
Deletion of Neuronal CuZnSOD Accelerates Age-Associated Muscle Mitochondria and Calcium Handling Dysfunction That Is Independent of Denervation and Precedes Sarcopenia

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
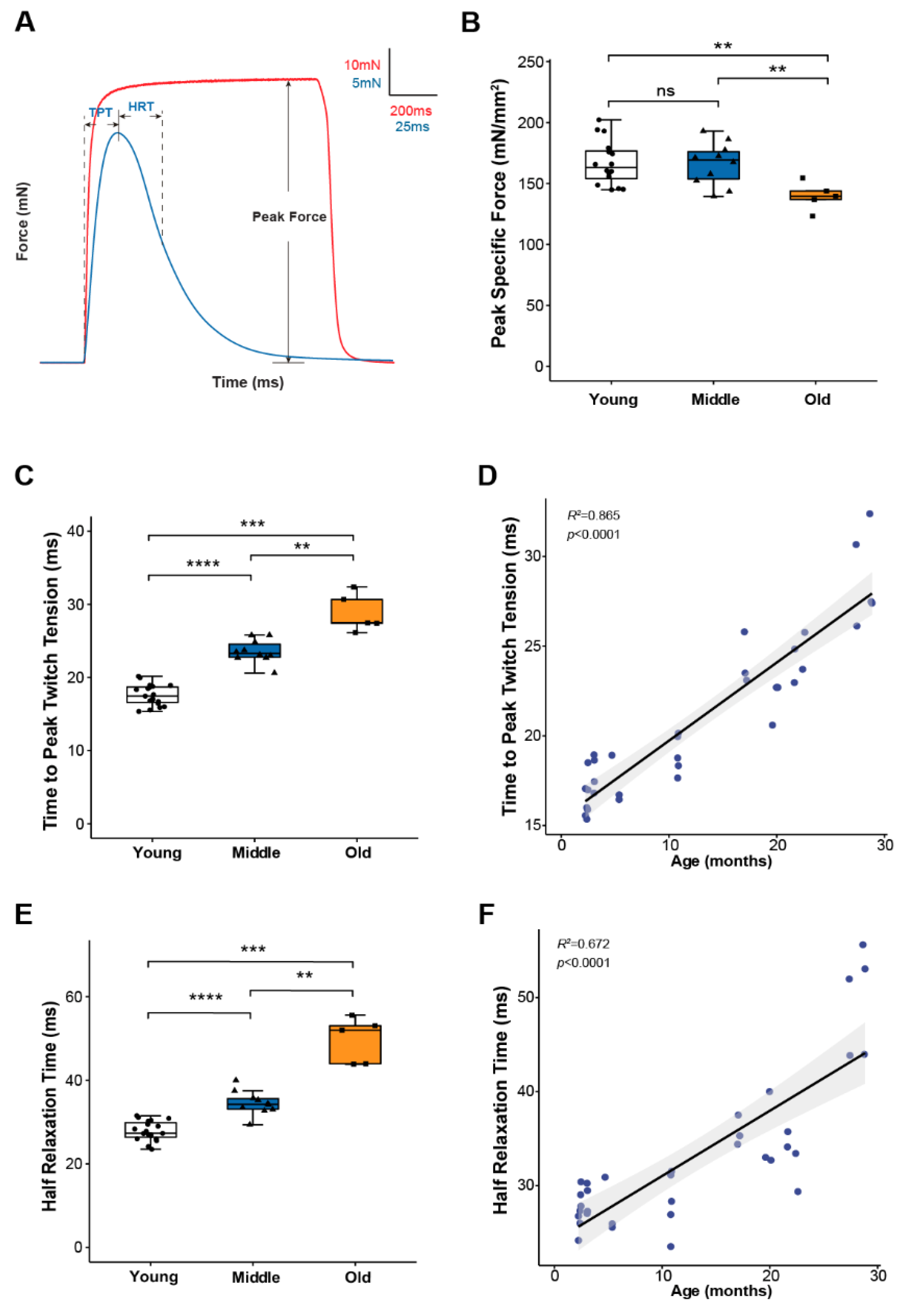
2.1. Lumbrical Muscle Contractile Properties Deteriorate with Aging, but Changes Occur Asynchronously for Different Parameters
2.2. Skeletal Muscle Calcium Handling Function Starts to Decline in Middle Age
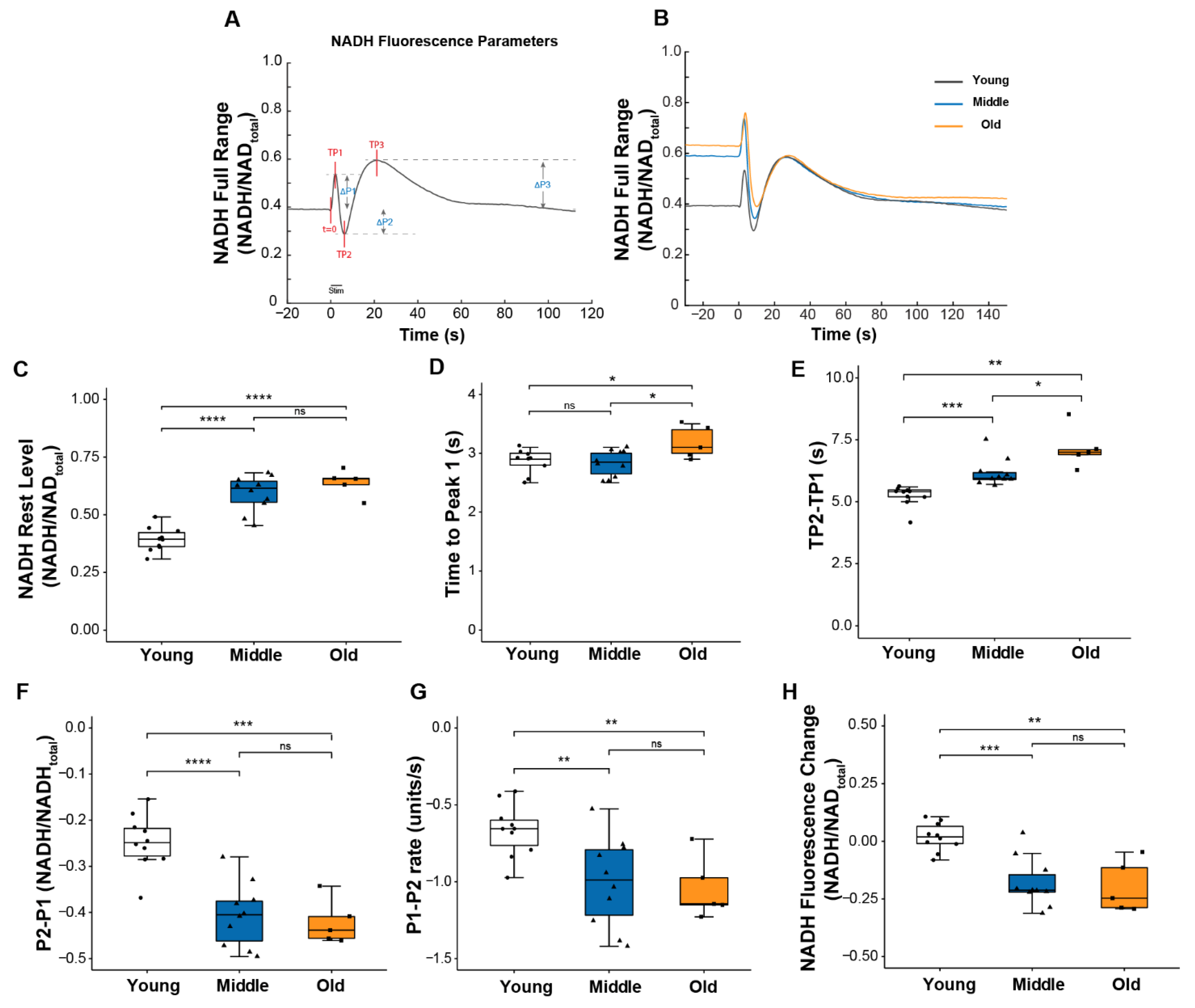
2.3. Skeletal Muscle Mitochondria Function Is Diminished by Middle Age
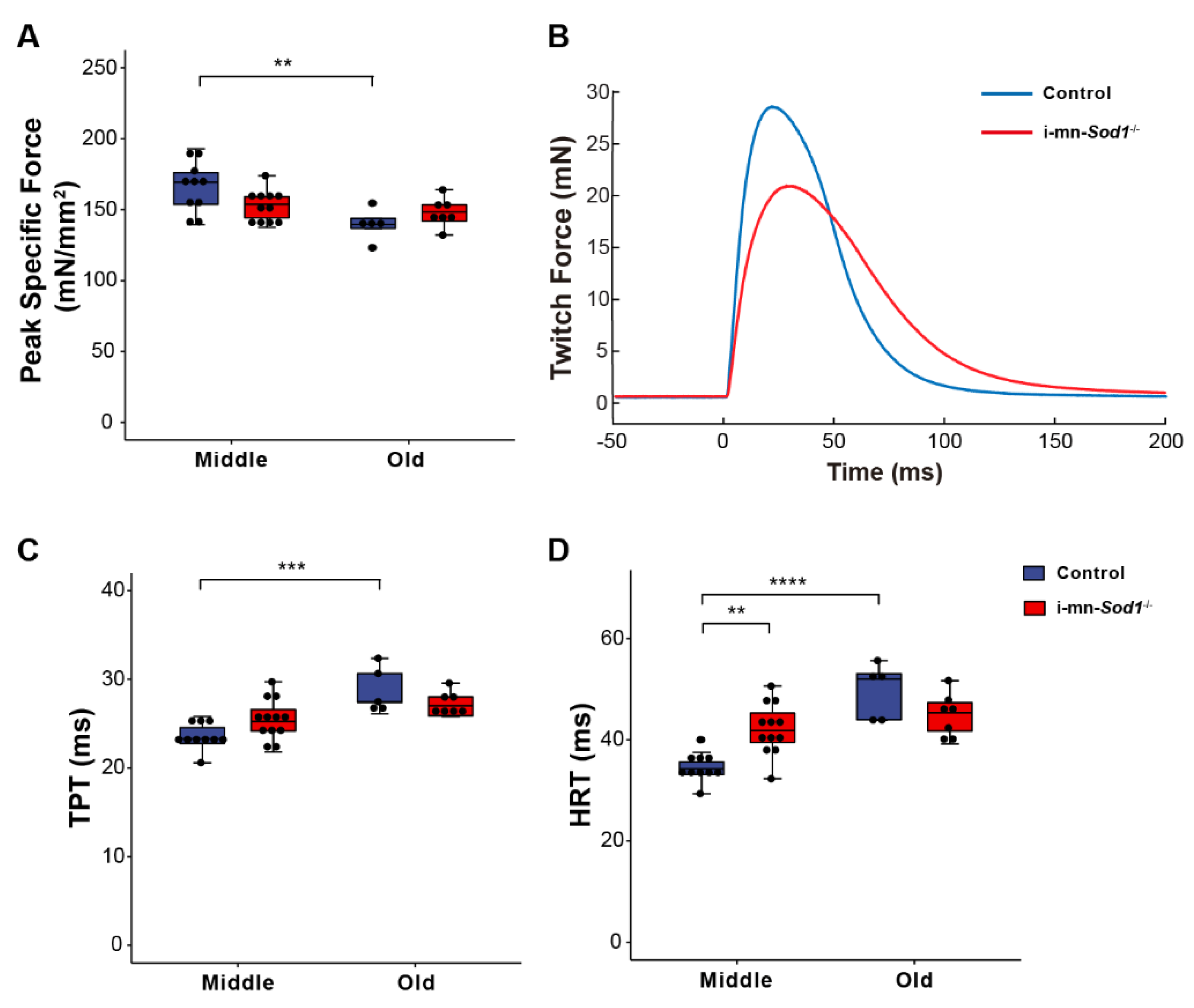
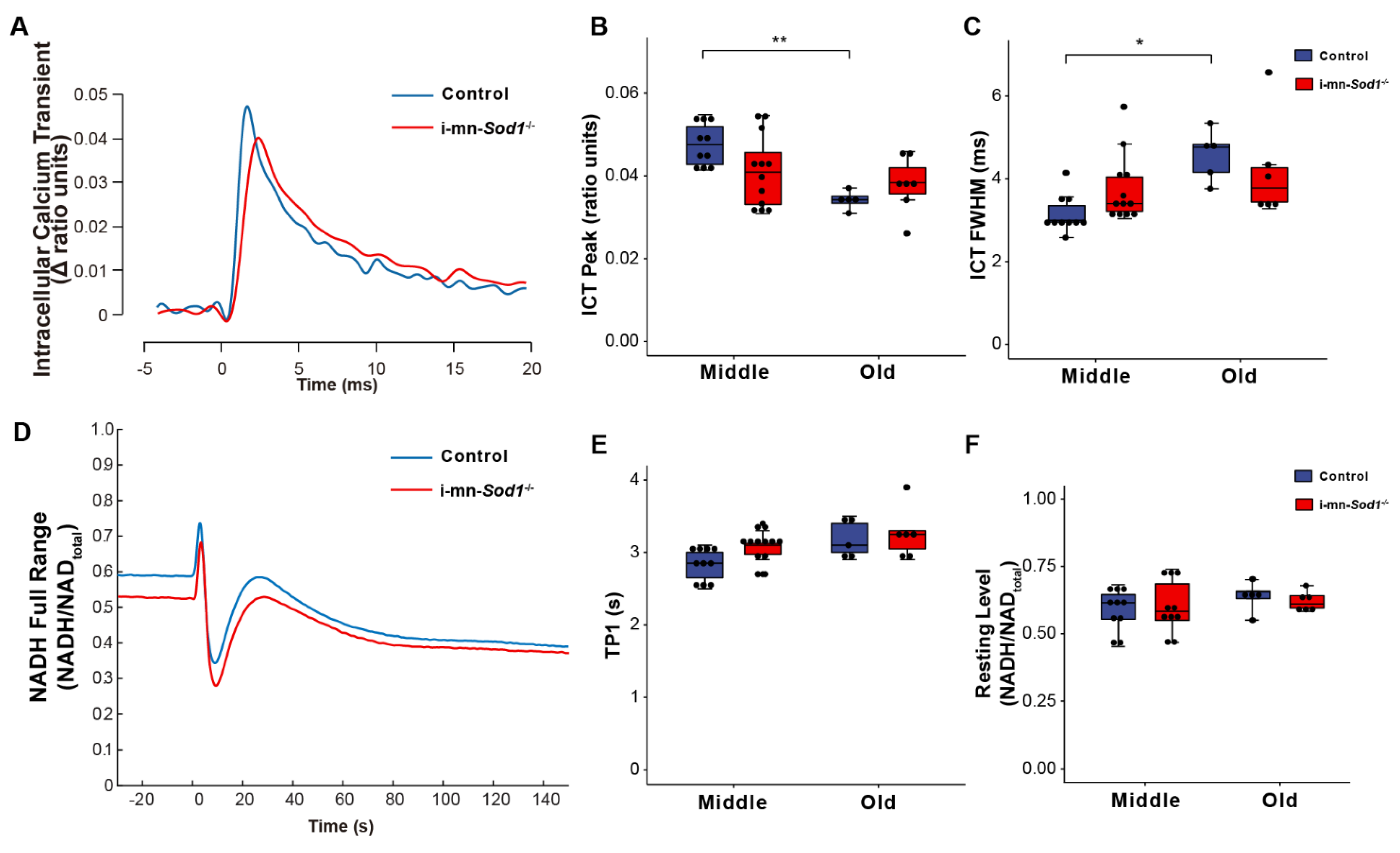
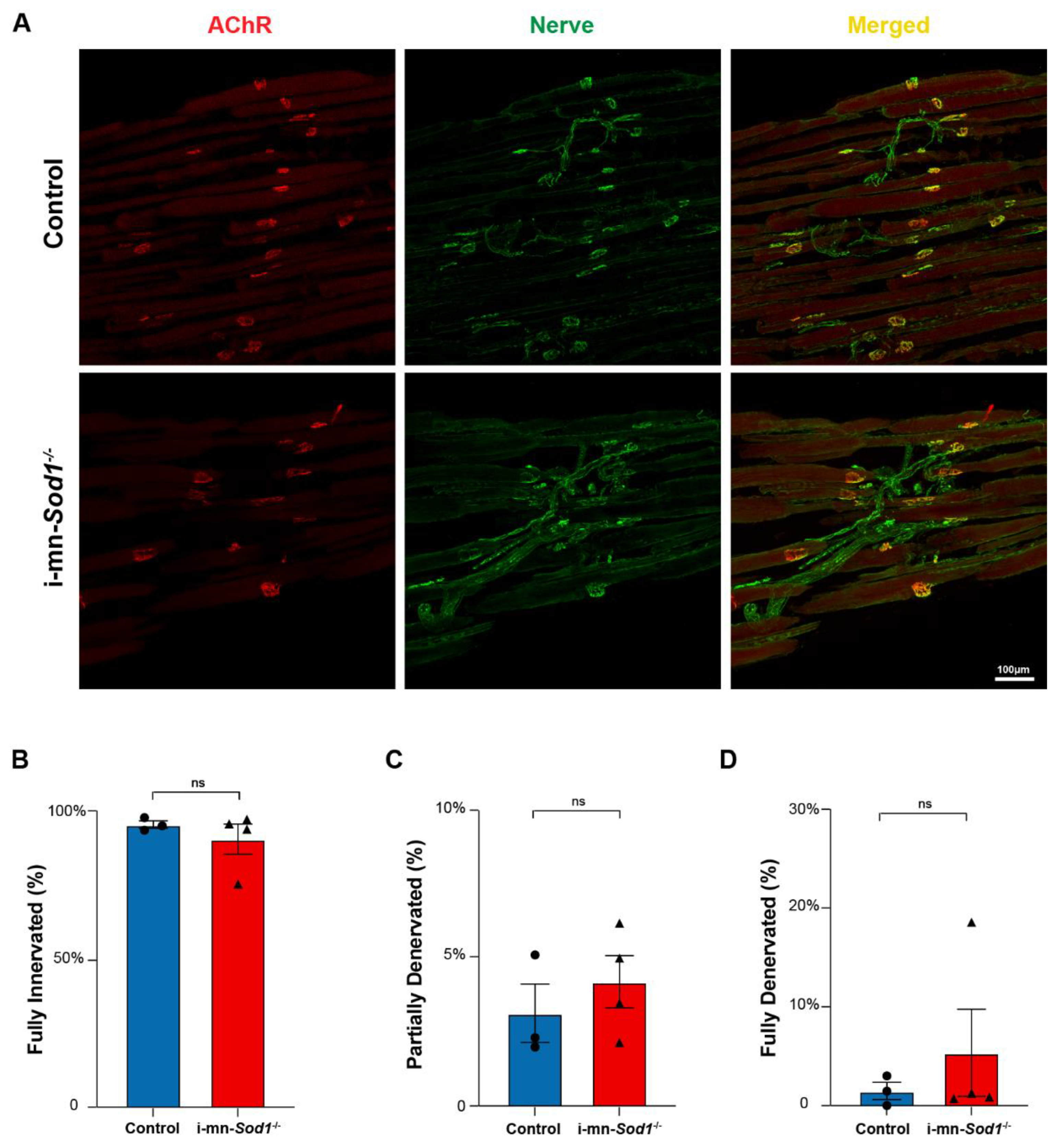
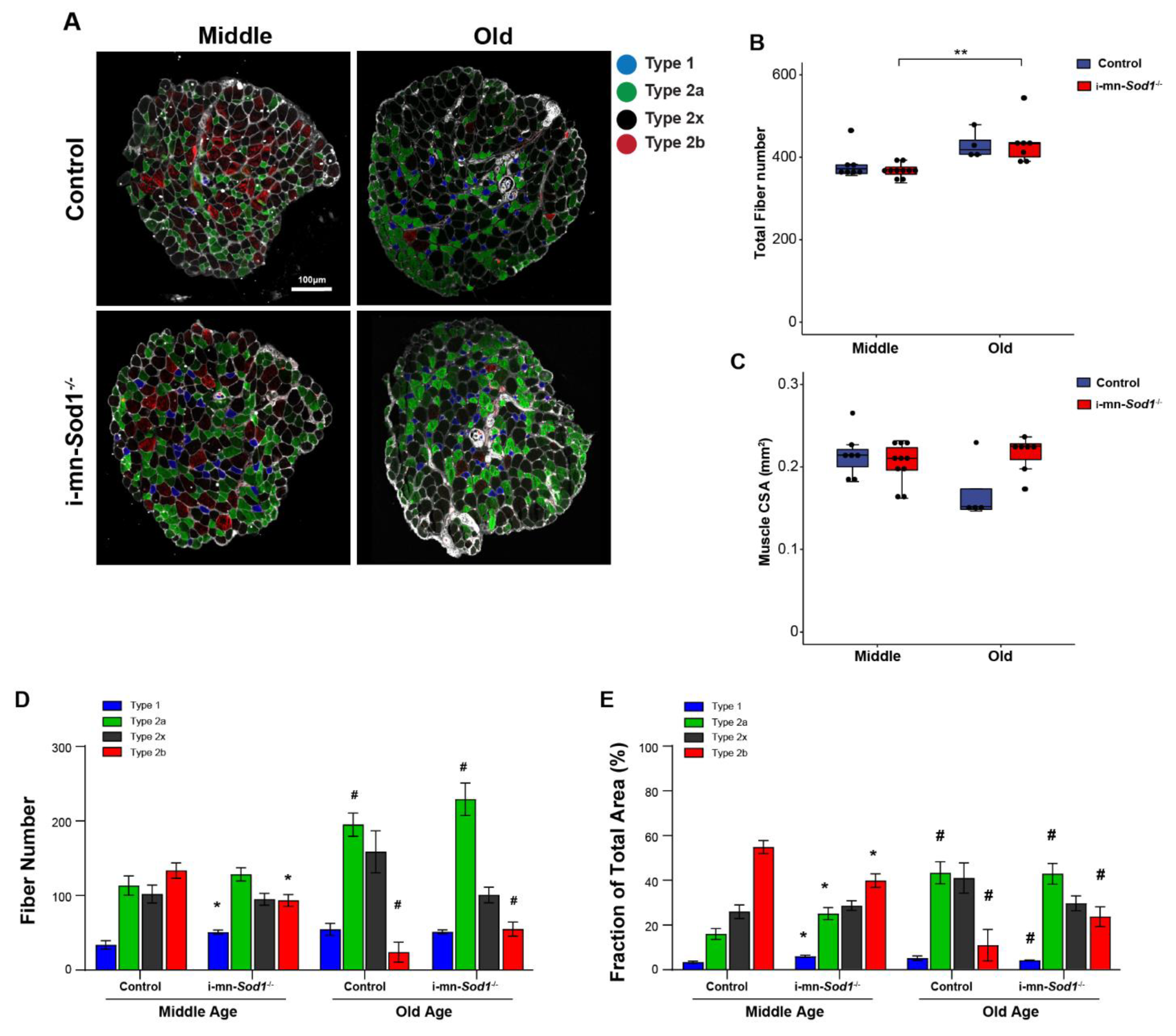
2.4. Knock Down of Sod1 in Motor Neurons Induces Post-Synaptic Changes in Myofiber Physiology without Evidence of Denervation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Contractile Properties
4.3. NADH Fluorescence
4.4. Intracellular Calcium Transients
4.5. Immunofluorescent Staining
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Barbiera, A.; Sica, G.; Scicchitano, B. Age-Related Alterations at Neuromuscular Junction: Role of Oxidative Stress and Epigenetic Modifications. Cells 2021, 10, 1307. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Van Remmen, H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011, 46, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepple, R.T. Mitochondrial Involvement and Impact in Aging Skeletal Muscle. Front. Aging Neurosci. 2014, 6, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbina-Varela, R.; Castillo, N.; Videla, L.A.; Del Campo, A. Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 7704. [Google Scholar] [CrossRef]
- Su, Y.; Ahn, B.; Macpherson, P.C.; Ranjit, R.; Claflin, D.R.; Van Remmen, H.; Brooks, S.V. Transgenic expression of SOD1 specifically in neurons of Sod1 deficient mice prevents defects in muscle mitochondrial function and calcium handling. Free Radic. Biol. Med. 2021, 165, 299–311. [Google Scholar] [CrossRef]
- Larkin, L.M.; Davis, C.S.; Robinson, C.; Kostrominova, T.Y.; Van Remmen, H.; Richardson, A.; Feldman, E.; Brooks, S.V. Skeletal muscle weakness due to deficiency of CuZn-superoxide dismutase is associated with loss of functional innervation. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R1400–R1407. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Davis, C.S.; Shi, Y.; Ivannikov, M.V.; Zhang, Y.; Vasilaki, A.; Macleod, G.T.; Richardson, A.; Van Remmen, H.; Jackson, M.J.; et al. Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice. FASEB J. 2014, 28, 1666–1681. [Google Scholar] [CrossRef] [Green Version]
- Scalabrin, M.; Pollock, N.; Staunton, C.A.; Brooks, S.V.; McArdle, A.; Jackson, M.J.; Vasilaki, A. Redox responses in skeletal muscle following denervation. Redox Biol. 2019, 26, 101294. [Google Scholar] [CrossRef]
- Spendiff, S.; Vuda, M.; Gouspillou, G.; Aare, S.; Perez, A.; Morais, J.A.; Jagoe, R.T.; Filion, M.-E.; Glicksman, R.; Kapchinsky, S.; et al. Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J. Physiol. 2016, 594, 7361–7379. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskaran, S.; Pollock, N.; MacPherson, P.C.; Ahn, B.; Piekarz, K.M.; Staunton, C.A.; Brown, J.L.; Qaisar, R.; Vasilaki, A.; Richardson, A.; et al. Neuron-specific deletion of CuZnSOD leads to an advanced sarcopenic phenotype in older mice. Aging Cell 2020, 19, e13225. [Google Scholar] [CrossRef] [PubMed]
- Delbono, O.; O’Rourke, K.; Ettinger, W. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J. Membr. Biol. 1995, 148, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Umanskaya, A.; Santulli, G.; Xie, W.; Andersson, D.; Reiken, S.R.; Marks, A.R. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc. Natl. Acad. Sci. USA 2014, 111, 15250–15255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boncompagni, S.; Pozzer, D.; Viscomi, C.; Ferreiro, A.; Zito, E. Physical and Functional Cross Talk between Endo-Sarcoplasmic Reticulum and Mitochondria in Skeletal Muscle. Antioxid. Redox Signal. 2020, 32, 873–883. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Zhou, S.; Crowley-McHattan, Z.; Wang, R.-Y.; Li, J.-P. A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function. Int. J. Environ. Res. Public Health 2021, 18, 3874. [Google Scholar] [CrossRef]
- Claflin, D.R.; Jackson, M.J.; Brooks, S. Age affects the contraction-induced mitochondrial redox response in skeletal muscle. Front. Physiol. 2015, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Sabri, M.I.; Ochs, S. Inhibition of glyceraldehyde-3-phosphate dehydrogenase in mammalian nerve by iodoacetic acid. J. Neurochem. 1971, 18, 1509–1514. [Google Scholar] [CrossRef]
- Brandes, R.; Bers, D. Simultaneous Measurements of Mitochondrial NADH and Ca2+ during Increased Work in Intact Rat Heart Trabeculae. Biophys. J. 2002, 83, 587–604. [Google Scholar] [CrossRef] [Green Version]
- Deschenes, M.R. Motor unit and neuromuscular junction remodeling with aging. Curr. Aging Sci. 2011, 4, 209–220. [Google Scholar] [CrossRef]
- Faulkner, J.A.; Larkin, L.M.; Claflin, D.R.; Brooks, S.V. Age-related changes in the structure and function of skeletal muscles. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1091–1096. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Davis, C.; Sakellariou, G.K.; Shi, Y.; Kayani, A.C.; Pulliam, D.; Bhattacharya, A.; Richardson, A.; Jackson, M.J.; McArdle, A.; et al. CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J. 2013, 27, 3536–3548. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ikeno, Y.; Bokov, A.; Gelfond, J.; Jaramillo, C.; Zhang, H.-M.; Liu, Y.; Qi, W.; Hubbard, G.; Richardson, A.; et al. Dietary restriction attenuates the accelerated aging phenotype of Sod1−/− mice. Free Radic. Biol. Med. 2013, 60, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leduc-Gaudet, J.-P.; Picard, M.; Pelletier, F.S.-J.; Sgarioto, N.; Auger, M.-J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, P.A.; Powers, S.K.; Ferreira, R.M.; Appell, H.J.; Duarte, J.A. Aging Impairs Skeletal Muscle Mitochondrial Bioenergetic Function. J. Gerontol. A Boil. Sci. Med. Sci. 2009, 64, 21–33. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial Structure and Function Are Disrupted by Standard Isolation Methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Wright, K.J.; Romestaing, C.; Thomas, M.M.; Rowan, S.L.; Taivassalo, T.; Hepple, R.T. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell 2010, 9, 1032–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [Green Version]
- Coen, P.M.; Musci, R.V.; Hinkley, J.M.; Miller, B.F. Mitochondria as a Target for Mitigating Sarcopenia. Front. Physiol. 2019, 9, 1883. [Google Scholar] [CrossRef] [Green Version]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef] [PubMed]
- Ábrigo, J.; Elorza, A.A.; Riedel, C.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of Oxidative Stress as Key Regulator of Muscle Wasting during Cachexia. Oxid. Med. Cell. Longev. 2018, 2018, 2063179. [Google Scholar] [CrossRef]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Picard, M.; Turnbull, D. The ageing neuromuscular system and sarcopenia: A mitochondrial perspective. J. Physiol. 2016, 594, 4499–4512. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ikeno, Y.; Qi, W.; Chaudhuri, A.; Li, Y.; Bokov, A.; Thorpe, S.R.; Baynes, J.W.; Epstein, C.; Richardson, A.; et al. Mice Deficient in Both Mn Superoxide Dismutase and Glutathione Peroxidase-1 Have Increased Oxidative Damage and a Greater Incidence of Pathology but No Reduction in Longevity. J. Gerontol. A Boil. Sci. Med. Sci. 2009, 64, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Ranjit, R.; Premkumar, P.; Pharaoh, G.; Piekarz, K.; Matsuzaki, S.; Claflin, D.; Riddle, K.; Judge, J.; Bhaskaran, S.; et al. Mitochondrial oxidative stress impairs contractile function but paradoxically increases muscle mass via fibre branching. J. Cachexia Sarcopenia Muscle 2019, 10, 411–428. [Google Scholar] [CrossRef] [Green Version]
- Zarse, K.; Schmeisser, S.; Groth, M.; Priebe, S.; Beuster, G.; Kuhlow, D.; Guthke, R.; Platzer, M.; Kahn, C.R.; Ristow, M. Impaired Insulin/IGF1 Signaling Extends Life Span by Promoting Mitochondrial L-Proline Catabolism to Induce a Transient ROS Signal. Cell Metab. 2012, 15, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, S.L.; Ventura, N.; Johnson, T.E. Relationship between Mitochondrial Electron Transport Chain Dysfunction, Development, and Life Extension in Caenorhabditis elegans. PLoS Biol. 2007, 5, e259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaisar, R.; Bhaskaran, S.; Ranjit, R.; Sataranatarajan, K.; Premkumar, P.; Huseman, K.; Van Remmen, H. Restoration of SERCA ATPase prevents oxidative stress-related muscle atrophy and weakness. Redox Biol. 2019, 20, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.L.; Chalmers, S.; McCarron, J.G. Mitochondrial organization and Ca2+ uptake. Biochem. Soc. Trans. 2012, 40, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as Sensors and Regulators of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Kadhiresan, V.A.; Hassett, C.A.; Faulkner, J.A. Properties of single motor units in medial gastrocnemius muscles of adult and old rats. J. Physiol. 1996, 493, 543–552. [Google Scholar] [CrossRef]
- Schiaffino, S.; Hanzlíková, V.; Pierobon, S. Relations between structure and function in rat skeletal muscle fibers. J. Cell Biol. 1970, 47, 107–119. [Google Scholar] [CrossRef]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- Anderson, E.J.; Neufer, P.D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am. J. Physiol.-Cell Physiol. 2006, 290, C844–C851. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Ritchie, D.; Thomas, M.M.; Wright, K.J.; Hepple, R.T. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell 2011, 10, 1047–1055. [Google Scholar] [CrossRef]
- Ham, D.J.; Börsch, A.; Lin, S.; Thürkauf, M.; Weihrauch, M.; Reinhard, J.R.; Delezie, J.; Battilana, F.; Wang, X.; Kaiser, M.S.; et al. The neuromuscular junction is a focal point of mTORC1 signaling in sarcopenia. Nat. Commun. 2020, 11, 4510. [Google Scholar] [CrossRef]
- Rowan, S.L.; Rygiel, K.; Purves-Smith, F.M.; Solbak, N.M.; Turnbull, D.M.; Hepple, R.T. Denervation Causes Fiber Atrophy and Myosin Heavy Chain Co-Expression in Senescent Skeletal Muscle. PLoS ONE 2012, 7, e29082. [Google Scholar] [CrossRef]
- Lexell, J. Evidence for Nervous System Degeneration with Advancing Age. J. Nutr. 1997, 127, 1011S–1013S. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, B.; Irving, D. The numbers of limb motor neurons in the human lumbosacral cord throughout life. J. Neurol. Sci. 1977, 34, 213–219. [Google Scholar] [CrossRef]
- Chai, R.J.; Vukovic, J.; Dunlop, S.; Grounds, M.; Shavlakadze, T. Striking Denervation of Neuromuscular Junctions without Lumbar Motoneuron Loss in Geriatric Mouse Muscle. PLoS ONE 2011, 6, e28090. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, N.; Castro, R.; Sutherland, N.M.; Vaughan, K.L.; Szarowicz, M.D.; De Cabo, R.; Mattison, J.A.; Valdez, G. α-Motor neurons are spared from aging while their synaptic inputs degenerate in monkeys and mice. Aging Cell 2018, 17, e12726. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Zhang, D.-Z.; Xi, Y.; Wu, C.-A. Protective effect of dexmedetomidine on neuronal hypoxic injury through inhibition of miR-134. Hum. Exp. Toxicol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; D’Angelo, M. Neuronal Cells Rearrangement during Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- Odnokoz, O.; Nakatsuka, K.; Wright, C.; Castellanos, J.; Klichko, V.I.; Kretzschmar, D.; Orr, W.C.; Radyuk, S.N. Mitochondrial Redox Signaling Is Critical to the Normal Functioning of the Neuronal System. Front. Cell Dev. Biol. 2021, 9, 613036. [Google Scholar] [CrossRef]
- Jagaraj, C.J.; Parakh, S.; Atkin, J.D. Emerging Evidence Highlighting the Importance of Redox Dysregulation in the Pathogenesis of Amyotrophic Lateral Sclerosis (ALS). Front. Cell. Neurosci. 2021, 14, 581950. [Google Scholar] [CrossRef]
- Hayes, L.R.; Asress, S.A.; Li, Y.; Galkin, A.; Stepanova, A.; Kawamata, H.; Manfredi, G.; Glass, J.D. Distal denervation in the SOD1 knockout mouse correlates with loss of mitochondria at the motor nerve terminal. Exp. Neurol. 2019, 318, 251–257. [Google Scholar] [CrossRef]
- Chance, B.; Jöbsis, F. Changes in Fluorescence in a Frog Sartorius Muscle Following a Twitch. Nature 1959, 184, 195–196. [Google Scholar] [CrossRef]
- Mayevsky, A.; Chance, B. Oxidation-reduction states of NADH in vivo: From animals to clinical use. Mitochondrion 2007, 7, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, P.C.D.; Farshi, P.; Goldman, D. Dach2-Hdac9 signaling regulates reinnervation of muscle endplates. Development 2015, 142, 4038–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, P.M.; Alway, S.E. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 2005, 565, 309–323. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Staining Target | Primary Antibody | Secondary Antibody |
|---|---|---|
| type 1 myosin heavy chain | BA-D5 (mouse IgG2b) (DSHB, Iowa City, IA, USA) (1/100) | Anti-mouse IgG2b 647nm, A21242 (Invitrogen, Carlsbad, CA, USA) (1/300) |
| type 2a myosin heavy chain | SC-71 (mouse IgG1) (DSHB, Iowa City, IA, USA) (1/100) | Anti-mouse IgG1 568nm, A21124 (Invitrogen, Carlsbad, CA, USA) (1/300) |
| type 2b myosin heavy chain | BF–F3 (mouse IgM) (DSHB, Iowa, USA) (1/100) | Anti-mouse IgM 488nm, A21042 (Invitrogen, Carlsbad, CA, USA) (1/300) |
| extracellular matrix | WGA CF405S conjugate, #29027 (Biotium, Hayward, CA, USA) (1/10) 1 h | |
| axons and presynaptic terminals | βIII-tubulin (Rabbit, BioLegend, San Diego, CA, USA, 802001, 1/1000) | Anti-rabbit Alexa 488 (1:2000 Thermo Fisher Scientific, Waltham, MA, USA A11034) |
| acetylcholine receptors | Alexa Fluor 594-conjugated α-bungarotoxin (α-Btx; 1:2000; Thermo Fisher Scientific, Waltham, MA, USA B-13423) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Y.; Claflin, D.R.; Huang, M.; Davis, C.S.; Macpherson, P.C.D.; Richardson, A.; Van Remmen, H.; Brooks, S.V. Deletion of Neuronal CuZnSOD Accelerates Age-Associated Muscle Mitochondria and Calcium Handling Dysfunction That Is Independent of Denervation and Precedes Sarcopenia. Int. J. Mol. Sci. 2021, 22, 10735. https://doi.org/10.3390/ijms221910735
Su Y, Claflin DR, Huang M, Davis CS, Macpherson PCD, Richardson A, Van Remmen H, Brooks SV. Deletion of Neuronal CuZnSOD Accelerates Age-Associated Muscle Mitochondria and Calcium Handling Dysfunction That Is Independent of Denervation and Precedes Sarcopenia. International Journal of Molecular Sciences. 2021; 22(19):10735. https://doi.org/10.3390/ijms221910735
Chicago/Turabian StyleSu, Yu, Dennis R. Claflin, Meixiang Huang, Carol S. Davis, Peter C. D. Macpherson, Arlan Richardson, Holly Van Remmen, and Susan V. Brooks. 2021. "Deletion of Neuronal CuZnSOD Accelerates Age-Associated Muscle Mitochondria and Calcium Handling Dysfunction That Is Independent of Denervation and Precedes Sarcopenia" International Journal of Molecular Sciences 22, no. 19: 10735. https://doi.org/10.3390/ijms221910735
APA StyleSu, Y., Claflin, D. R., Huang, M., Davis, C. S., Macpherson, P. C. D., Richardson, A., Van Remmen, H., & Brooks, S. V. (2021). Deletion of Neuronal CuZnSOD Accelerates Age-Associated Muscle Mitochondria and Calcium Handling Dysfunction That Is Independent of Denervation and Precedes Sarcopenia. International Journal of Molecular Sciences, 22(19), 10735. https://doi.org/10.3390/ijms221910735