JAZF1, A Novel p400/TIP60/NuA4 Complex Member, Regulates H2A.Z Acetylation at Regulatory Regions

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
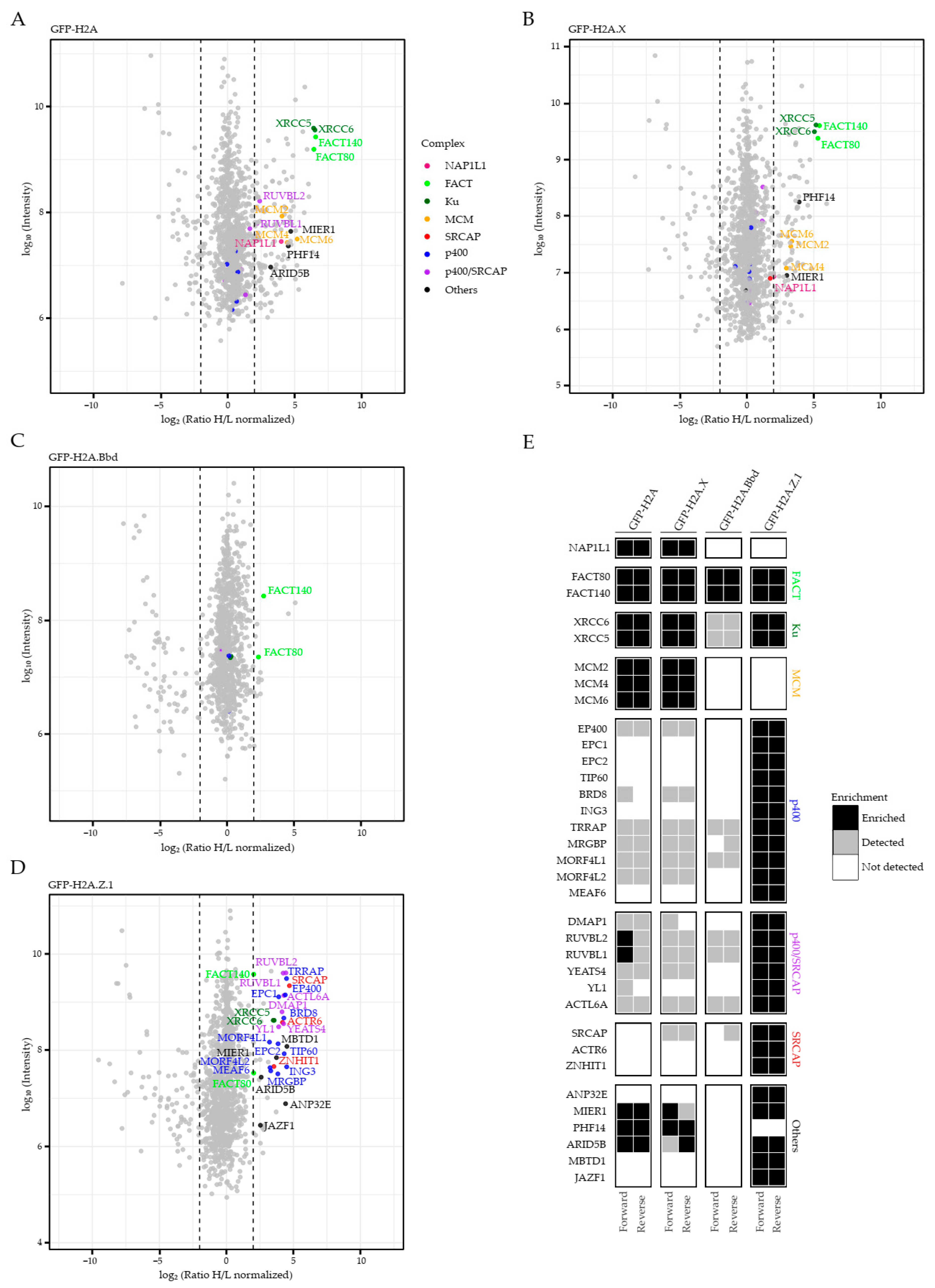
2.1. Identification of Chaperone/Remodeling Complexes of Human H2A Variants
2.2. JAZF1 Is a Member of an MBTD1-Containing But ANP32E-Excluding H2A.Z-Specific p400 Chaperone Sub-Complex
2.3. JAZF1 Depletion Leads to Deregulation of Genes Involved in Ribosome Biogenesis
2.4. JAZF1 Depletion Does Not Influence H2A.Z Occupancy But Leads to Reduced H2A.Z Acetylation Levels at Intronic Enhancer Sites
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Transfection and Flow Cytometry
4.2. Plasmids
4.3. SILAC Followed by Mass Spectrometric (MS) Identification
4.4. Antibodies
4.5. Immunoprecipitation of GFP-Tagged Proteins
4.6. Immunoblotting
4.7. siRNA Transfections
4.8. RNA Isolation, cDNA Preparation and RT-qPCR
4.9. RNA-Seq and Data Analysis
4.10. Immunofluorescence (IF) Microscopy
4.11. Histone Extraction
4.12. Chromatin Immunoprecipitation Followed by Sequencing (ChIP-Seq)
4.12.1. Chromatin Immunoprecipitation (ChIP)
4.12.2. Illumina Sequencing
4.12.3. ChIP-Seq Analysis
4.13. ChIP–qPCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARID5B | AT rich interactive domain 5B |
| BSA | Bovines Serum Albumin |
| BP | Biological pathway |
| CC | Cellular compartment |
| ChIP-seq | Chromatin Immunoprecipitation followed by sequencing |
| DR1 | Direct repeat DNA element 1 |
| ETS | E26 transformation-specific |
| FACT | Facilitates chromatin transcription |
| FDR | False Discovery Rate |
| GFP | Green Fluorescent Protein |
| GSEA | Gene set enrichment analysis |
| GO | Gene Ontology |
| HPRT1 | Hypoxanthine Phosphoribosyltransferase 1 |
| IF | Immunofluorescence |
| IP | Immunoprecipitation |
| JAZF1 | Juxtaposed with another zinc finger 1 |
| KAT | Lysine Acetyltransferase |
| MBTD1 | MBT domain protein 1 |
| MIER1 | Mesoderm induction early response 1 |
| MS | Mass spectrometry |
| NES | Normalized Enrichment Score |
| PCA | Principle component analysis |
| RNA-seq | RNA-sequencing |
| rRNA | ribosomal RNA |
| RT-qPCR | Reverse Transcriptase-quantitative Polymerase Chain Reaction |
| SD | Standard Deviation |
| SILAC | Stable Isotope Labeling by Amino Acids in Cell Culture |
| siNTC | Non-target control siRNA |
| siJAZF1 | JAZF1-specific siRNA |
| siTIP60 | TIP60-specific siRNA |
| siRNA | small interfering RNA |
| snoRNP | small nucleolar Ribonuclear Protein |
| T2D | Type 2 Diabetes |
| TIP60 | TAT-Interactive Protein 60 kDa |
| TSS | Transcriptional Start Site |
References
- Van Holde, K.E. Chromatin; Springer: New York, NY, USA, 1988. [Google Scholar]
- Bonisch, C.; Nieratschker, S.M.; Orfanos, N.K.; Hake, S.B. Chromatin proteomics and epigenetic regulatory circuits. Expert Rev. Proteom. 2008, 5, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef]
- Zink, L.M.; Hake, S.B. Histone variants: Nuclear function and disease. Curr. Opin. Genet. Dev. 2016, 37, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Iouzalen, N.; Moreau, J.; Mechali, M. H2A.ZI, a new variant histone expressed during Xenopus early development exhibits several distinct features from the core histone H2A. Nucleic Acids Res. 1996, 24, 3947–3952. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.S.; Kalashnikova, T.; Smith, M.M. Histone H2A.Z regulats transcription and is partially redundant with nucleosome remodeling complexes. Cell 2000, 103, 411–422. [Google Scholar] [CrossRef]
- Meneghini, M.D.; Wu, M.; Madhani, H.D. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 2003, 112, 725–736. [Google Scholar] [CrossRef]
- Rangasamy, D.; Greaves, I.; Tremethick, D.J. RNA interference demonstrates a novel role for H2A.Z in chromosome segregation. Nat. Struct. Mol. Biol. 2004, 11, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Kalocsay, M.; Hiller, N.J.; Jentsch, S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell 2009, 33, 335–343. [Google Scholar] [CrossRef]
- Weber, C.M.; Henikoff, J.G.; Henikoff, S. H2A.Z nucleosomes enriched over active genes are homotypic. Nat. Struct. Mol. Biol. 2010, 17, 1500–1507. [Google Scholar] [CrossRef]
- Zovkic, I.B.; Paulukaitis, B.S.; Day, J.J.; Etikala, D.M.; Sweatt, J.D. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature 2014, 515, 582–586. [Google Scholar] [CrossRef]
- Svotelis, A.; Gevry, N.; Grondin, G.; Gaudreau, L. H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle 2010, 9, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Dryhurst, D.; McMullen, B.; Fazli, L.; Rennie, P.S.; Ausio, J. Histone H2A.Z prepares the prostate specific antigen (PSA) gene for androgen receptor-mediated transcription and is upregulated in a model of prostate cancer progression. Cancer Lett. 2012, 315, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Mora, F.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Nair, S.S.; Patterson, K.I.; Tremethick, D.J.; Stirzaker, C.; Clark, S.J. Acetylation of H2A.Z is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res. 2012, 22, 307–321. [Google Scholar] [CrossRef]
- Kim, K.; Punj, V.; Choi, J.; Heo, K.; Kim, J.M.; Laird, P.W.; An, W. Gene dysregulation by histone variant H2A.Z in bladder cancer. Epigenet. Chromatin 2013, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Punzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone Variant H2A.Z.2 Mediates Proliferation and Drug Sensitivity of Malignant Melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Eirin-Lopez, J.M.; Gonzalez-Romero, R.; Dryhurst, D.; Ishibashi, T.; Ausio, J. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol. Biol. 2009, 9, 31. [Google Scholar] [CrossRef]
- Dryhurst, D.; Ishibashi, T.; Rose, K.L.; Eirin-Lopez, J.M.; McDonald, D.; Silva-Moreno, B.; Veldhoen, N.; Helbing, C.C.; Hendzel, M.J.; Shabanowitz, J.; et al. Characterization of the histone H2A.Z-1 and H2A.Z-2 isoforms in vertebrates. BMC Biol. 2009, 7, 86. [Google Scholar] [CrossRef]
- Bonisch, C.; Schneider, K.; Punzeler, S.; Wiedemann, S.M.; Bielmeier, C.; Bocola, M.; Eberl, H.C.; Kuegel, W.; Neumann, J.; Kremmer, E.; et al. H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization. Nucleic Acids Res. 2012, 40, 5951–5964. [Google Scholar] [CrossRef] [PubMed]
- Billon, P.; Cote, J. Precise deposition of histone H2A.Z in chromatin for genome expression and maintenance. Biochim. Biophys. Acta 2011, 1819, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Brahma, S.; Udugama, M.I.; Kim, J.; Hada, A.; Bhardwaj, S.K.; Hailu, S.G.; Lee, T.H.; Bartholomew, B. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Obri, A.; Ouararhni, K.; Papin, C.; Diebold, M.L.; Padmanabhan, K.; Marek, M.; Stoll, I.; Roy, L.; Reilly, P.T.; Mak, T.W.; et al. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 2014, 505, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Conerly, M.L.; Teves, S.S.; Diolaiti, D.; Ulrich, M.; Eisenman, R.N.; Henikoff, S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res. 2010, 20, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.R.; Roh, T.Y.; Schones, D.E.; Wang, Z.B.; Wei, G.; Chepelev, I.; Zhao, K.J. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Millau, J.F.; Gaudreau, L. CTCF, cohesin, and histone variants: Connecting the genome. Biochem. Cell Biol. Biochim. Biol. Cell. 2011, 89, 505–513. [Google Scholar] [CrossRef]
- Rangasamy, D.; Berven, L.; Ridgway, P.; Tremethick, D.J. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003, 22, 1599–1607. [Google Scholar] [CrossRef]
- Greaves, I.K.; Rangasamy, D.; Ridgway, P.; Tremethick, D.J. H2A.Z contributes to the unique 3D structure of the centromere. Proc. Natl. Acad. Sci. USA 2007, 104, 525–530. [Google Scholar] [CrossRef]
- Zink, L.M.; Delbarre, E.; Eberl, H.C.; Keilhauer, E.C.; Bonisch, C.; Punzeler, S.; Bartkuhn, M.; Collas, P.; Mann, M.; Hake, S.B. H3.Y discriminates between HIRA and DAXX chaperone complexes and reveals unexpected insights into human DAXX-H3.3-H4 binding and deposition requirements. Nucleic Acids Res. 2017, 45, 5691–5706. [Google Scholar] [CrossRef]
- Orphanides, G.; Wu, W.H.; Lane, W.S.; Hampsey, M.; Reinberg, D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999, 400, 284–288. [Google Scholar] [CrossRef]
- Hondele, M.; Stuwe, T.; Hassler, M.; Halbach, F.; Bowman, A.; Zhang, E.T.; Nijmeijer, B.; Kotthoff, C.; Rybin, V.; Amlacher, S.; et al. Structural basis of histone H2A-H2B recognition by the essential chaperone FACT. Nature 2013, 499, 111–114. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, K.D.; Zhang, N.F.; Wei, H.; Tan, Y.Z.; Zhang, Z.N.; Carragher, B.; Potter, C.S.; D’Arcy, S.; Luger, K. FACT caught in the act of manipulating the nucleosome. Nature 2020, 577, 426–431. [Google Scholar] [CrossRef]
- Aguilar-Gurrieri, C.; Larabi, A.; Vinayachandran, V.; Patel, N.A.; Yen, K.Y.; Reja, R.; Ebong, I.O.; Schoehn, G.; Robinson, C.V.; Pugh, B.F.; et al. Structural evidence for Nap1-dependent H2A-H2B deposition and nucleosome assembly. EMBO J. 2016, 35, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Onesti, S. The MCM complex: (Just) A replicative helicase? Biochem. Soc. Trans. 2008, 36, 136–140. [Google Scholar] [CrossRef]
- Latreille, D.; Bluy, L.; Benkirane, M.; Kiernan, R.E. Identification of histone 3 variant 2 interacting factors. Nucleic Acids Res. 2014, 42, 3542–3550. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Difilippantonio, S.; Difilippantonio, M.J.; Fernandez-Capetillo, O.; Pilch, D.R.; Sedelnikova, O.A.; Eckhaus, M.; Ried, T.; Bonner, W.M.; Nussenzweig, A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003, 114, 371–383. [Google Scholar] [CrossRef]
- Piquet, S.; Le Parc, F.; Bai, S.K.; Chevallier, O.; Adam, S.; Polo, S.E. The Histone Chaperone FACT Coordinates H2A.X-Dependent Signaling and Repair of DNA Damage. Mol. Cell 2018, 72, 888–901. [Google Scholar] [CrossRef]
- Sansoni, V.; Casas-Delucchi, C.S.; Rajan, M.; Schmidt, A.; Bonisch, C.; Thomae, A.W.; Staege, M.S.; Hake, S.B.; Cardoso, M.C.; Imhof, A. The histone variant H2A.Bbd is enriched at sites of DNA synthesis. Nucleic Acids Res. 2014, 42, 6405–6420. [Google Scholar] [CrossRef]
- Bonisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef]
- Mao, Z.; Pan, L.; Wang, W.X.; Sun, J.; Shan, S.; Dong, Q.; Liang, X.P.; Dai, L.C.; Ding, X.J.; Chen, S.; et al. Anp32e, a higher eukaryotic histone chaperone directs preferential recognition for H2A.Z. Cell Res. 2014, 24, 389–399. [Google Scholar] [CrossRef]
- Gursoy-Yuzugullu, O.; Ayrapetov, M.K.; Price, B.D. Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 7507–7512. [Google Scholar] [CrossRef]
- Murphy, K.E.; Meng, F.W.; Makowski, C.E.; Murphy, P.J. Genome-wide chromatin accessibility is restricted by ANP32E. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Fujino, S.; Nakanishi, G.; Kim, Y.S.; Jetten, A.M. TIP27: A novel repressor of the nuclear orphan receptor TAK1/TR4. Nucleic Acids Res. 2004, 32, 4194–4204. [Google Scholar] [CrossRef] [PubMed]
- Eryilmaz, J.; Pan, P.; Amaya, M.F.; Allali-Hassani, A.; Dong, A.P.; Adams-Cioaba, M.A.; MacKenzie, F.; Vedadi, M.; Min, J.R. Structural Studies of a Four-MBT Repeat Protein MBTD1. PLoS ONE 2009, 4, e7274. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, K.; Fradet-Turcotte, A.; Avvakumov, N.; Lambert, J.P.; Roques, C.; Pandita, R.K.; Paquet, E.; Herst, P.; Gingras, A.C.; Pandita, T.K.; et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 2016, 62, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Ueda, T.; Nagata, S.; Fukunaga, R. Essential Role of p400/mDomino Chromatin-remodeling ATPase in Bone Marrow Hematopoiesis and Cell-cycle Progression. J. Biol. Chem. 2010, 285, 30214–30223. [Google Scholar] [CrossRef] [PubMed]
- Kobiita, A.; Godbersen, S.; Araldi, E.; Ghoshdastider, U.; Schmid, M.W.; Spinas, G.; Moch, H.; Stoffel, M. The Diabetes Gene JAZF1 Is Essential for the Homeostatic Control of Ribosome Biogenesis and Function in Metabolic Stress. Cell Rep. 2020, 32, 107846. [Google Scholar] [CrossRef]
- Stanek, D.; Neugebauer, K.M. The Cajal body: A meeting place for spliceosomal snRNPs in the nuclear maze. Chromosoma 2006, 115, 343–354. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L.; Sleeman, J.E. The Cajal body and the nucleolus: “In a relationship” or “It’s complicated”? RNA Biol. 2017, 14, 739–751. [Google Scholar] [CrossRef]
- Couture, J.P.; Nolet, G.; Beaulieu, E.; Blouin, R.; Gevry, N. The p400/Brd8 Chromatin Remodeling Complex Promotes Adipogenesis by Incorporating Histone Variant H2A.Z at PPAR gamma Target Genes. Endocrinology 2012, 153, 5796–5808. [Google Scholar] [CrossRef]
- Narkaj, K.; Stefanelli, G.; Wahdan, M.; Azam, A.B.; Ramzan, F.; Steininger, C.F.D.J.; Walters, B.J.; Zovkic, I.B. Blocking H2A.Z Incorporation via Tip60 Inhibition Promotes Systems Consolidation of Fear Memory in Mice. Eneuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Giaimo, B.D.; Ferrante, F.; Vallejo, D.M.; Hein, K.; Gutierrez-Perez, I.; Nist, A.; Stiewe, T.; Mittler, G.; Herold, S.; Zimmermann, T.; et al. Histone variant H2A.Z deposition and acetylation directs the canonical Notch signaling response. Nucleic Acids Res. 2018, 46, 8197–8215. [Google Scholar] [CrossRef] [PubMed]
- Punzeler, S.; Link, S.; Wagner, G.; Keilhauer, E.C.; Kronbeck, N.; Spitzer, R.M.M.; Leidescher, S.; Markaki, Y.; Mentele, E.; Regnard, C.; et al. Multivalent binding of PWWP2A to H2A.Z regulates mitosis and neural crest differentiation. EMBO J. 2017, 36, 2263–2279. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; Spitzer, R.M.M.; Sana, M.; Torrado, M.; Volker-Albert, M.C.; Keilhauer, E.C.; Burgold, T.; Punzeler, S.; Low, J.K.K.; Lindstrom, I.; et al. PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Sun, Z.; Filipescu, D.; Andrade, J.; Gaspar-Maia, A.; Ueberheide, B.; Bernstein, E. Transcription-associated histone pruning demarcates macroH2A chromatin domains. Nat. Struct. Mol. Biol. 2018, 25, 958–970. [Google Scholar] [CrossRef]
- Chang, L.; Loranger, S.S.; Mizzen, C.; Ernst, S.G.; Allis, C.D.; Annunziato, A.T. Histones in transit: Cytosolic histone complexes and diacetylation of H4 during nucleosome assembly in human cells. Biochemistry 1997, 36, 469–480. [Google Scholar] [CrossRef]
- Adams, C.R.; Kamakaka, R.T. Chromatin assembly: Biochemical identities and genetic redundancy. Curr. Opin. Genet. Dev. 1999, 9, 185–190. [Google Scholar] [CrossRef]
- Paterno, G.D.; Li, Y.; Luchman, H.A.; Ryan, P.J.; Gillespie, L.L. cDNA cloning of a novel, developmentally regulated immediate early gene activated by fibroblast growth factor and encoding a nuclear protein. J. Biol. Chem. 1997, 272, 25591–25595. [Google Scholar] [CrossRef]
- Whitson, R.H.; Huang, T.; Itakura, K. The novel Mrf-2 DNA-binding domain recognizes a five-base core sequence through major and minor-groove contacts. Biochem. Biophys. Res. Commun. 1999, 258, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Smith, E.R.; Morgan, M.A.J.; Ugarenko, M.; Khaltyan, N.; Helmin, K.A.; Ryan, C.A.; Murray, D.C.; Rickels, R.A.; Yilmaz, B.D.; et al. CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv. 2019, 5, eaax2887. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Putative involvement of the histone acetyltransferase Tip60 in ribosomal gene transcription. Nucleic Acids Res. 2004, 32, 1654–1665. [Google Scholar] [CrossRef]
- Yuasa, K.; Aoki, N.; Hijikata, T. JAZF1 promotes proliferation of C2C12 cells, but retards their myogenic differentiation through transcriptional repression of MEF2C and MRF4-Implications for the role of Jazf1 variants in oncogenesis and type 2 diabetes. Exp. Cell Res. 2015, 336, 287–297. [Google Scholar] [CrossRef]
- Meng, F.P.; Lin, Y.; Yang, M.; Li, M.Y.; Yang, G.Y.; Hao, P.; Li, L. JAZF1 Inhibits Adipose Tissue Macrophages and Adipose Tissue Inflammation in Diet-Induced Diabetic Mice. Biomed. Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.Z.; Wang, Y.D.; Qi, X.Y.; Xiao, X.H. JAZF1, a relevant metabolic regulator in type 2 diabetes. Diabetes-Metab. Res. 2019, 35, e3148. [Google Scholar] [CrossRef]
- Yuan, L.; Luo, X.; Zeng, M.; Zhang, Y.; Yang, M.; Zhang, L.; Liu, R.; Boden, G.; Liu, H.; Ma, Z.A.; et al. Transcription factor TIP27 regulates glucose homeostasis and insulin sensitivity in a PI3-kinase/Akt-dependent manner in mice. Int. J. Obes. 2015, 39, 949–958. [Google Scholar] [CrossRef]
- Sung, Y.; Park, S.; Park, S.J.; Jeong, J.; Choi, M.; Lee, J.; Kwon, W.; Jang, S.; Lee, M.H.; Kim, D.J.; et al. Jazf1 promotes prostate cancer progression by activating JNK/Slug. Oncotarget 2018, 9, 755–765. [Google Scholar] [CrossRef]
- Hattori, T.; Coustry, F.; Stephens, S.; Eberspaecher, H.; Takigawa, M.; Yasuda, H.; de Crombrugghe, B. Transcriptional regulation of chondrogenesis by coactivator Tip60 via chromatin association with Sox9 and Sox5. Nucleic Acids Res. 2008, 36, 3011–3024. [Google Scholar] [CrossRef]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative Interaction Proteomics and Genome-Wide Profiling of Epigenetic Histone Marks and Their Readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.M.P.; van Schaik, F.M.A.; Varier, R.A.; Baltissen, M.P.A.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T.M. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- R Core Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2020. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Backman, T.W.H.; Girke, T. systemPipeR: NGS workflow and report generation environment. BMC Bioinform. 2016, 17, 388. [Google Scholar] [CrossRef] [PubMed]
- Reimers, M.; Carey, V.J. Bioconductor: An open source framework for bioinformatics and computational biology. Method Enzymol. 2006, 411, 119–134. [Google Scholar]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F. Trim Galore: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files; Babraham Institute: Cambridge, UK, 2015; Volume 516, p. 517. [Google Scholar]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with 667 Enhanced Colouring and Labeling; R Package Version 1.6. 0. 668. 2020. Available online: https://github.com/kevinblighe (accessed on 10 December 2020).
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wiedemann, S.M.; Mildner, S.N.; Bonisch, C.; Israel, L.; Maiser, A.; Matheisl, S.; Straub, T.; Merkl, R.; Leonhardt, H.; Kremmer, E.; et al. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell Biol. 2010, 190, 777–791. [Google Scholar] [CrossRef]
- Hake, S.B.; Garcia, B.A.; Kauer, M.; Baker, S.P.; Shabanowitz, J.; Hunt, D.F.; Allis, C.D. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc. Natl. Acad. Sci. USA 2005, 102, 6344–6349. [Google Scholar] [CrossRef]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Procida, T.; Friedrich, T.; Jack, A.P.M.; Peritore, M.; Bönisch, C.; Eberl, H.C.; Daus, N.; Kletenkov, K.; Nist, A.; Stiewe, T.; et al. JAZF1, A Novel p400/TIP60/NuA4 Complex Member, Regulates H2A.Z Acetylation at Regulatory Regions. Int. J. Mol. Sci. 2021, 22, 678. https://doi.org/10.3390/ijms22020678
Procida T, Friedrich T, Jack APM, Peritore M, Bönisch C, Eberl HC, Daus N, Kletenkov K, Nist A, Stiewe T, et al. JAZF1, A Novel p400/TIP60/NuA4 Complex Member, Regulates H2A.Z Acetylation at Regulatory Regions. International Journal of Molecular Sciences. 2021; 22(2):678. https://doi.org/10.3390/ijms22020678
Chicago/Turabian StyleProcida, Tara, Tobias Friedrich, Antonia P. M. Jack, Martina Peritore, Clemens Bönisch, H. Christian Eberl, Nadine Daus, Konstantin Kletenkov, Andrea Nist, Thorsten Stiewe, and et al. 2021. "JAZF1, A Novel p400/TIP60/NuA4 Complex Member, Regulates H2A.Z Acetylation at Regulatory Regions" International Journal of Molecular Sciences 22, no. 2: 678. https://doi.org/10.3390/ijms22020678
APA StyleProcida, T., Friedrich, T., Jack, A. P. M., Peritore, M., Bönisch, C., Eberl, H. C., Daus, N., Kletenkov, K., Nist, A., Stiewe, T., Borggrefe, T., Mann, M., Bartkuhn, M., & Hake, S. B. (2021). JAZF1, A Novel p400/TIP60/NuA4 Complex Member, Regulates H2A.Z Acetylation at Regulatory Regions. International Journal of Molecular Sciences, 22(2), 678. https://doi.org/10.3390/ijms22020678