Y.; Tatsumi, K.; et al. Consensus statement for the diagnosis and treatment of drug-induced lung injuries. Respir. Investig. 2013, 51, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Min, J.H.; Lee, H.Y.; Lim, H.; Ahn, M.J.; Park, K.; Chung, M.P.; Lee, K.S. Drug-induced interstitial lung disease in tyrosine kinase inhibitor therapy for non-small cell lung cancer: A review on current insight. Cancer Chemother. Pharmacol. 2011, 68, 1099–1109. [Google Scholar] [CrossRef]
- Stanam, A.; Gibson-Corley, K.N.; Love-Homan, L.; Ihejirika, N.; Simons, A.L. Interleukin-1 blockade overcomes erlotinib resistance in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 76087–76100. [Google Scholar] [CrossRef]
- Gelfo, V.; Rodia, M.T.; Pucci, M.; Dall’Ora, M.; Santi, S.; Solmi, R.; Roth, L.; Lindzen, M.; Bonafe, M.; Bertotti, A.; et al. A module of inflammatory cytokines defines resistance of colorectal cancer to EGFR inhibitors. Oncotarget 2016, 7, 72167–72183. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, J.; Chen, J.; Jin, S.; Yao, J.; Yu, T.; Wang, W.; Guo, R. IL-22 Confers EGFR-TKI Resistance in NSCLC via the AKT and ERK Signaling Pathways. Front. Oncol. 2019, 9, 1167. [Google Scholar] [CrossRef]
- Liu, Y.N.; Chang, T.H.; Tsai, M.F.; Wu, S.G.; Tsai, T.H.; Chen, H.Y.; Yu, S.L.; Yang, J.C.; Shih, J.Y. IL-8 confers resistance to EGFR inhibitors by inducing stem cell properties in lung cancer. Oncotarget 2015, 6, 10415–10431. [Google Scholar] [CrossRef]
- Umekawa, K.; Kimura, T.; Kudoh, S.; Suzumura, T.; Oka, T.; Nagata, M.; Mitsuoka, S.; Matsuura, K.; Nakai, T.; Yoshimura, N.; et al. Plasma RANTES, IL-10, and IL-8 levels in non-small-cell lung cancer patients treated with EGFR-TKIs. BMC Res. Notes 2013, 6, 139. [Google Scholar] [CrossRef]
- Ando, K.; Ohmori, T.; Inoue, F.; Kadofuku, T.; Hosaka, T.; Ishida, H.; Shirai, T.; Okuda, K.; Hirose, T.; Horichi, N.; et al. Enhancement of sensitivity to tumor necrosis factor alpha in non-small cell lung cancer cells with acquired resistance to gefitinib. Clin. Cancer Res. 2005, 11, 8872–8879. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Guo, G.; Gerber, D.E.; Gao, B.; Peyton, M.; Huang, C.; Minna, J.D.; Hatanpaa, K.J.; Kernstine, K.; Cai, L.; et al. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J. Clin. Investig. 2018, 128, 2500–2518. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Suh, C.H.; Park, H.S.; Kim, K.W.; Pyo, J.; Hatabu, H.; Nishino, M. Pneumonitis in advanced non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitor: Meta-analysis of 153 cohorts with 15,713 patients: Meta-analysis of incidence and risk factors of EGFR-TKI pneumonitis in NSCLC. Lung Cancer 2018, 123, 60–69. [Google Scholar] [CrossRef]
- Atagi, S.; Katakami, N.; Yoshioka, H.; Fukuoka, M.; Kudoh, S.; Ogiwara, A.; Imai, M.; Ueda, M.; Matsui, S. Nested case control study of proteomic biomarkers for interstitial lung disease in Japanese patients with non-small-cell lung cancer treated with erlotinib: A multicenter phase IV study (JO21661). Clin. Lung Cancer 2013, 14, 407–417. [Google Scholar] [CrossRef]
- OECD Health Statistics. 2020. Available online: https://www.oecd.org/health/health-data.htm (accessed on 4 December 2020).
- Raghu, G.; Remy-Jardin, M.; Myers, J.; Richeldi, L.; Wilson, K.C. The 2018 Diagnosis of Idiopathic Pulmonary Fibrosis Guidelines: Surgical Lung Biopsy for Radiological Pattern of Probable Usual Interstitial Pneumonia is Not Mandatory. Am. J. Respir. Crit. Care Med. 2019, 200, 1089–1092. [Google Scholar] [CrossRef]
- Shi, L.; Tang, J.; Tong, L.; Liu, Z. Risk of interstitial lung disease with gefitinib and erlotinib in advanced non-small cell lung cancer: A systematic review and meta-analysis of clinical trials. Lung Cancer 2014, 83, 231–239. [Google Scholar] [CrossRef]
- Hosomi, Y.; Morita, S.; Sugawara, S.; Kato, T.; Fukuhara, T.; Gemma, A.; Takahashi, K.; Fujita, Y.; Harada, T.; Minato, K.; et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer with Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J. Clin. Oncol. 2020, 38, 115–123. [Google Scholar] [CrossRef]
- Noronha, V.; Patil, V.M.; Joshi, A.; Menon, N.; Chougule, A.; Mahajan, A.; Janu, A.; Purandare, N.; Kumar, R.; More, S.; et al. Gefitinib Versus Gefitinib Plus Pemetrexed and Carboplatin Chemotherapy in EGFR-Mutated Lung Cancer. J. Clin. Oncol. 2020, 38, 124–136. [Google Scholar] [CrossRef]
- Saito, H.; Fukuhara, T.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): Interim analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Oncol. 2019, 20, 625–635. [Google Scholar] [CrossRef]
- Herbst, R.S.; Ansari, R.; Bustin, F.; Flynn, P.; Hart, L.; Otterson, G.A.; Vlahovic, G.; Soh, C.H.; O’Connor, P.; Hainsworth, J. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): A double-blind, placebo-controlled, phase 3 trial. Lancet 2011, 377, 1846–1854. [Google Scholar] [CrossRef]
- Seto, T.; Kato, T.; Nishio, M.; Goto, K.; Atagi, S.; Hosomi, Y.; Yamamoto, N.; Hida, T.; Maemondo, M.; Nakagawa, K.; et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): An open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014, 15, 1236–1244. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Ponce Aix, S.; Paz-Ares, L.; Chiu, C.H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Yang, J.C.; Yu, H.; Kim, S.W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.L.; White, D.A.; Jiang, H.; Gemma, A. Diagnosis and management of drug-associated interstitial lung disease. Br. J. Cancer 2004, 91 (Suppl. S2), S24–S30. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Margaritopoulos, G.A.; Tomassetti, S.; Bonella, F.; Costabel, U.; Poletti, V. Interstitial lung disease. Eur. Respir. Rev. 2014, 23, 40–54. [Google Scholar] [CrossRef]
- Endo, M.; Johkoh, T.; Kimura, K.; Yamamoto, N. Imaging of gefitinib-related interstitial lung disease: Multi-institutional analysis by the West Japan Thoracic Oncology Group. Lung Cancer 2006, 52, 135–140. [Google Scholar] [CrossRef]
- Chang, H.L.; Chen, Y.H.; Taiwan, H.C.; Yang, C.J. EGFR Tyrosine Kinase Inhibitor-Associated Interstitial Lung Disease during the Coronavirus Disease 2019 Pandemic. J. Thorac. Oncol. 2020, 15, e129–e131. [Google Scholar] [CrossRef]
- Kohno, N.; Kyoizumi, S.; Awaya, Y.; Fukuhara, H.; Yamakido, M.; Akiyama, M. New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest 1989, 96, 68–73. [Google Scholar] [CrossRef]
- Kohno, N.; Hamada, H.; Fujioka, S.; Hiwada, K.; Yamakido, M.; Akiyama, M. Circulating antigen KL-6 and lactate dehydrogenase for monitoring irradiated patients with lung cancer. Chest 1992, 102, 117–122. [Google Scholar] [CrossRef]
- Ohnishi, H.; Yokoyama, A.; Yasuhara, Y.; Watanabe, A.; Naka, T.; Hamada, H.; Abe, M.; Nishimura, K.; Higaki, J.; Ikezoe, J.; et al. Circulating KL-6 levels in patients with drug induced pneumonitis. Thorax 2003, 58, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Kawase, S.; Hattori, N.; Ishikawa, N.; Horimasu, Y.; Fujitaka, K.; Furonaka, O.; Isobe, T.; Miyoshi, S.; Hamada, H.; Yamane, T.; et al. Change in serum KL-6 level from baseline is useful for predicting life-threatening EGFR-TKIs induced interstitial lung disease. Respir. Res. 2011, 12, 97. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C.; Raghu, G.; Baughman, R.P.; Brown, K.K.; Costabel, U.; du Bois, R.M.; Drent, M.; Haslam, P.L.; Kim, D.S.; Nagai, S.; et al. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Tani, T.; Naoki, K.; Asakura, T.; Hirano, T.; Suzuki, S.; Masuzawa, K.; Hasegawa, H.; Kuroda, A.; Yasuda, H.; Ishii, M.; et al. Successful treatment of non-small-cell lung cancer with afatinib and a glucocorticoid following gefitinib- and erlotinib-induced interstitial lung disease: A case report. Mol. Clin. Oncol. 2016, 5, 488–490. [Google Scholar] [CrossRef]
- Kuo, L.C.; Lin, P.C.; Wang, K.F.; Yuan, M.K.; Chang, S.C. Successful treatment of gefitinib-induced acute interstitial pneumonitis with high-dose corticosteroid: A case report and literature review. Med. Oncol. 2011, 28, 79–82. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef]
- González-Gay, M.A.; Loricera, J.; Blanco, R. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 1493. [Google Scholar] [CrossRef]
- Toniati, P.; Piva, S.; Cattalini, M.; Garrafa, E.; Regola, F.; Castelli, F.; Franceschini, F.; Airò, P.; Bazzani, C.; Beindorf, E.A.; et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun. Rev. 2020, 19, 102568. [Google Scholar] [CrossRef]
- Michot, J.M.; Albiges, L.; Chaput, N.; Saada, V.; Pommeret, F.; Griscelli, F.; Balleyguier, C.; Besse, B.; Marabelle, A.; Netzer, F.; et al. Tocilizumab, an anti-IL-6 receptor antibody, to treat COVID-19-related respiratory failure: A case report. Ann. Oncol. 2020, 31, 961–964. [Google Scholar] [CrossRef]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS((R)) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef]
- Kanayama, M.; Mori, M.; Matsumiya, H.; Taira, A.; Shinohara, S.; Kuwata, T.; Imanishi, N.; Yoneda, K.; Kuroda, K.; Tanaka, F. Perioperative pirfenidone treatment for lung cancer patients with idiopathic pulmonary fibrosis. Surg. Today 2020, 50, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yano, Y.; Kuge, T.; Okabe, F.; Ishijima, M.; Uenami, T.; Kanazu, M.; Akazawa, Y.; Yamaguchi, T.; Mori, M. Safety and effectiveness of pirfenidone combined with carboplatin-based chemotherapy in patients with idiopathic pulmonary fibrosis and non-small cell lung cancer: A retrospective cohort study. Thorac. Cancer 2020, 11, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Otsubo, K.; Kishimoto, J.; Kenmotsu, H.; Minegishi, Y.; Ichihara, E.; Shiraki, A.; Kato, T.; Atagi, S.; Horinouchi, H.; Ando, M.; et al. Treatment Rationale and Design for J-SONIC: A Randomized Study of Carboplatin Plus Nab-paclitaxel With or Without Nintedanib for Advanced Non-Small-cell Lung Cancer with Idiopathic Pulmonary Fibrosis. Clin. Lung Cancer 2018, 19, e5–e9. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Rash | Diarrhea | AST/ALT Elevation | ILD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Trial | Drugs | n | ALL (%) | ≥G3 (%) | ALL (%) | ≥G3 (%) | ALL (%) | ≥G3 (%) | ALL (%) | ≥G3 (%) |
| NEJ002 | Gefitinib | 144 | 71.0 | 5.3 | 34.2 | 0.9 | 55.2 | 26.3 | 5.3 | 2.6 |
| Carbo/Pacli 1 | 144 | 19.2 | 2.7 | 6.1 | 0 | 32.4 | 0.9 | 0 | 0 | |
| WJOG 5108L | Gefitinib | 277 | 74.7 | 2.2 | 42.6 | 2.2 | 42.2/50.9 | 6.1/13.0 | 4.3 | 0.4 |
| Erlotinib | 276 | 92.4 | 18.1 | 51.1 | 3.3 | 34.9/38.2 | 2.2/3.3 | 4.0 | 1.4 | |
| LUX Lung7 | Afatinib | 160 | 88.0 | 12.0 | 91.0 | 13.0 | 10.0 | 0 | 1.3 | 0.6 |
| Gefitinib | 159 | 61.0 | 1.0 | 61.0 | 1.0 | 25.0 | 9.0 | 0 | 0 | |
| FLAURA | Osimertinib | 279 | 58 | 1 | 58 | 2 | 9/6 | 1/1 | 4.0 | NA 3 |
| Gef. or Erlo 2 | 277 | 78 | 7 | 57 | 2 | 25/27 | 4/8 | 2.2 | NA 3 | |
| Author | Patients | Treatments | ILD Incidence (%) | Japanese ILD Incidence (%) | Reference |
|---|---|---|---|---|---|
| Suh et al. | 15,713 | Gef. 1, Erlo. 2, Afa. 3, Osim. 4 | 1.1 | 4.8 | [64] |
| Shi et al. | 8609 | Gef. 1, Erlo. 2 | 1.2 | 3.3 | [68] |
| Takeda et al. | 1468 | Gef. 1, Erlo. 2, Afa. 3 | 0.6–2.2 | 3.8 | [22] |
| ILD | ||||
|---|---|---|---|---|
| Trial | Drugs | ALL (%) | ≥G3 (%) | Reference |
| NEJ009 | CBDCA 1 + PEM 2 + Gefitinib | 11/170 (6.5%) | 4/170 (2.4%) | [69] |
| Gefitinib | 6/171 (3.5%) | 1/171 (0.6%) | ||
| NEJ026 | Erlotinib + Bevacizumab | 0/112 (0%) | 0/121 (0%) | [71] |
| Erlotinib | 5/114 (4.4%) | 0/114 (0%) | ||
| RELAY | Erlotinib + Ramucirumab | 3/221 (1.4%) | 1/221 (0.5%) | [74] |
| Erlotinib | 4/225 (1.8%) | 2/225 (0.9%) | ||
| TATTON | Osimertinib + Durvalumab | 5/23 (22%) | 2/23 (8.7%) | [75] |
| Pattern | Findings on Roentgenography | Manifestations on CT Scan | Corresponding Pattern of ILD | Incidence (%) | Mortality (%) |
|---|---|---|---|---|---|
| A | Diffuse and faint opacity without volume loss | Non-specific area: Ground-glass opacity | NSIP 1 | 47.1 | 31.0 |
| B | Peripheral consolidation | Multifocal area: Airspace consolidation | OP/BOOP 2 | 13.7 | 28.6 |
| C | Patchy or diffuse faint, liner opacities | Patchy distribution area: Ground-glass opacity interlobular septal thickening | AEP 3 | 2.0 | 0.0 |
| D | Diffuse faint opacity or consolidation with volume loss | Extensive bilateral area: Ground-glass opacities; air space consolidation with traction bronchiectasis | AIP 4 | 23.5 | 75.0 |
| Others | Non-specific | Non-specific | N/A | 13.7 | 45.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohmori, T.; Yamaoka, T.; Ando, K.; Kusumoto, S.; Kishino, Y.; Manabe, R.; Sagara, H. Molecular and Clinical Features of EGFR-TKI-Associated Lung Injury. Int. J. Mol. Sci. 2021, 22, 792. https://doi.org/10.3390/ijms22020792
Ohmori T, Yamaoka T, Ando K, Kusumoto S, Kishino Y, Manabe R, Sagara H. Molecular and Clinical Features of EGFR-TKI-Associated Lung Injury. International Journal of Molecular Sciences. 2021; 22(2):792. https://doi.org/10.3390/ijms22020792
Chicago/Turabian StyleOhmori, Tohru, Toshimitsu Yamaoka, Koichi Ando, Sojiro Kusumoto, Yasunari Kishino, Ryou Manabe, and Hironori Sagara. 2021. "Molecular and Clinical Features of EGFR-TKI-Associated Lung Injury" International Journal of Molecular Sciences 22, no. 2: 792. https://doi.org/10.3390/ijms22020792
APA StyleOhmori, T., Yamaoka, T., Ando, K., Kusumoto, S., Kishino, Y., Manabe, R., & Sagara, H. (2021). Molecular and Clinical Features of EGFR-TKI-Associated Lung Injury. International Journal of Molecular Sciences, 22(2), 792. https://doi.org/10.3390/ijms22020792