Myosins, an Underestimated Player in the Infectious Cycle of Pathogenic Bacteria


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
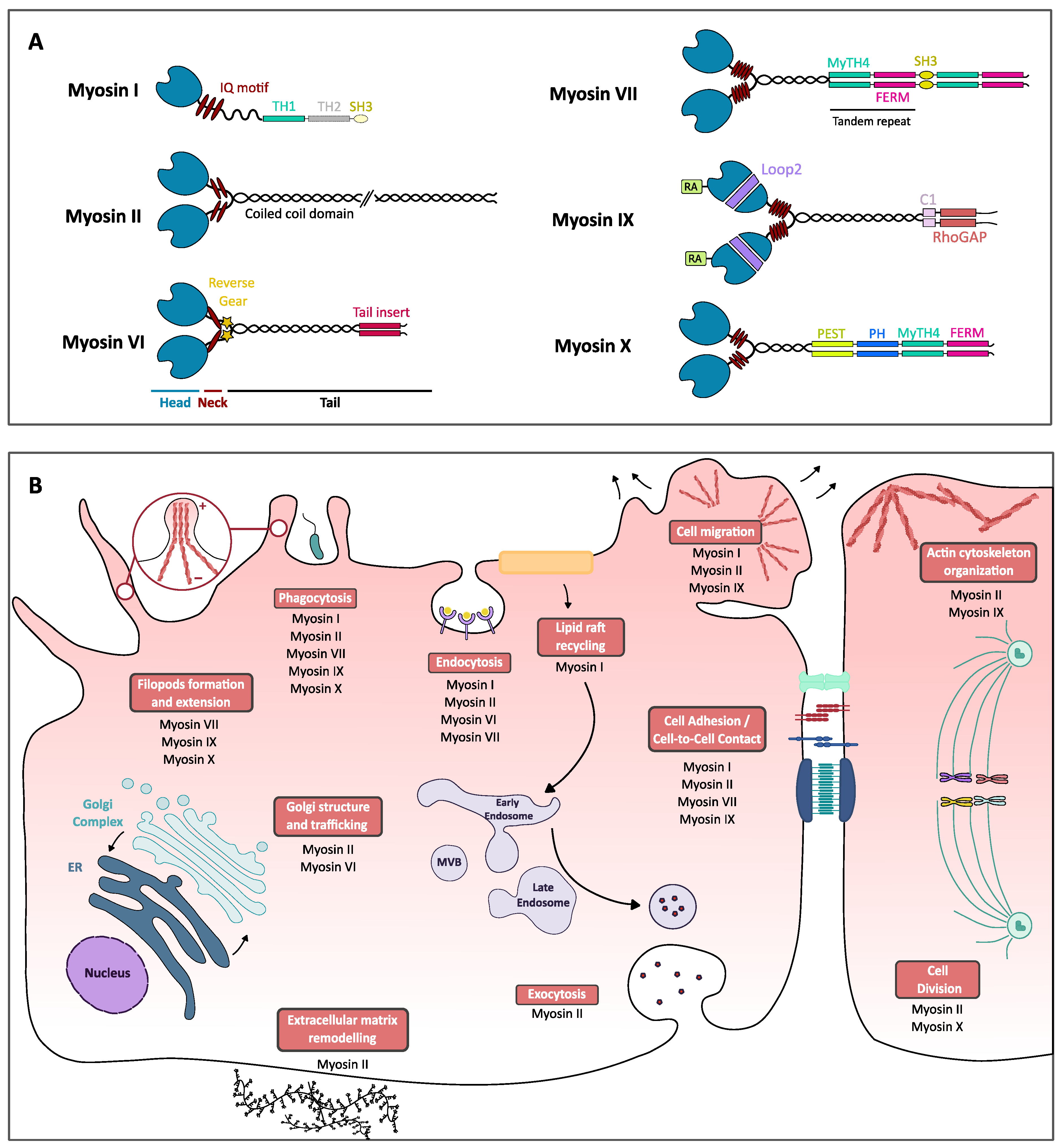
2. Structural and Functional Insights on Myosins I, II, VI, VII, IX and X
2.1. Myosins I
2.2. Myosins II
2.3. Myosins VI
2.4. Myosins VII
2.5. Myosins IX
2.6. Myosins X
3. Myosins and Their Role in Pathogenic Bacteria Infectious Cycle
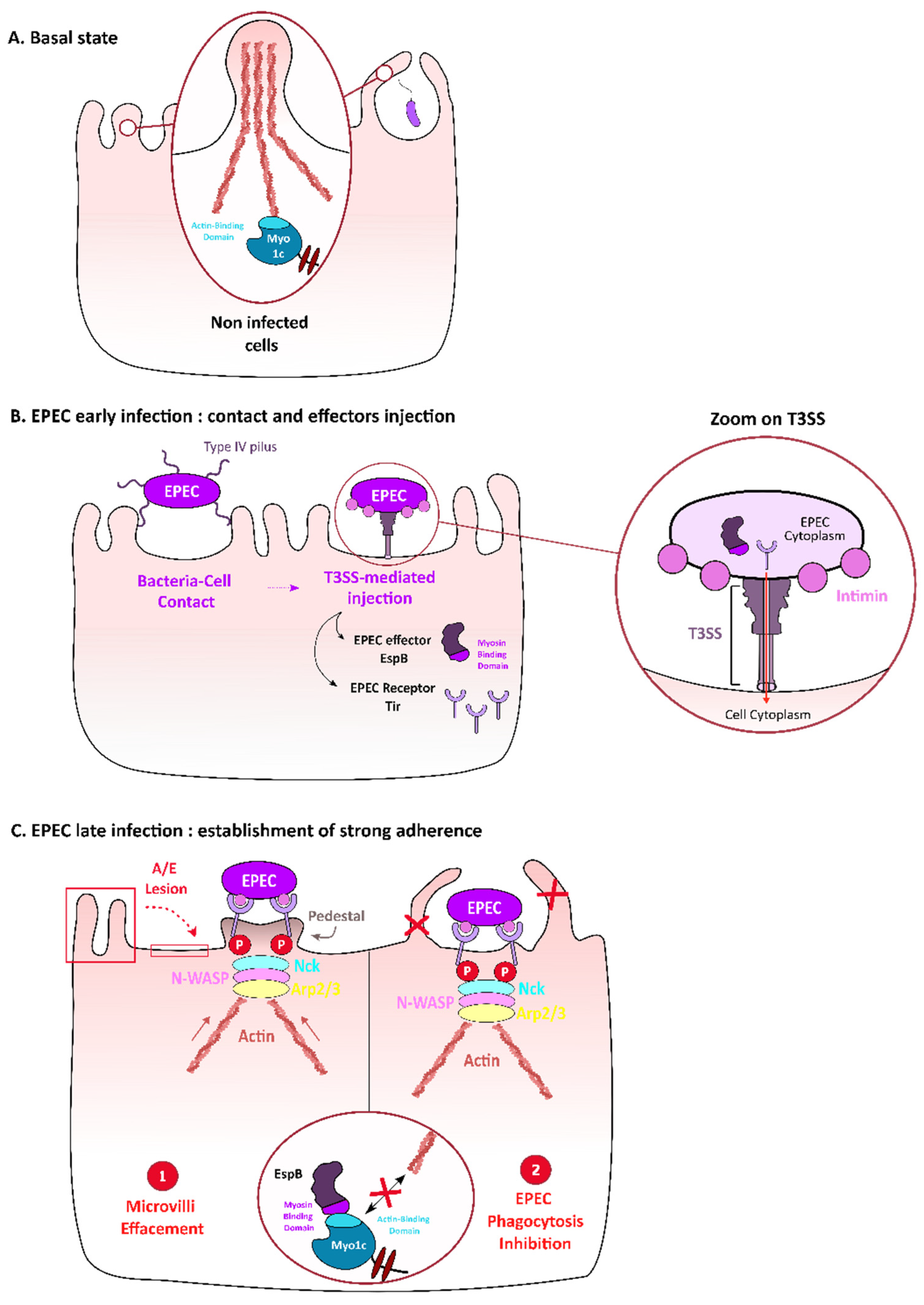
3.1. Myosin I—Targeting by Enteropathogenic E. coli (EPEC) to Contribute to Adhesion and to Evade Macrophages Phagocytosis
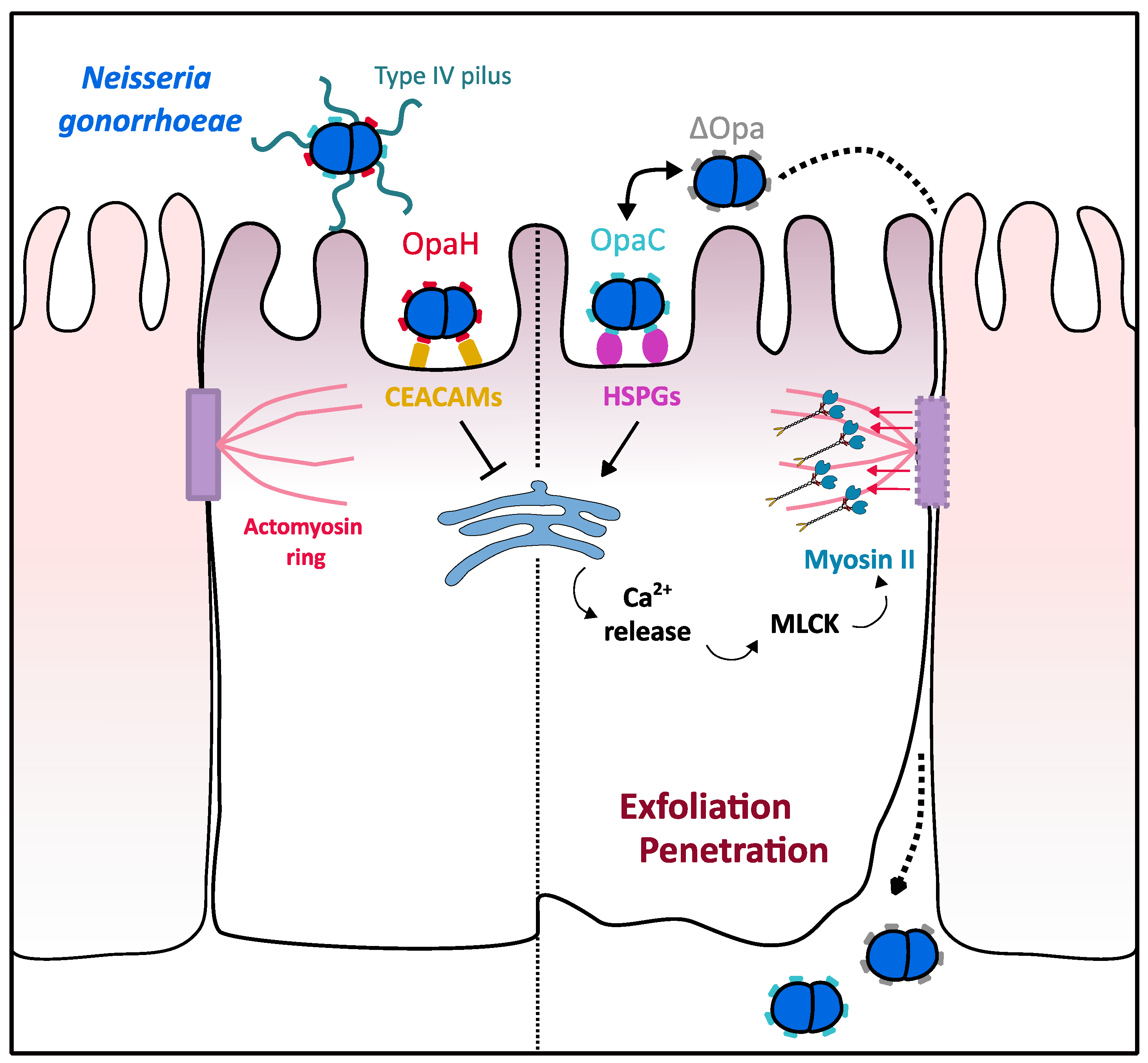
3.2. Myosin II Contribution to N. gonorrhoeae Crossing over the Epithelial Barrier
3.3. Myosins I, II and VI Contribution to S. typhimurium Invasion and Replication in Epithelial Cells
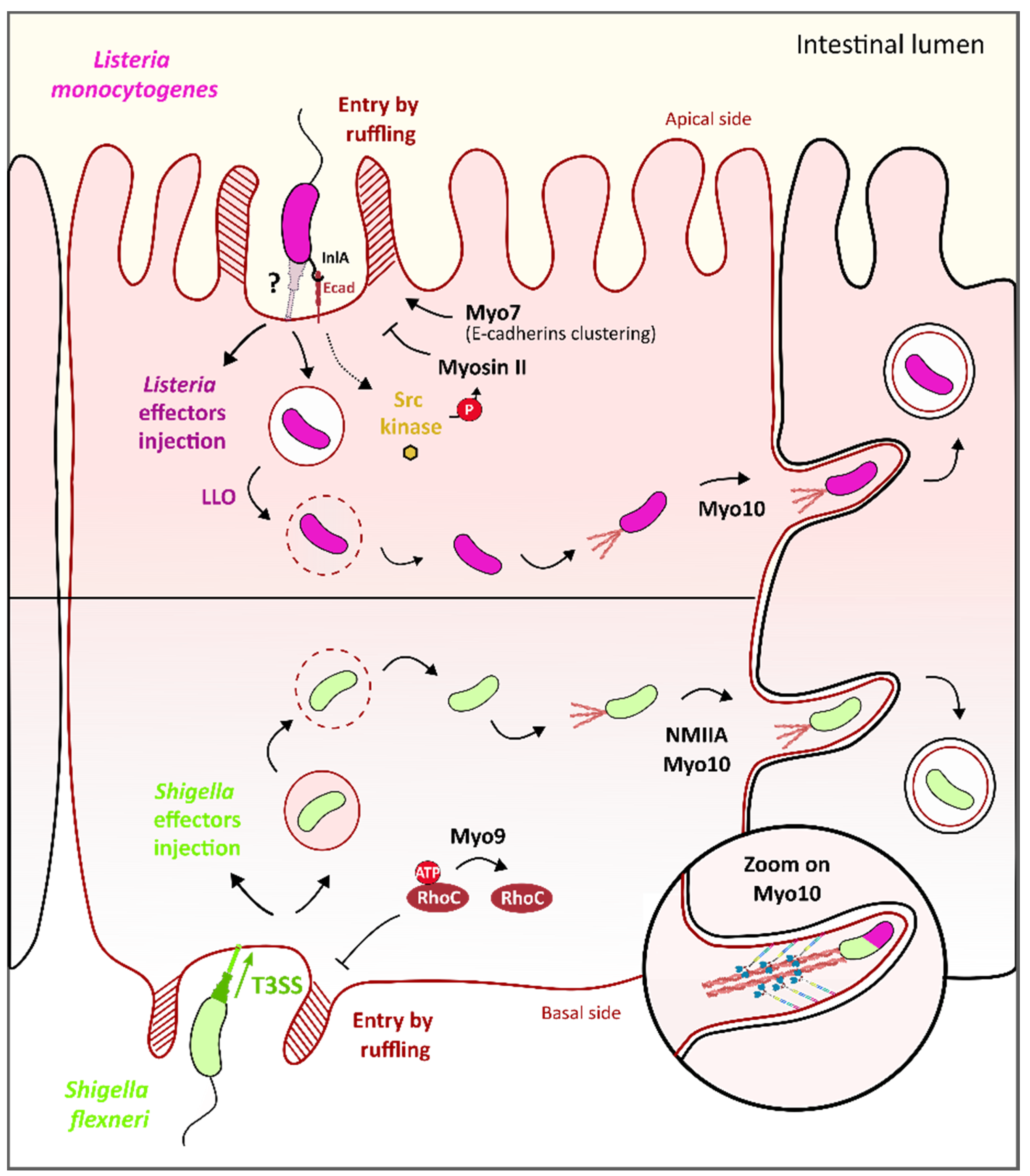
3.4. Myosin IX and Myosins II/X Contribution to S. flexneri Epithelial Cell Invasion and Promotion of Cell-to-Cell Spreading
3.5. Myosin VII and Myosins IIA/X Contribution to L. monocytogenes Epithelial Cell Invasion and Promotion of Cell-to-Cell Spreading
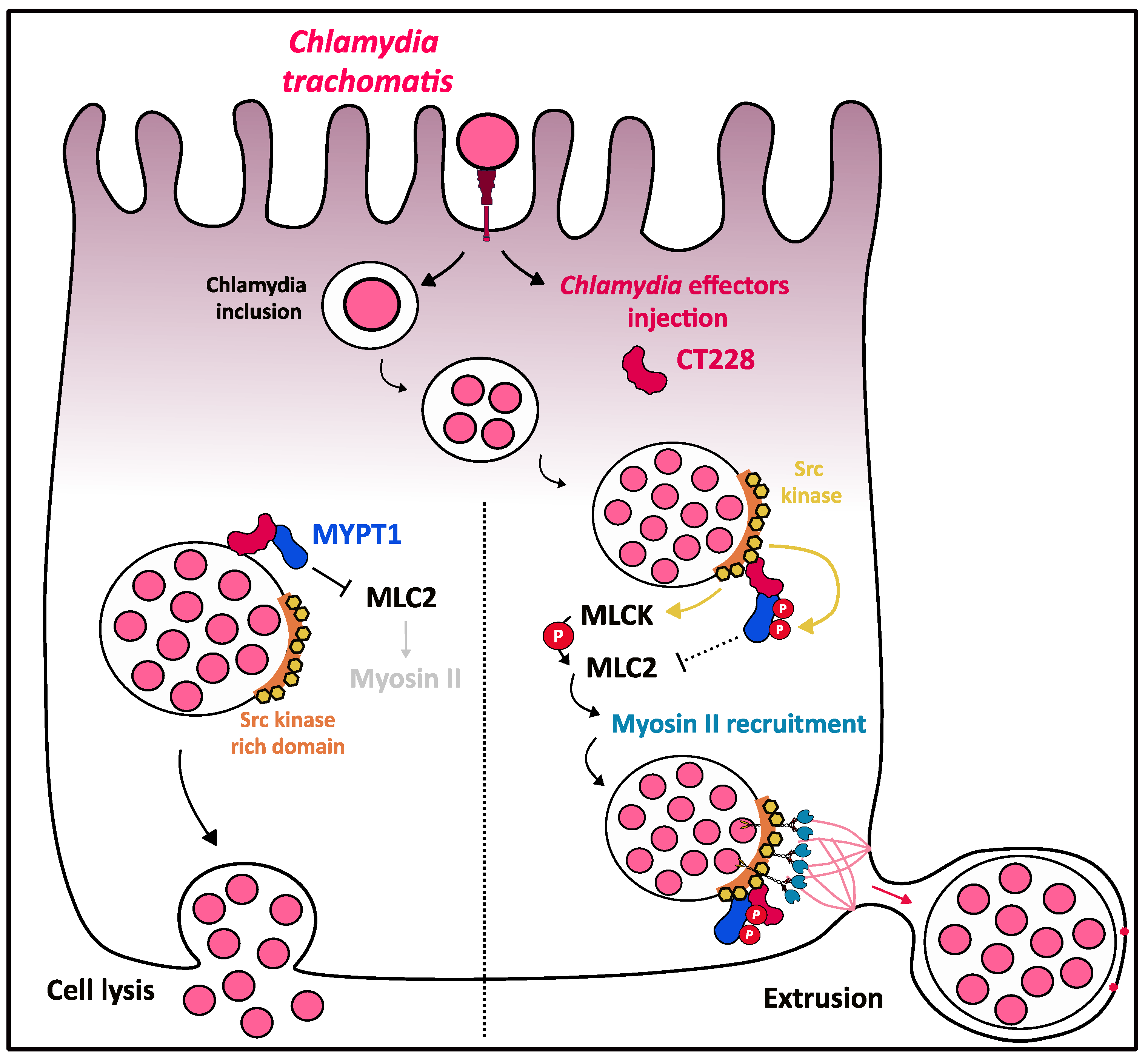
3.6. Myosin II Contribution to C. trachomatis Exit out of the Host Cell
4. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Lymn, R.W.; Taylor, E.W. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 1971, 10, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Rayment, I.; Holden, H.M.; Whittaker, M.; Yohn, C.B.; Lorenz, M.; Holmes, K.C.; Milligan, R.A. Structure of the actin-myosin complex and its implications for muscle contraction. Science 1993, 261, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Sellers, J.R.; Pato, M.D.; Adelstein, R.S. Reversible phosphorylation of smooth muscle myosin, heavy meromyosin, and platelet myosin. J. Biol. Chem. 1981, 256, 13137–13142. [Google Scholar] [CrossRef]
- Uyeda, T.Q.; Abramson, P.D.; Spudich, J.A. The neck region of the myosin motor domain acts as a lever arm to generate movement. Proc. Natl. Acad. Sci. USA 1996, 93, 4459–4464. [Google Scholar] [CrossRef]
- Eisenberg, E.; Hill, T.L. Muscle contraction and free energy transduction in biological systems. Science 1985, 227, 999–1006. [Google Scholar] [CrossRef]
- Scholey, J.M.; Taylor, K.A.; Kendrick-Jones, J. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature 1980, 287, 233–235. [Google Scholar] [CrossRef]
- Adelstein, R.S.; Conti, M.A.; Pato, M.D. Regulation of myosin light chain kinase by reversible phosphorylation and calcium-calmodulin. Ann. N. Y. Acad. Sci. 1980, 356, 142–150. [Google Scholar] [CrossRef]
- Oliver, T.N.; Berg, J.S.; Cheney, R.E. Tails of unconventional myosins. CMLS Cell. Mol. Life Sci. 1999, 56, 243–257. [Google Scholar] [CrossRef]
- Thompson, R.F.; Langford, G.M. Myosin superfamily evolutionary history. Anat. Rec. 2002, 268, 276–289. [Google Scholar] [CrossRef]
- Mermall, V.; Post, P.L.; Mooseker, M.S. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science 1998, 279, 527–533. [Google Scholar] [CrossRef]
- Krendel, M.; Mooseker, M.S. Myosins: Tails (and heads) of functional diversity. Physiology 2005, 20, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Doberstein, S.K.; Pollard, T.D. Localization and specificity of the phospholipid and actin binding sites on the tail of Acanthamoeba myosin IC. J. Cell Biol. 1992, 117, 1241–1249. [Google Scholar] [CrossRef]
- Adams, R.J.; Pollard, T.D. Binding of myosin I to membrane lipids. Nature 1989, 340, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Mamane, A.; Lee-Tin-Wah, J.; Di Cicco, A.; Prévost, C.; Lévy, D.; Joanny, J.-F.; Coudrier, E.; Bassereau, P. Catch-bond behaviour facilitates membrane tubulation by non-processive myosin 1b. Nat. Commun. 2014, 5, 3624. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Eswarappa, S.M.; Hitomi, M.; Fox, P.L. Myo1c facilitates G-actin transport to the leading edge of migrating endothelial cells. J. Cell Biol. 2012, 198, 47–55. [Google Scholar] [CrossRef]
- Cheng, J.; Grassart, A.; Drubin, D.G. Myosin 1E coordinates actin assembly and cargo trafficking during clathrin-mediated endocytosis. Mol. Biol. Cell 2012, 23, 2891–2904. [Google Scholar] [CrossRef]
- Dart, A.E.; Tollis, S.; Bright, M.D.; Frankel, G.; Endres, R.G. The motor protein myosin 1G functions in FcγR-mediated phagocytosis. J. Cell Sci. 2012, 125, 6020–6029. [Google Scholar] [CrossRef]
- Brandstaetter, H.; Kendrick-Jones, J.; Buss, F. Myo1c regulates lipid raft recycling to control cell spreading, migration and Salmonella invasion. J. Cell Sci. 2012, 125, 1991–2003. [Google Scholar] [CrossRef]
- Niederman, R.; Pollard, T.D. Human platelet myosin. II. In vitro assembly and structure of myosin filaments. J. Cell Biol. 1975, 67, 72–92. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Zareno, J.; Whitmore, L.; Choi, C.K.; Horwitz, A.F. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 2007, 176, 573–580. [Google Scholar] [CrossRef]
- Kolega, J. Cytoplasmic dynamics of myosin IIA and IIB: Spatial “sorting” of isoforms in locomoting cells. J. Cell Sci. 1998, 111, 2085–2095. [Google Scholar] [PubMed]
- Chandrasekar, I.; Goeckeler, Z.M.; Turney, S.G.; Wang, P.; Wysolmerski, R.B.; Adelstein, R.S.; Bridgman, P.C. Nonmuscle myosin II is a critical regulator of clathrin mediated endocytosis. Traffic 2014, 15, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Müsch, A.; Cohen, D.; Rodriguez-Boulan, E. Myosin II is involved in the production of constitutive transport vesicles from the TGN. J. Cell Biol. 1997, 138, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Olazabal, I.M.; Caron, E.; May, R.C.; Schilling, K.; Knecht, D.A.; Machesky, L.M. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcgammaR, phagocytosis. Curr. Biol. 2002, 12, 1413–1418. [Google Scholar] [CrossRef]
- Patel, P.C.; Harrison, R.E. Membrane Ruffles Capture C3bi-opsonized Particles in Activated Macrophages. MBoC 2008, 19, 4628–4639. [Google Scholar] [CrossRef]
- Wells, A.L.; Lin, A.W.; Chen, L.-Q.; Safer, D.; Cain, S.M.; Hasson, T.; Carragher, B.O.; Milligan, R.A.; Sweeney, H.L. Myosin VI is an actin-based motor that moves backwards. Nature 1999, 401, 505–508. [Google Scholar] [CrossRef]
- Bahloul, A.; Chevreux, G.; Wells, A.L.; Martin, D.; Nolt, J.; Yang, Z.; Chen, L.-Q.; Potier, N.; Van Dorsselaer, A.; Rosenfeld, S.; et al. The unique insert in myosin VI is a structural calcium-calmodulin binding site. Proc. Natl. Acad. Sci. USA 2004, 101, 4787–4792. [Google Scholar] [CrossRef]
- Park, H.; Li, A.; Chen, L.-Q.; Houdusse, A.; Selvin, P.R.; Sweeney, H.L. The unique insert at the end of the myosin VI motor is the sole determinant of directionality. Proc. Natl. Acad. Sci. USA 2007, 104, 778–783. [Google Scholar] [CrossRef]
- Buss, F.; Arden, S.D.; Lindsay, M.; Luzio, J.P.; Kendrick-Jones, J. Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J. 2001, 20, 3676–3684. [Google Scholar] [CrossRef]
- Mooseker, M.S.; Pollard, T.D.; Wharton, K.A. Nucleated polymerization of actin from the membrane-associated ends of microvillar filaments in the intestinal brush border. J. Cell Biol. 1982, 95, 223–233. [Google Scholar] [CrossRef]
- Biancospino, M.; Buel, G.R.; Niño, C.A.; Maspero, E.; Scotto di Perrotolo, R.; Raimondi, A.; Redlingshöfer, L.; Weber, J.; Brodsky, F.M.; Walters, K.J.; et al. Clathrin light chain A drives selective myosin VI recruitment to clathrin-coated pits under membrane tension. Nat. Commun. 2019, 10, 4974. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, L.; Lee, T.; Hasson, T. Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol. Biol. Cell 2003, 14, 2728–2743. [Google Scholar] [CrossRef] [PubMed]
- Warner, C.L.; Stewart, A.; Luzio, J.P.; Steel, K.P.; Libby, R.T.; Kendrick-Jones, J.; Buss, F. Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell’s waltzer mice. EMBO J. 2003, 22, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Sahlender, D.A.; Roberts, R.C.; Arden, S.D.; Spudich, G.; Taylor, M.J.; Luzio, J.P.; Kendrick-Jones, J.; Buss, F. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J. Cell Biol. 2005, 169, 285–295. [Google Scholar] [CrossRef]
- Inoue, A.; Ikebe, M. Characterization of the motor activity of mammalian myosin VIIA. J. Biol. Chem. 2003, 278, 5478–5487. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Hasson, T.; Kelley, P.M.; Schwender, B.J.; Schwartz, M.F.; Ramakrishnan, M.; Kimberling, W.J.; Mooseker, M.S.; Corey, D.P. Molecular cloning and domain structure of human Myosin-VIIa, the gene product defective in usher syndrome 1B. Genomics 1996, 36, 440–448. [Google Scholar] [CrossRef]
- Tuxworth, R.I.; Weber, I.; Wessels, D.; Addicks, G.C.; Soll, D.R.; Gerisch, G.; Titus, M.A. A role for myosin VII in dynamic cell adhesion. Curr. Biol. 2001, 11, 318–329. [Google Scholar] [CrossRef]
- Tuxworth, R.I.; Stephens, S.; Ryan, Z.C.; Titus, M.A. Identification of a myosin VII-Talin complex. J. Biol. Chem. 2005, 280, 26557–26564. [Google Scholar] [CrossRef]
- Bosmani, C.; Leuba, F.; Hanna, N.; Bach, F.; Burdet, F.; Pagni, M.; Hagedorn, M.; Soldati, T. Vacuolins and myosin VII are required for phagocytic uptake and phagosomal membrane recycling in Dictyostelium discoideum. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Chieregatti, E.; Gärtner, A.; Stöffler, H.E.; Bähler, M. Myr 7 is a novel myosin IX-RhoGAP expressed in rat brain. J. Cell Sci. 1998, 111, 3597–3608. [Google Scholar]
- Liao, W.; Elfrink, K.; Bähler, M. Head of myosin IX binds calmodulin and moves processively toward the plus-end of actin filaments. J. Biol. Chem. 2010, 285, 24933–24942. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, J.; Scheel, A.A.; Diekmann, D.; Hall, A.; Ruppert, C.; Bähler, M. A novel type of myosin implicated in signalling by rho family GTPases. EMBO J. 1995, 14, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Post, P.L.; Tyska, M.J.; O’Connell, C.B.; Johung, K.; Hayward, A.; Mooseker, M.S. Myosin-IXb is a single-headed and processive motor. J. Biol. Chem. 2002, 277, 11679–11683. [Google Scholar] [CrossRef] [PubMed]
- Saczko-Brack, D.; Warchol, E.; Rogez, B.; Kröss, M.; Heissler, S.M.; Sellers, J.R.; Batters, C.; Veigel, C. Self-organization of actin networks by a monomeric myosin. Proc. Natl. Acad. Sci. USA 2016, 113, E8387–E8395. [Google Scholar] [CrossRef] [PubMed]
- Elfrink, K.; Liao, W.; Pieper, U.; Oeding, S.J.; Bähler, M. The Loop2 insertion of Type IX myosin acts as an electrostatic actin tether that permits processive movement. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Nalavadi, V.; Nyitrai, M.; Bertolini, C.; Adamek, N.; Geeves, M.A.; Bähler, M. Kinetic mechanism of myosin IXB and the contributions of two class IX-specific regions. J. Biol. Chem. 2005, 280, 38957–38968. [Google Scholar] [CrossRef]
- Kambara, T.; Ikebe, M. A unique ATP hydrolysis mechanism of single-headed processive myosin, myosin IX. J. Biol. Chem. 2006, 281, 4949–4957. [Google Scholar] [CrossRef]
- Van den Boom, F.; Düssmann, H.; Uhlenbrock, K.; Abouhamed, M.; Bähler, M. The myosin IXb motor activity targets the myosin IXb RhoGAP domain as cargo to sites of actin polymerization. Mol. Biol. Cell 2007, 18, 1507–1518. [Google Scholar] [CrossRef]
- Omelchenko, T.; Hall, A. Myosin-IXA regulates collective epithelial cell migration by targeting RhoGAP activity to cell-cell junctions. Curr. Biol. 2012, 22, 278–288. [Google Scholar] [CrossRef]
- Knight, P.J.; Thirumurugan, K.; Xu, Y.; Wang, F.; Kalverda, A.P.; Stafford, W.F.; Sellers, J.R.; Peckham, M. The predicted coiled-coil domain of myosin 10 forms a novel elongated domain that lengthens the head. J. Biol. Chem. 2005, 280, 34702–34708. [Google Scholar] [CrossRef]
- Berg, J.S.; Derfler, B.H.; Pennisi, C.M.; Corey, D.P.; Cheney, R.E. Myosin-X, a novel myosin with pleckstrin homology domains, associates with regions of dynamic actin. J. Cell Sci. 2000, 113, 3439–3451. [Google Scholar] [PubMed]
- Berg, J.S.; Cheney, R.E. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 2002, 4, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Bohil, A.B.; Robertson, B.W.; Cheney, R.E. Myosin-X is a molecular motor that functions in filopodia formation. Proc. Natl. Acad. Sci. USA 2006, 103, 12411–12416. [Google Scholar] [CrossRef] [PubMed]
- Tokuo, H.; Mabuchi, K.; Ikebe, M. The motor activity of myosin-X promotes actin fiber convergence at the cell periphery to initiate filopodia formation. J. Cell Biol. 2007, 179, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berg, J.S.; Li, Z.; Wang, Y.; Lång, P.; Sousa, A.D.; Bhaskar, A.; Cheney, R.E.; Strömblad, S. Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat. Cell Biol. 2004, 6, 523–531. [Google Scholar] [CrossRef]
- Toyoshima, F.; Nishida, E. Integrin-mediated adhesion orients the spindle parallel to the substratum in an EB1- and myosin X-dependent manner. EMBO J. 2007, 26, 1487–1498. [Google Scholar] [CrossRef]
- Cox, D.; Berg, J.S.; Cammer, M.; Chinegwundoh, J.O.; Dale, B.M.; Cheney, R.E.; Greenberg, S. Myosin X is a downstream effector of PI(3)K during phagocytosis. Nat. Cell Biol. 2002, 4, 469–477. [Google Scholar] [CrossRef]
- Moon, H.W.; Whipp, S.C.; Argenzio, R.A.; Levine, M.M.; Giannella, R.A. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 1983, 41, 1340–1351. [Google Scholar] [CrossRef]
- McDaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 1995, 92, 1664–1668. [Google Scholar] [CrossRef]
- Rosenshine, I.; Ruschkowski, S.; Stein, M.; Reinscheid, D.J.; Mills, S.D.; Finlay, B.B. A pathogenic bacterium triggers epithelial signals to form a functional bacterial receptor that mediates actin pseudopod formation. EMBO J. 1996, 15, 2613–2624. [Google Scholar] [CrossRef]
- Hartland, E.L.; Daniell, S.J.; Delahay, R.M.; Neves, B.C.; Wallis, T.; Shaw, R.K.; Hale, C.; Knutton, S.; Frankel, G. The type III protein translocation system of enteropathogenic Escherichia coli involves EspA-EspB protein interactions. Mol. Microbiol. 2000, 35, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Tacket, C.O.; Sztein, M.B.; Losonsky, G.; Abe, A.; Finlay, B.B.; McNamara, B.P.; Fantry, G.T.; James, S.P.; Nataro, J.P.; Levine, M.M.; et al. Role of EspB in experimental human enteropathogenic Escherichia coli infection. Infect. Immun. 2000, 68, 3689–3695. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Akeda, Y.; Kono, G.; Takahashi, A.; Imura, K.; Iida, T.; Honda, T. The EspB protein of enterohaemorrhagic Escherichia coli interacts directly with α-catenin. Cell. Microbiol. 2002, 4, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Iizumi, Y.; Sagara, H.; Kabe, Y.; Azuma, M.; Kume, K.; Ogawa, M.; Nagai, T.; Gillespie, P.G.; Sasakawa, C.; Handa, H. The enteropathogenic E. coli effector EspB facilitates microvillus effacing and antiphagocytosis by inhibiting myosin function. Cell Host Microbe 2007, 2, 383–392. [Google Scholar] [CrossRef]
- Swanson, J.A.; Johnson, M.T.; Beningo, K.; Post, P.; Mooseker, M.; Araki, N. A contractile activity that closes phagosomes in macrophages. J. Cell Sci. 1999, 112, 307–316. [Google Scholar] [PubMed]
- Källström, H.; Liszewski, M.K.; Atkinson, J.P.; Jonsson, A.B. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol. Microbiol. 1997, 25, 639–647. [Google Scholar] [CrossRef]
- Wang, L.-C.; Yu, Q.; Edwards, V.; Lin, B.; Qiu, J.; Turner, J.R.; Stein, D.C.; Song, W. Neisseria gonorrhoeae infects the human endocervix by activating non-muscle myosin II-mediated epithelial exfoliation. PLoS Pathog. 2017, 13, e1006269. [Google Scholar] [CrossRef]
- Utech, M.; Ivanov, A.I.; Samarin, S.N.; Bruewer, M.; Turner, J.R.; Mrsny, R.J.; Parkos, C.A.; Nusrat, A. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: Myosin II-dependent vacuolarization of the apical plasma membrane. MBoC 2005, 16, 5040–5052. [Google Scholar] [CrossRef]
- Makino, S.; van Putten, J.P.; Meyer, T.F. Phase variation of the opacity outer membrane protein controls invasion by Neisseria gonorrhoeae into human epithelial cells. EMBO J. 1991, 10, 1307–1315. [Google Scholar] [CrossRef]
- Hardt, W.-D.; Chen, L.-M.; Schuebel, K.E.; Bustelo, X.R.; Galán, J.E. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 1998, 93, 815–826. [Google Scholar] [CrossRef]
- Stender, S.; Friebel, A.; Linder, S.; Rohde, M.; Mirold, S.; Hardt, W.-D. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol. Microbiol. 2000, 36, 1206–1221. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, L.-M.; Hernandez, L.; Shears, S.B.; Galán, J.E. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol. Microbiol. 2001, 39, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Hänisch, J.; Ehinger, J.; Ladwein, M.; Rohde, M.; Derivery, E.; Bosse, T.; Steffen, A.; Bumann, D.; Misselwitz, B.; Hardt, W.-D.; et al. Molecular dissection of Salmonella-induced membrane ruffling versus invasion. Cell. Microbiol. 2010, 12, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Hänisch, J.; Kölm, R.; Wozniczka, M.; Bumann, D.; Rottner, K.; Stradal, T.E.B. Activation of a RhoA/Myosin II-Dependent but Arp2/3 Complex-Independent Pathway Facilitates Salmonella Invasion. Cell Host Microbe 2011, 9, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Norris, F.A.; Wilson, M.P.; Wallis, T.S.; Galyov, E.E.; Majerus, P.W. SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc. Natl. Acad. Sci. USA 1998, 95, 14057–14059. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.B.E.; Humphreys, D.; Singh, V.; Davidson, A.C.; Arden, S.D.; Buss, F.; Koronakis, V. MYO6 is targeted by Salmonella virulence effectors to trigger PI3-kinase signaling and pathogen invasion into host cells. Proc. Natl. Acad. Sci. USA 2017, 114, 3915–3920. [Google Scholar] [CrossRef]
- Nichols, C.D.; Casanova, J.E. Salmonella-directed recruitment of new membrane to invasion foci via the host exocyst complex. Curr. Biol. 2010, 20, 1316–1320. [Google Scholar] [CrossRef]
- Deiwick, J.; Salcedo, S.P.; Boucrot, E.; Gilliland, S.M.; Henry, T.; Petermann, N.; Waterman, S.R.; Gorvel, J.-P.; Holden, D.W.; Méresse, S. The translocated salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect. Immun. 2006, 74, 6965–6972. [Google Scholar] [CrossRef]
- Abrahams, G.L.; Müller, P.; Hensel, M. Functional dissection of SseF, a type III effector protein involved in positioning the salmonella-containing vacuole. Traffic 2006, 7, 950–965. [Google Scholar] [CrossRef]
- Henry, T.; Couillault, C.; Rockenfeller, P.; Boucrot, E.; Dumont, A.; Schroeder, N.; Hermant, A.; Knodler, L.A.; Lecine, P.; Steele-Mortimer, O.; et al. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. USA 2006, 103, 13497–13502. [Google Scholar] [CrossRef]
- Beuzón, C.R.; Méresse, S.; Unsworth, K.E.; Ruíz-Albert, J.; Garvis, S.; Waterman, S.R.; Ryder, T.A.; Boucrot, E.; Holden, D.W. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 2000, 19, 3235–3249. [Google Scholar] [CrossRef] [PubMed]
- Steele-Mortimer, O. The Salmonella-containing vacuole—Moving with the times. Curr. Opin. Microbiol. 2008, 11, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Wasylnka, J.A.; Bakowski, M.A.; Szeto, J.; Ohlson, M.B.; Trimble, W.S.; Miller, S.I.; Brumell, J.H. Role for myosin II in regulating positioning of salmonella-containing vacuoles and intracellular replication. Infect. Immun. 2008, 76, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.; Paterson, H.F.; Marshall, C.J. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat. Cell Biol. 2005, 7, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; van Nhieu, G.T.; Égile, C. Rupture of the intestinal epithelial barrier and mucosal invasion by shigella flexneri. Clin. Infect. Dis. 1999, 28, 466–475. [Google Scholar] [CrossRef][Green Version]
- Tran Van Nhieu, G.; Caron, E.; Hall, A.; Sansonetti, P.J. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 1999, 18, 3249–3262. [Google Scholar] [CrossRef]
- Handa, Y.; Suzuki, M.; Ohya, K.; Iwai, H.; Ishijima, N.; Koleske, A.J.; Fukui, Y.; Sasakawa, C. Shigella IpgB1 promotes bacterial entry through the ELMO–Dock180 machinery. Nat. Cell Biol. 2007, 9, 121–128. [Google Scholar] [CrossRef]
- Mounier, J.; Popoff, M.R.; Enninga, J.; Frame, M.C.; Sansonetti, P.J.; van Nhieu, G.T. The IpaC carboxyterminal effector domain mediates src-dependent actin polymerization during shigella invasion of epithelial cells. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef]
- Sansonetti, P.J.; Ryter, A.; Clerc, P.; Maurelli, A.T.; Mounier, J. Multiplication of Shigella flexneri within HeLa cells: Lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect. Immun. 1986, 51, 461–469. [Google Scholar] [CrossRef]
- Schuch, R.; Sandlin, R.C.; Maurelli, A.T. A system for identifying post-invasion functions of invasion genes: Requirements for the Mxi-Spa type III secretion pathway of Shigella flexneri in intercellular dissemination. Mol. Microbiol. 1999, 34, 675–689. [Google Scholar] [CrossRef]
- Graf, B.; Bähler, M.; Hilpelä, P.; Böwe, C.; Adam, T. Functional role for the class IX myosin myr5 in epithelial cell infection by Shigella flexneri. Cell. Microbiol. 2000, 2, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Mounier, J.; Laurent, V.; Hall, A.; Fort, P.; Carlier, M.F.; Sansonetti, P.J.; Egile, C. Rho family GTPases control entry of Shigella flexneri into epithelial cells but not intracellular motility. J. Cell Sci. 1999, 112, 2069–2080. [Google Scholar] [PubMed]
- Rathman, M.; de Lanerolle, P.; Ohayon, H.; Gounon, P.; Sansonetti, P. Myosin light chain kinase plays an essential role in S. flexneri dissemination. J. Cell Sci. 2000, 113, 3375–3386. [Google Scholar]
- Lum, M.; Morona, R. Myosin IIA is essential for Shigella flexneri cell-to-cell spread. Pathog. Dis. 2014, 72, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Bishai, E.A.; Sidhu, G.S.; Li, W.; Dhillon, J.; Bohil, A.B.; Cheney, R.E.; Hartwig, J.H.; Southwick, F.S. Myosin-X facilitates Shigella-induced membrane protrusions and cell-to-cell spread. Cell. Microbiol. 2013, 15, 353–367. [Google Scholar] [CrossRef]
- Cossart, P.; Pizarro-Cerdá, J.; Lecuit, M. Invasion of mammalian cells by Listeria monocytogenes: Functional mimicry to subvert cellular functions. Trends Cell Biol. 2003, 13, 23–31. [Google Scholar] [CrossRef]
- Sousa, S.; Cabanes, D.; Bougnères, L.; Lecuit, M.; Sansonetti, P.; Tran-Van-Nhieu, G.; Cossart, P. Src, cortactin and Arp2/3 complex are required for E-cadherin-mediated internalization of Listeria into cells. Cell. Microbiol. 2007, 9, 2629–2643. [Google Scholar] [CrossRef]
- Bonazzi, M.; Veiga, E.; Pizarro-Cerdá, J.; Cossart, P. Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell. Microbiol. 2008, 10, 2208–2222. [Google Scholar] [CrossRef]
- Küssel-Andermann, P.; El-Amraoui, A.; Safieddine, S.; Nouaille, S.; Perfettini, I.; Lecuit, M.; Cossart, P.; Wolfrum, U.; Petit, C. Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin–catenins complex. EMBO J. 2000, 19, 6020–6029. [Google Scholar] [CrossRef]
- Almeida, M.T.; Mesquita, F.S.; Cruz, R.; Osório, H.; Custódio, R.; Brito, C.; Vingadassalom, D.; Martins, M.; Leong, J.M.; Holden, D.W.; et al. Src-dependent tyrosine phosphorylation of non-muscle myosin heavy chain-iia restricts listeria monocytogenes cellular infection. J. Biol. Chem. 2015, 290, 8383–8395. [Google Scholar] [CrossRef]
- Jiwani, S.; Ohr, R.J.; Fischer, E.R.; Hackstadt, T.; Alvarado, S.; Romero, A.; Jewett, T.J. Chlamydia trachomatis Tarp cooperates with the Arp2/3 complex to increase the rate of actin polymerization. Biochem. Biophys. Res. Commun. 2012, 420, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Hackstadt, T.; Fischer, E.R.; Scidmore, M.A.; Rockey, D.D.; Heinzen, R.A. Origins and functions of the chlamydial inclusion. Trends Microbiol. 1997, 5, 288–293. [Google Scholar] [CrossRef]
- Hybiske, K.; Stephens, R.S. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 2007, 104, 11430–11435. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.; Kirker, K.; Zuck, M.; James, G.; Hybiske, K. Actin recruitment to the chlamydia inclusion is spatiotemporally regulated by a mechanism that requires host and bacterial factors. PLoS ONE 2012, 7, e46949. [Google Scholar] [CrossRef] [PubMed]
- Lutter, E.I.; Barger, A.C.; Nair, V.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep. 2013, 3, 1921–1931. [Google Scholar] [CrossRef]
- Feng, J.; Ito, M.; Ichikawa, K.; Isaka, N.; Nishikawa, M.; Hartshorne, D.J.; Nakano, T. Inhibitory phosphorylation site for rho-associated kinase on smooth muscle myosin phosphatase. J. Biol. Chem. 1999, 274, 37385–37390. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Blikslager, A.T. The regulation of intestinal mucosal barrier by myosin light chain kinase/rho kinases. Int. J. Mol. Sci. 2020, 21, 3550. [Google Scholar] [CrossRef]
- Stradal, T.E.B.; Schelhaas, M. Actin dynamics in host–pathogen interaction. FEBS Lett. 2018, 592, 3658–3669. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, H.; Park, Y.H. Molecular mechanisms of host cytoskeletal rearrangements by shigella invasins. Int. J. Mol. Sci. 2014, 15, 18253–18266. [Google Scholar] [CrossRef]
- Truong, D.; Copeland, J.W.; Brumell, J.H. Bacterial subversion of host cytoskeletal machinery: Hijacking formins and the Arp2/3 complex. BioEssays 2014, 36, 687–696. [Google Scholar] [CrossRef]
- Pizarro-Cerdá, J.; Cossart, P. Listeria monocytogenes: Cell biology of invasion and intracellular growth. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.V.; Cruz, L.; Araújo, N.S.; Angeloni, M.B.; Fonseca, B.B.; Gomes, A.O.; Carvalho, F.R.; Gonçalves, A.L.R.; Barbosa, B.F. A glance at Listeria and Salmonella cell invasion: Different strategies to promote host actin polymerization. Int. J. Med. Microbiol. 2012, 302, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Weddle, E.; Agaisse, H. Principles of intracellular bacterial pathogen spread from cell to cell. PLoS Pathog. 2018, 14, e1007380. [Google Scholar] [CrossRef] [PubMed]
- Kocks, C.; Gouin, E.; Tabouret, M.; Berche, P.; Ohayon, H.; Cossart, P.L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 1992, 68, 521–531. [Google Scholar] [CrossRef]
- Kocks, C.; Marchand, J.-B.; Gouin, E.; D’Hauteville, H.; Sansonetti, P.J.; Carlier, M.-F.; Cossart, P. The unrelated surface proteins ActA of Listeria monocytogenes and lcsA of Shigella flexneri are sufficient to confer actin-based motility on Listeria innocua and Escherichia coli respectively. Mol. Microbiol. 1995, 18, 413–423. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pillon, M.; Doublet, P. Myosins, an Underestimated Player in the Infectious Cycle of Pathogenic Bacteria. Int. J. Mol. Sci. 2021, 22, 615. https://doi.org/10.3390/ijms22020615
Pillon M, Doublet P. Myosins, an Underestimated Player in the Infectious Cycle of Pathogenic Bacteria. International Journal of Molecular Sciences. 2021; 22(2):615. https://doi.org/10.3390/ijms22020615
Chicago/Turabian StylePillon, Margaux, and Patricia Doublet. 2021. "Myosins, an Underestimated Player in the Infectious Cycle of Pathogenic Bacteria" International Journal of Molecular Sciences 22, no. 2: 615. https://doi.org/10.3390/ijms22020615
APA StylePillon, M., & Doublet, P. (2021). Myosins, an Underestimated Player in the Infectious Cycle of Pathogenic Bacteria. International Journal of Molecular Sciences, 22(2), 615. https://doi.org/10.3390/ijms22020615