
Functional Role of Non-Muscle Myosin II in Microglia: An Updated Review




Abstract
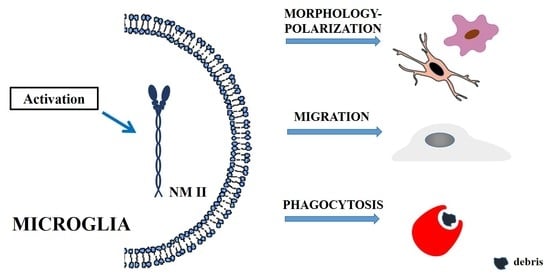
1. Introduction
2. Non-Muscle Myosin II
3. Non-Muscle Myosin II in Microglia
3.1. Microglia
3.2. NM II in Microglia Morphology and Polarization
3.3. NM II in Microglia Migration
3.4. NM II in Microglial Phagocytosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berg, J.S.; Powell, B.C.; Cheney, R.E. A millennial myosin census. Mol. Biol. Cell. 2001, 12, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Kollmar, M.; Mühlhausen, S. Myosin repertoire expansion coincides with eukaryotic diversification in the Mesoproterozoic era. BMC Evol Biol. 2017, 17, 562. [Google Scholar] [CrossRef] [PubMed]
- Szent-Györgyi, A.G. The early history of the biochemistry of muscle contraction. J. Gen. Physiol. 2004, 123, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Korn, E.D. Acanthamoeba myosin. I. Isolation from Acanthamoeba castellanii of an enzyme similar to muscle myosin. J. Biol. Chem. 1973, 248, 4682–4690. [Google Scholar] [CrossRef]
- McIntosh, B.B.; Ostap, E.M. Myosin-I molecular motors at a glance. J. Cell Sci. 2016, 129, 2689–2695. [Google Scholar] [CrossRef]
- Fili, N.; Toseland, C.P. Unconventional Myosins: How Regulation Meets Function. Int. J. Mol. Sci. 2019, 21, 67. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.A.; Finan, D.; Sivaramakrishnan, S.; Spudich, J.A. Principles of unconventional myosin function and targeting. Annu. Rev. Cell Dev. Biol. 2011, 27, 133–155. [Google Scholar] [CrossRef]
- Javier-Torrent, M.; Saura, C.A. Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration. Cells 2020, 9, 1926. [Google Scholar] [CrossRef]
- Girón-Pérez, D.A.; Piedra-Quintero, Z.L.; Santos-Argumedo, L. Class I myosins: Highly versatile proteins with specific functions in the immune system. J. Leukoc. Biol. 2019, 105, 973–981. [Google Scholar] [CrossRef]
- Li, J.; Lu, Q.; Zhang, M. Structural Basis of Cargo Recognition by Unconventional Myosins in Cellular Trafficking. Traffic 2016, 17, 822–838. [Google Scholar] [CrossRef]
- De Lanerolle, P.; Serebryannyy, L. Nuclear actin and myosins: Life without filaments. Nat. Cell Biol. 2011, 13, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Keeling, M.C.; Flores, L.R.; Dodhy, A.H.; Murray, E.R.; Gavara, N. Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci. Rep. 2017, 7, 5219. [Google Scholar] [CrossRef]
- Brown, M.E.; Bridgman, P.C. Myosin function in nervous and sensory systems. J. Neurobiol. 2004, 58, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.R.; Sousa, M.M. Non-Muscle Myosin II in Axonal Cell Biology: From the Growth Cone to the Axon Initial Segment. Cells 2020, 9, 1961. [Google Scholar] [CrossRef]
- Mehra, A.; Guérit, S.; Macrez, R.; Gosselet, F.; Sevin, E.; Lebas, H.; Maubert, E.; de Vries, H.E.; Bardou, I.; Vivien, D.; et al. Nonionotropic Action of Endothelial NMDA Receptors on Blood-Brain Barrier Permeability via Rho/ROCK-Mediated Phosphorylation of Myosin. J. Neurosci. 2020, 40, 1778–1787. [Google Scholar] [CrossRef]
- Thal, S.C.; Neuhaus, W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch. Med. Res. 2014, 45, 698–710. [Google Scholar] [CrossRef]
- Domingues, H.S.; Urbanski, M.M.; Macedo-Ribeiro, S.; Almaktari, A.; Irfan, A.; Hernandez, Y.; Wang, H.; Relvas, J.B.; Rubinstein, B.; Melendez-Vasquez, C.V.; et al. Pushing myelination—Developmental regulation of myosin expression drives oligodendrocyte morphological differentiation. J. Cell Sci. 2020, 133, jcs232264. [Google Scholar] [CrossRef]
- Jansen, S.; Gudi, V.; Prajeeth, C.K.; Singh, V.; Stahl, K.; Heckers, S.; Skripuletz, T.; Pul, R.; Trebst, C.; Tsiavaliaris, G.; et al. A pivotal role of nonmuscle myosin II during microglial activation. Exp. Neurol. 2014, 261, 666–676. [Google Scholar] [CrossRef]
- Billington, N.; Wang, A.; Mao, J.; Adelstein, R.S.; Sellers, J.R. Characterization of three full-length human nonmuscle myosin II paralogs. J. Biol. Chem. 2013, 288, 33398–33410. [Google Scholar] [CrossRef]
- Hartman, M.A.; Spudich, J.A. The myosin superfamily at a glance. J. Cell Sci. 2012, 125, 1627–1632. [Google Scholar] [CrossRef]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Sousa, S. Non-Muscle Myosin 2A (NM2A): Structure, Regulation and Function. Cells 2020, 9, 1590. [Google Scholar] [CrossRef]
- Wang, A.; Ma, X.; Conti, M.A.; Adelstein, R.S. Distinct and redundant roles of the non-muscle myosin II isoforms and functional domains. Biochem. Soc. Trans. 2011, 39, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Heissler, S.M.; Manstein, D.J. Nonmuscle myosin-2: Mix and match. Cell. Mol. Life Sci. 2013, 70, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.V.; Nag, S.; Spudich, A.; Ruppel, K.M.; Spudich, J.A. The Myosin Family of Mechanoenzymes: From Mechanisms to Therapeutic Approaches. Annu. Rev. Biochem. 2020, 89, 667–693. [Google Scholar] [CrossRef] [PubMed]
- Golomb, E.; Ma, X.; Jana, S.S.; Preston, Y.A.; Kawamoto, S.; Shoham, N.G.; Goldin, E.; Conti, M.A.; Sellers, J.R.; Adelstein, R.S. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 2004, 279, 2800–2808. [Google Scholar] [CrossRef]
- Nagy, A.; Takagi, Y.; Billington, N.; Sun, S.A.; Hong, D.K.T.; Homsher, E.; Wang, A.; Sellers, J.R. Kinetic characterization of nonmuscle myosin IIb at the single molecule level. J. Biol. Chem. 2013, 288, 709–722. [Google Scholar] [CrossRef]
- Sandquist, J.C.; Means, A.R. The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol. Biol. Cell. 2008, 19, 5156–5167. [Google Scholar] [CrossRef]
- Heissler, S.M.; Sellers, J.R. Kinetic Adaptations of Myosins for Their Diverse Cellular Functions. Traffic 2016, 17, 839–859. [Google Scholar] [CrossRef]
- Masters, T.A.; Kendrick-Jones, J.; Buss, F. Myosins: Domain Organisation, Motor Properties, Physiological Roles and Cellular Functions. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2017; Volume 235, pp. 77–122. [Google Scholar] [CrossRef]
- Heissler, S.M.; Sellers, J.R. Various Themes of Myosin Regulation. J. Mol. Biol. 2016, 428, 1927–1946. [Google Scholar] [CrossRef]
- Yamashiro, S.; Totsukawa, G.; Yamakita, Y.; Sasaki, Y.; Madaule, P.; Ishizaki, T.; Narumiya, S.; Matsumura, F. Citron kinase, a Rho-dependent kinase, induces di-phosphorylation of regulatory light chain of myosin II. Mol. Biol. Cell. 2003, 14, 1745–1756. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Zhang, H.; Zeng, W.; Li, S.; Chen, C.; Song, X.; Sun, J.; Sun, Z.; Cui, C.; et al. ZIPK mediates endothelial cell contraction through myosin light chain phosphorylation and is required for ischemic-reperfusion injury. FASEB J. 2019, 33, 9062–9074. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Qiao, Y.N.; Tao, T.; Zhao, W.; Wei, L.S.; Li, Y.Q.; Wang, W.; Wang, Y.; Zhou, Y.W.; Zheng, Y.Y.; et al. Distinct Roles of Smooth Muscle and Non-muscle Myosin Light Chain-Mediated Smooth Muscle Contraction. Front. Physiol. 2020, 11, 593966. [Google Scholar] [CrossRef] [PubMed]
- Goeckeler, Z.M.; Masaracchia, R.A.; Zeng, Q.; Chew, T.L.; Gallagher, P.; Wysolmerski, R.B. Phosphorylation of myosin light chain kinase by p21-activated kinase PAK2. J. Biol. Chem. 2000, 275, 18366–18374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bhetwal, B.P.; Gunst, S.J. Rho kinase collaborates with p21-activated kinase to regulate actin polymerization and contraction in airway smooth muscle. J. Physiol. 2018, 596, 3617–3635. [Google Scholar] [CrossRef] [PubMed]
- Artamonov, M.V.; Sonkusare, S.K.; Good, M.E.; Momotani, K.; Eto, M.; Isakson, B.E.; Le, T.H.; Cope, E.L.; Derewenda, Z.S.; Derewenda, U.; et al. RSK2 contributes to myogenic vasoconstriction of resistance arteries by activating smooth muscle myosin and the Na+/H+ exchanger. Sci. Signal. 2018, 11, eaar3924. [Google Scholar] [CrossRef] [PubMed]
- Tan, I.; Yong, J.; Dong, J.M.; Lim, L.; Leung, T. A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 2008, 135, 123–136. [Google Scholar] [CrossRef]
- Kassianidou, E.; Hughes, J.H.; Kumar, S. Activation of ROCK and MLCK tunes regional stress fiber formation and mechanics via preferential myosin light chain phosphorylation. Mol. Biol. Cell. 2017, 28, 3832–3843. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.C.; Tatenhorst, L.; Roser, A.E.; Saal, K.A.; Tönges, L.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef]
- Bao, J.; Jana, S.S.; Adelstein, R.S. Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J. Biol. Chem. 2005, 280, 19594–19599. [Google Scholar] [CrossRef]
- Rochlin, M.W.; Itoh, K.; Adelstein, R.S.; Bridgman, P.C. Localization of myosin II A and B isoforms in cultured neurons. J. Cell Sci. 1995, 108, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jana, S.S.; Conti, M.A.; Kawamoto, S.; Claycomb, W.C.; Adelstein, R.S. Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol. Biol. Cell. 2010, 21, 3952–3962. [Google Scholar] [CrossRef]
- Hodges, J.L.; Newell-Litwa, K.; Asmussen, H.; Vicente-Manzanares, M.; Horwitz, A.R. Myosin IIb activity and phosphorylation status determines dendritic spine and post-synaptic density morphology. PLoS ONE 2011, 6, e24149. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Endo, M.; Hata, K.; Taniguchi, J.; Kitajo, K.; Tomura, S.; Yamaguchi, A.; Mueller, B.K.; Yamashita, T. Myosin IIA is required for neurite outgrowth inhibition produced by repulsive guidance molecule. J. Neurochem. 2008, 105, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Leo-Macias, A.; Yuen, S.; Khatri, L.; Pfennig, S.; Zhang, Y.; Agullo-Pascual, E.; Caillol, G.; Zhu, M.S.; Rothenberg, E.; et al. Localized myosin II activity regulates assembly and plasticity of the axon initial segment. Neuron 2018, 97, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Srivastava, K.; Shao, B.; Bayraktutan, U. PKC-β exacerbates in vitro brain barrier damage in hyperglycemic settings via regulation of RhoA/Rho-kinase/MLC2 pathway. J. Cereb. Blood Flow Metab. 2013, 33, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rusielewicz, T.; Tewari, A.; Leitman, E.M.; Einheber, S.; Melendez-Vasquez, C.V. Myosin II is a negative regulator of oligodendrocyte morphological differentiation. J. Neurosci. Res. 2012, 90, 1547–1556. [Google Scholar] [CrossRef]
- Wang, H.; Tewari, A.; Einheber, S.; Salzer, J.L.; Melendez-Vasquez, C.V. Myosin II has distinct functions in PNS and CNS myelin sheath formation. J. Cell Biol. 2008, 182, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Liu, W.; Ruan, Z.; Xu, Z.; Fu, L. Myosin IIA Regulated Tight Junction in Oxygen Glucose-Deprived Brain Endothelial Cells Via Activation of TLR4/PI3K/Akt/JNK1/2/14-3-3ε/NF-κB/MMP9 Signal Transduction Pathway. Cell Mol. Neurobiol. 2019, 39, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Chauhan, V.P.; Elzinga, M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: Filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry 1998, 37, 1989–2003. [Google Scholar] [CrossRef]
- Even-Faitelson, L.; Ravid, S. PAK1 and aPKCzeta regulate myosin II-B phosphorylation: A novel signaling pathway regulating filament assembly. Mol. Biol. Cell 2006, 17, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Beach, J.R.; Licate, L.S.; Crish, J.F.; Egelhoff, T.T. Analysis of the role of Ser1/Ser2/Thr9 phosphorylation on myosin II assembly and function in live cells. BMC Cell Biol. 2011, 12, 1252. [Google Scholar] [CrossRef] [PubMed]
- Mosser, C.A.; Baptista, S.; Arnoux, I.; Audinat, E. Microglia in CNS development: Shaping the brain for the future. Prog. Neurobiol. 2017, 149, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gomez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. Neuroprotective function of microglia in the developing brain. Neuronal Signal 2021, 5, 200024. [Google Scholar] [CrossRef]
- Diaz-Aparicio, I.; Beccari, S.; Abiega, O.; Sierra, A. Clearing the corpses: Regulatory mechanisms, novel tools, and therapeutic potential of harnessing microglial phagocytosis in the diseased brain. Neural Regen. Res. 2016, 11, 1533–1539. [Google Scholar] [CrossRef]
- Rodriguez-Iglesias, N.; Sierra, A.; Valero, J. Rewiring of Memory Circuits: Connecting Adult Newborn Neurons with the Help of Microglia. Front. Cell Dev. Biol. 2019, 7, 24. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Cengiz, P.; Zafer, D.; Chandrashekhar, J.H.; Chanana, V.; Bogost, J.; Waldman, A.; Novak, B.; Kintner, D.B.; Ferrazzano, P.A. Developmental differences in microglia morphology and gene expression during normal brain development and in response to hypoxia-ischemia. Neurochem. Int. 2019, 127, 137–147. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. Role of Microglia in Neurological Disorders and Their Potentials as a Therapeutic Target. Mol. Neurobiol. 2017, 54, 7567–7584. [Google Scholar] [CrossRef] [PubMed]
- Trotta, T.; Panaro, M.A.; Cianciulli, A.; Mori, G.; di Benedetto, A.; Porro, C. Microglia-derived extracellular vesicles in Alzheimer’s Disease: A double-edged sword. Biochem. Pharmacol. 2018, 148, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Panaro, M.A.; Lofrumento, D.D.; Hasalla, E.; Trotta, T. The multiple roles of exosomes in Parkinson’s disease: An overview. Immunopharmacol. Immunotoxicol. 2019, 41, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef]
- Raj, D.D.A.; Jaarsma, D.; Holtman, I.R.; Olah, M.; Ferreira, F.M.; Schaafsma, W.; Brouwer, N.; Meijer, M.M.; de Waard, M.C.; van der Pluijm, I.; et al. Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol. Aging 2014, 35, 2147–2160. [Google Scholar] [CrossRef]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef]
- Lombardi, M.; Gabrielli, M.; Adinolfi, E.; Verderio, C. Role of ATP in Extracellular Vesicle Biogenesis and Dynamics. Front. Pharmacol. 2021, 12, 654023. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Trotta, T.; Panaro, M.A. Microvesicles in the brain: Biomarker, messenger or mediator? J. Neuroimmunol. 2015, 288, 70–78. [Google Scholar] [CrossRef]
- Dozio, V.; Sanchez, J.C. Characterisation of extracellular vesicle-subsets derived from brain endothelial cells and analysis of their protein cargo modulation after TNF exposure. J. Extracell. Vesicles 2017, 6, 1302705. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; de Almeida, L.P. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control. Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.A.; Even-Ram, S.; Liu, C.; Yamada, K.M.; Adelstein, R.S. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J. Biol. Chem. 2004, 279, 41263–41266. [Google Scholar] [CrossRef]
- Scheiblich, H.; Bicker, G. Regulation of Microglial Phagocytosis by RhoA/ROCK-Inhibiting Drugs. Cell Mol. Neurobiol. 2017, 37, 461–473. [Google Scholar] [CrossRef]
- Rousseau, M.; Gaugler, M.H.; Rodallec, A.; Bonnaud, S.; Paris, F.; Corre, I. RhoA GTPase regulates radiation-induced alterations in endothelial cell adhesion and migration. Biochem. Biophys. Res. Commun. 2011, 414, 750–755. [Google Scholar] [CrossRef]
- Zhou, Q.; Gensch, C.; Liao, J.K. Rho-associated coiled-coil-forming kinases (ROCKs): Potential targets for the treatment of atherosclerosis and vascular disease. Trends Pharmacol. Sci. 2011, 32, 167–173. [Google Scholar] [CrossRef]
- Fu, P.C.; Tang, R.H.; Yu, Z.Y.; Xie, M.J.; Wang, W.; Luo, X. The Rho-associated kinase inhibitors Y27632 and fasudil promote microglial migration in the spinal cord via the ERK signaling pathway. Neural Regen. Res. 2018, 13, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, E.; Yamanaka, H.; Kobayashi, K.; Yagi, H.; Sakagami, M.; Noguchi, K. RhoA/ROCK pathway mediates p38 MAPK activation and morphological changes downstream of P2Y12/13 receptors in spinal microglia in neuropathic pain. GLIA 2015, 63, 216–228. [Google Scholar] [CrossRef]
- Gitik, M.; Reichert, F.; Rotshenker, S. Cytoskeleton plays a dual role of activation and inhibition in myelin and zymosan phagocytosis by microglia. FASEB J. 2010, 24, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sun, Z.; Jin, M.; Tu, Y.; Wang, S.; Yang, X.; Chen, Q.; Zhang, X.; Han, Y.; Pi, R. Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J. Neuroimmunol. 2017, 305, 108–114. [Google Scholar] [CrossRef]
- Pistono, C.; Bister, N.; Stanová, I.; Malm, T. Glia-Derived Extracellular Vesicles: Role in Central Nervous System Communication in Health and Disease. Front. Cell Dev. Biol. 2021, 8, 623771. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef]
- Nawaz, M.; Camussi, G.; Valadi, H.; Nazarenko, I.; Ekström, K.; Wang, X.; Principe, S.; Shah, N.; Ashraf, N.M.; Fatima, F.; et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 2014, 11, 688–701. [Google Scholar] [CrossRef]
- Hugel, B.; Martínez, M.C.; Kunzelmann, C.; Freyssinet, J.M. Membrane microparticles: Two sides of the coin. Physiology 2005, 20, 22–27. [Google Scholar] [CrossRef]
- Muhsin-Sharafaldine, M.R.; McLellan, A.D. Tumor-Derived Apoptotic Vesicles: With Death They Do Part. Front. Immunol. 2018, 9, 957. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef]
- Faas, M.M.; Sáez, T.; de Vos, P. Extracellular ATP and adenosine: The Yin and Yang in immune responses? Mol. Aspects Med. 2017, 55, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Gonnord, P.; Delarasse, C.; Auger, R.; Benihoud, K.; Prigent, M.; Cuif, M.H.; Lamaze, C.; Kanellopoulos, J.M. Palmitoylation of the P2X7 receptor, an ATP-gated channel, controls its expression and association with lipid rafts. FASEB J. 2009, 23, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.J.; Rathsam, C.; Stokes, L.; McGeachie, A.B.; Wiley, J.S. Extracellular ATP dissociates nonmuscle myosin from P2X(7) complex: This dissociation regulates P2X(7) pore formation. Am. J. Physiol. Cell Physiol. 2009, 297, C430–C439. [Google Scholar] [CrossRef] [PubMed]
- Smolders, S.M.T.; Kessels, S.; Vangansewinkel, T.; Rigo, J.M.; Legendre, P.; Brône, B. Microglia: Brain cells on the move. Prog. Neurobiol. 2019, 178, 101612. [Google Scholar] [CrossRef]
- Fan, Y.; Xie, L.; Chung, C.Y. Signaling Pathways Controlling Microglia Chemotaxis. Mol. Cells 2017, 40, 163–168. [Google Scholar] [CrossRef]
- Borm, B.; Requardt, R.P.; Herzog, V.; Kirfel, G. Membrane ruffles in cell migration: Indicators of inefficient lamellipodia adhesion and compartments of actin filament reorganization. Exp. Cell Res. 2005, 302, 83–95. [Google Scholar] [CrossRef]
- Block, M.R.; Badowski, C.; Millon-Fremillon, A.; Bouvard, D.; Bouin, A.P.; Faurobert, E.; Gerber-Scokaert, D.; Planus, E.; Albiges-Rizo, C. Podosome-type adhesions and focal adhesions, so alike yet so different. Eur. J. Cell Biol. 2008, 87, 491–506. [Google Scholar] [CrossRef]
- Siddiqui, T.A.; Lively, S.; Vincent, C.; Schlichter, L.C. Regulation of podosome formation, microglial migration and invasion by Ca(2+)-signaling molecules expressed in podosomes. J. Neuroinflamm. 2012, 9, 250. [Google Scholar] [CrossRef]
- Rafiq, N.B.M.; Grenci, G.; Lim, C.K.; Kozlov, M.M.; Jones, G.E.; Viasnoff, V.; Bershadsky, A.D. Forces and constraints controlling podosome assembly and disassembly. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180228. [Google Scholar] [CrossRef]
- Shutova, M.S.; Asokan, S.B.; Talwar, S.; Assoian, R.K.; Bear, J.E.; Svitkina, T.M. Self-sorting of nonmuscle myosins IIA and IIB polarizes the cytoskeleton and modulates cell motility. J. Cell Biol. 2017, 216, 2877–2889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, P.; Wang, D.; Liu, Y.; Cao, L.; Wang, Y.; Xu, Y.; Zhu, C. Involvement of RhoA/ROCK Signaling in Aβ-Induced Chemotaxis, Cytotoxicity and Inflammatory Response of Microglial BV2 Cells. Cell. Mol. Neurobiol. 2019, 39, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Ros, C.M.; Annese, V.; Carrillo-de Sauvage, M.A.; Ros-Bernal, F.; Gómez, A.; Yuste, J.E.; Campuzano, C.M.; de Pablos, V.; Fernandez-Villalba, E.; et al. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci. Rep. 2012, 2, 809. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Duan, M.; Yang, L.; Buch, S. Nonmuscle myosin light-chain kinase mediates microglial migration induced by HIV Tat: Involvement of β1 integrins. FASEB J. 2013, 27, 1532–1548. [Google Scholar] [CrossRef]
- Westman, J.; Grinstein, S.; Marques, P.E. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front. Immunol. 2020, 10, 3030. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef]
- ElAli, A.; Rivest, S. Microglia Ontology and Signaling. Front. Cell Dev. Biol. 2016, 4, 72. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Alvarado, G.; Li, W. Tubby regulates microglial phagocytosis through MerTK. J. Neuroimmunol. 2012, 252, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [PubMed]
- van Zon, J.S.; Tzircotis, G.; Caron, E.; Howard, M. A mechanical bottleneck explains the variation in cup growth during FcgammaR phagocytosis. Mol. Syst. Biol. 2009, 5, 298. [Google Scholar] [CrossRef]
- Herant, M.; Lee, C.Y.; Dembo, M.; Heinrich, V. Protrusive push versus enveloping embrace: Computational model of phagocytosis predicts key regulatory role of cytoskeletal membrane anchors. PLoS Comput. Biol. 2011, 7, e1001068. [Google Scholar] [CrossRef]
- Prass, M.; Jacobson, K.; Mogilner, A.; Radmacher, M. Direct measurement of the lamellipodial protrusive force in a migrating cell. J. Cell Biol. 2006, 174, 767–772. [Google Scholar] [CrossRef]
- Mueller, J.; Szep, G.; Nemethova, M.; de Vries, I.; Lieber, A.D.; Winkler, C.; Kruse, K.; Small, J.V.; Schmeiser, C.; Keren, K.; et al. Load Adaptation of Lamellipodial Actin Networks. Cell 2017, 171, 188–200. [Google Scholar] [CrossRef]
- Bieling, P.; Li, T.D.; Weichsel, J.; McGorty, R.; Jreij, P.; Huang, B.; Fletcher, D.A.; Mullins, R.D. Force Feedback Controls Motor Activity and Mechanical Properties of Self-Assembling Branched Actin Networks. Cell 2016, 164, 115–127. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.; Condeelis, J. The cofilin activity cycle in lamellipodia and invadopodia. J. Cell Biochem. 2009, 108, 1252–1262. [Google Scholar] [CrossRef]
- Olazabal, I.M.; Caron, E.; May, R.C.; Schilling, K.; Knecht, D.A.; Machesky, L.M. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcgammaR, phagocytosis. Curr. Biol. 2002, 12, 1413–1418. [Google Scholar] [CrossRef]
- Hall, A.B.; Gakidis, M.A.M.; Glogauer, M.; Wilsbacher, J.L.; Gao, S.; Swat, W.; Brugge, J.S. Requirements for Vav guanine nucleotide exchange factors and Rho GTPases in FcgammaR- and complement-mediated phagocytosis. Immunity 2006, 24, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Tsuchida, M.A.; Allen, G.M.; Barnhart, E.L.; Applegate, K.T.; Yam, P.T.; Ji, L.; Keren, K.; Danuser, G.; Theriot, J.A. Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature 2010, 465, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.Y.; Schulze, U.; Salbreux, G.; Roensch, J.; Joanny, J.F.; Paluch, E. Role of cortical tension in bleb growth. Proc. Natl. Acad. Sci. USA 2009, 106, 18581–18586. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Zareno, J.; Whitmore, L.; Choi, C.K.; Horwitz, A.F. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 2007, 176, 573–580. [Google Scholar] [CrossRef]
- Cai, Y.; Biais, N.; Giannone, G.; Tanase, M.; Jiang, G.; Hofman, J.M.; Wiggins, C.H.; Silberzan, P.; Buguin, A.; Ladoux, B.; et al. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys. J. 2006, 91, 3907–3920. [Google Scholar] [CrossRef]
- Bufi, N.; Saitakis, M.; Dogniaux, S.; Buschinger, O.; Bohineust, A.; Richert, A.; Maurin, M.; Hivroz, C.; Asnacios, A. Human Primary Immune Cells Exhibit Distinct Mechanical Properties that Are Modified by Inflammation. Biophys. J. 2015, 108, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021. [Google Scholar] [CrossRef]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018, 285, 3566–3575. [Google Scholar] [CrossRef]
- Kataoka, A.; Koga, Y.; Uesugi, A.; Tozaki-Saitoh, H.; Tsuda, M.; Inoue, K. Involvement of vasodilator-stimulated phosphoprotein in UDP-induced microglial actin aggregation via PKC- and Rho-dependent pathways. Purinergic Signal. 2011, 7, 403–411. [Google Scholar] [CrossRef][Green Version]
- Wen, R.X.; Shen, H.; Huang, S.X.; Wang, L.P.; Li, Z.W.; Peng, P.; Mamtilahun, M.; Tang, Y.H.; Shen, F.X.; Tian, H.L.; et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci. Ther. 2020, 26, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, W.; Liu, Y.; Xu, P.; Li, Z.; Wu, R.; Shi, X.; Tang, Y. P2Y6 Receptor-Mediated Microglial Phagocytosis in Radiation-Induced Brain Injury. Mol. Neurobiol. 2016, 53, 3552–3564. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.J.; Saunders, B.M.; Jursik, C.; Wiley, J.S. The P2X7-nonmuscle myosin membrane complex regulates phagocytosis of nonopsonized particles and bacteria by a pathway attenuated by extracellular ATP. Blood 2010, 115, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.J.; Huang, X.; Ou, A.; Rembach, A.; Fowler, C.; Avula, P.K.; Horton, A.; Doecke, J.D.; Villemagne, V.L.; Macaulay, S.L.; et al. Innate phagocytosis by peripheral blood monocytes is altered in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 377–389. [Google Scholar] [CrossRef]
- Francistiova, L.; Bianchi, C.; Di Lauro, C.; Sebastián-Serrano, A.; de Diego-Garcia, L.; Kobolák, J.; Dinnyées, A.; Diaz-Hernandez, M. The Role of P2X7 Receptor in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 94. [Google Scholar] [CrossRef]
- Gu, B.J.; Wiley, J.S. P2X7 as a scavenger receptor for innate phagocytosis in the brain. Br. J. Pharmacol. 2018, 175, 4195–4208. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Effect on NM II. | Kinases | References |
|---|---|---|
| Activation at Thr18 and Ser19 | MLCK | [39] |
| ROCK | [39] | |
| CRIK | [32] | |
| DAPK3 | [33] | |
| MRCK | [38] | |
| ZIPK | [33] | |
| Activation at Ser19 | PAK | [35] |
| Activation at Ser1943 | CKII | [21,52] |
| Activation at Ser1916 and Ser1937 | PKC | [21,52,53] |
| Inactivation at Thr9, Ser1 and Ser2 | PKC | [21,54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porro, C.; Pennella, A.; Panaro, M.A.; Trotta, T. Functional Role of Non-Muscle Myosin II in Microglia: An Updated Review. Int. J. Mol. Sci. 2021, 22, 6687. https://doi.org/10.3390/ijms22136687
Porro C, Pennella A, Panaro MA, Trotta T. Functional Role of Non-Muscle Myosin II in Microglia: An Updated Review. International Journal of Molecular Sciences. 2021; 22(13):6687. https://doi.org/10.3390/ijms22136687
Chicago/Turabian StylePorro, Chiara, Antonio Pennella, Maria Antonietta Panaro, and Teresa Trotta. 2021. "Functional Role of Non-Muscle Myosin II in Microglia: An Updated Review" International Journal of Molecular Sciences 22, no. 13: 6687. https://doi.org/10.3390/ijms22136687
APA StylePorro, C., Pennella, A., Panaro, M. A., & Trotta, T. (2021). Functional Role of Non-Muscle Myosin II in Microglia: An Updated Review. International Journal of Molecular Sciences, 22(13), 6687. https://doi.org/10.3390/ijms22136687