
Charting the N-Terminal Acetylome: A Comprehensive Map of Human NatA Substrates


Abstract
:1. Introduction
2. Results
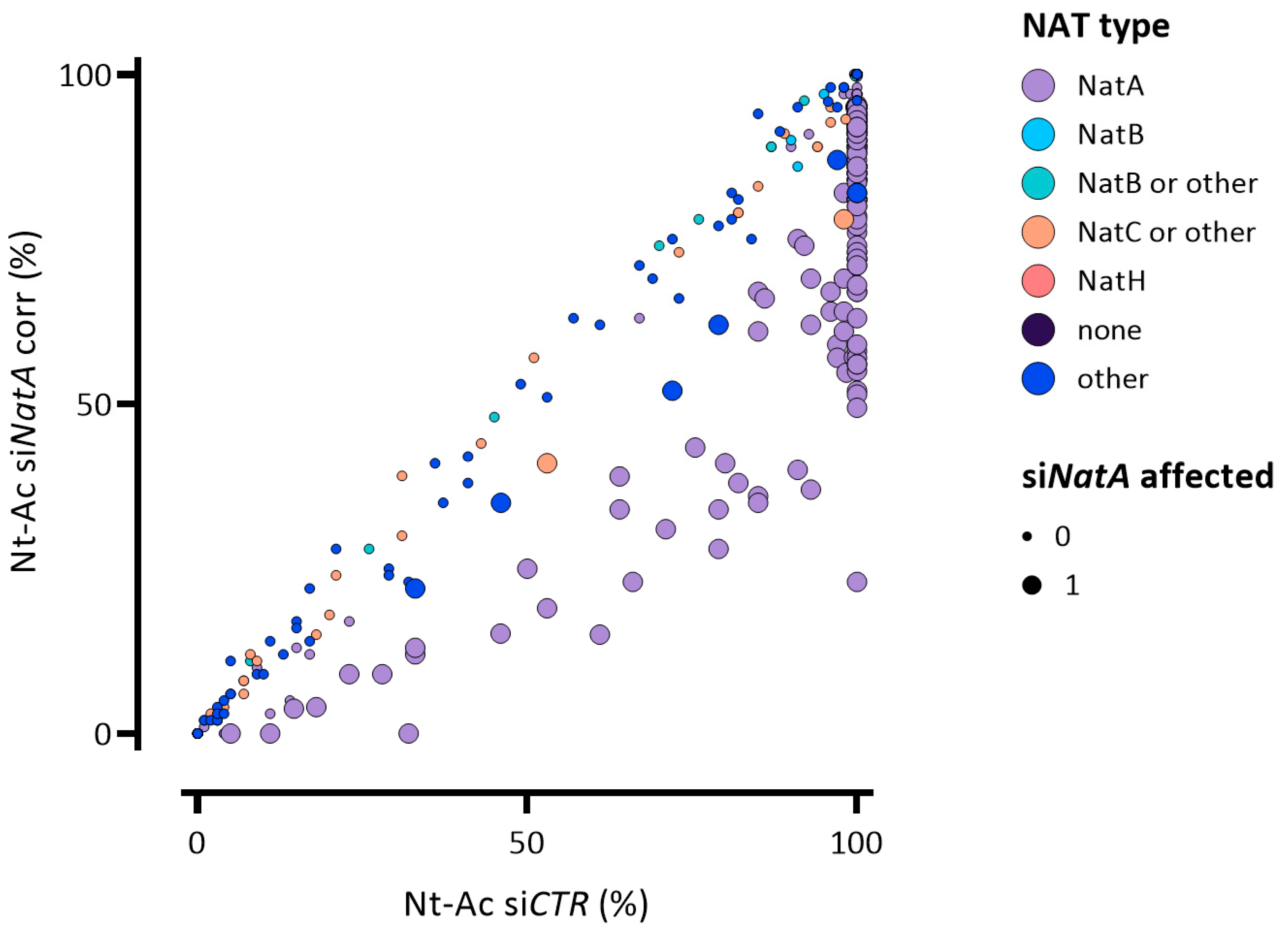
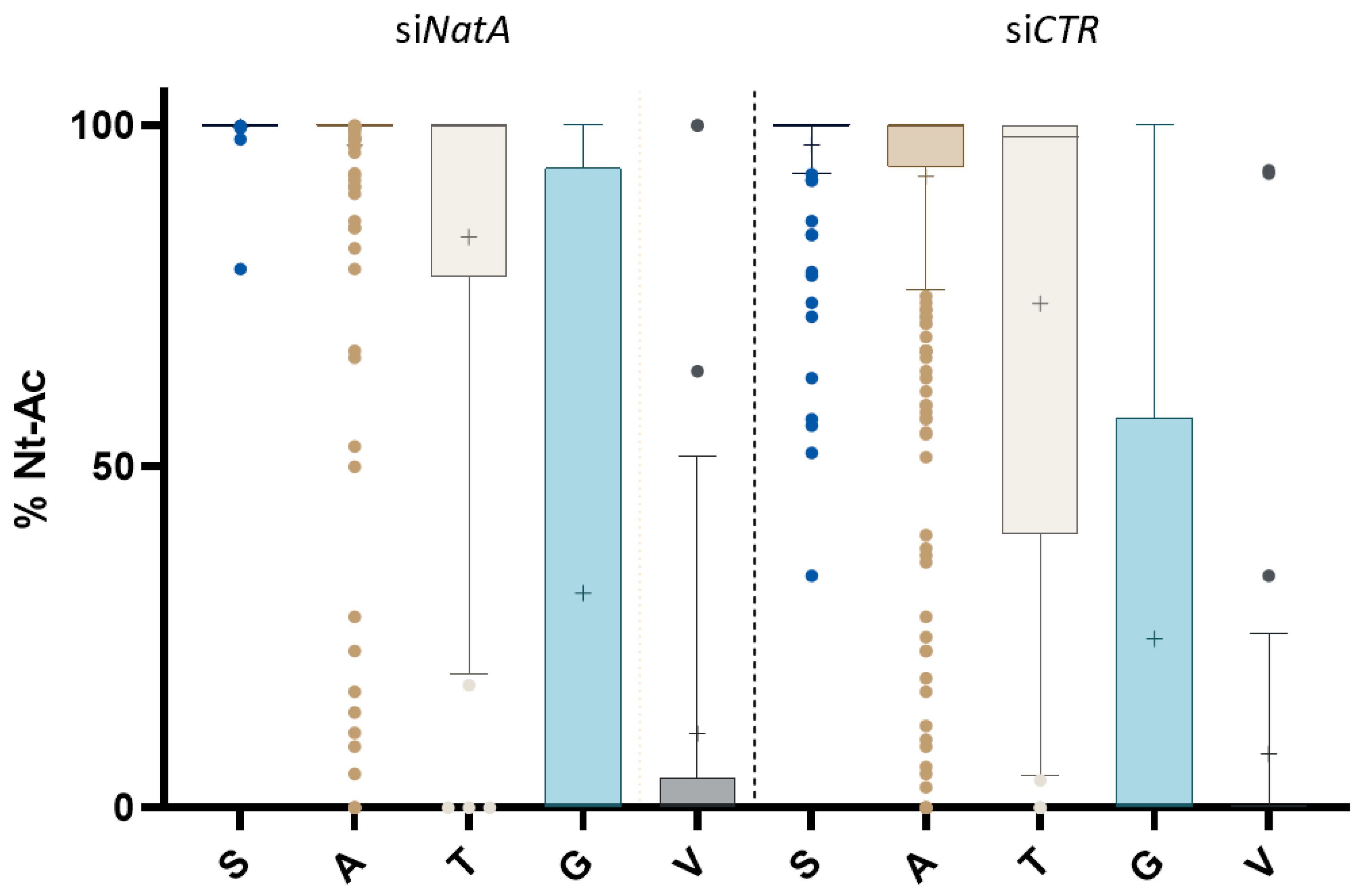
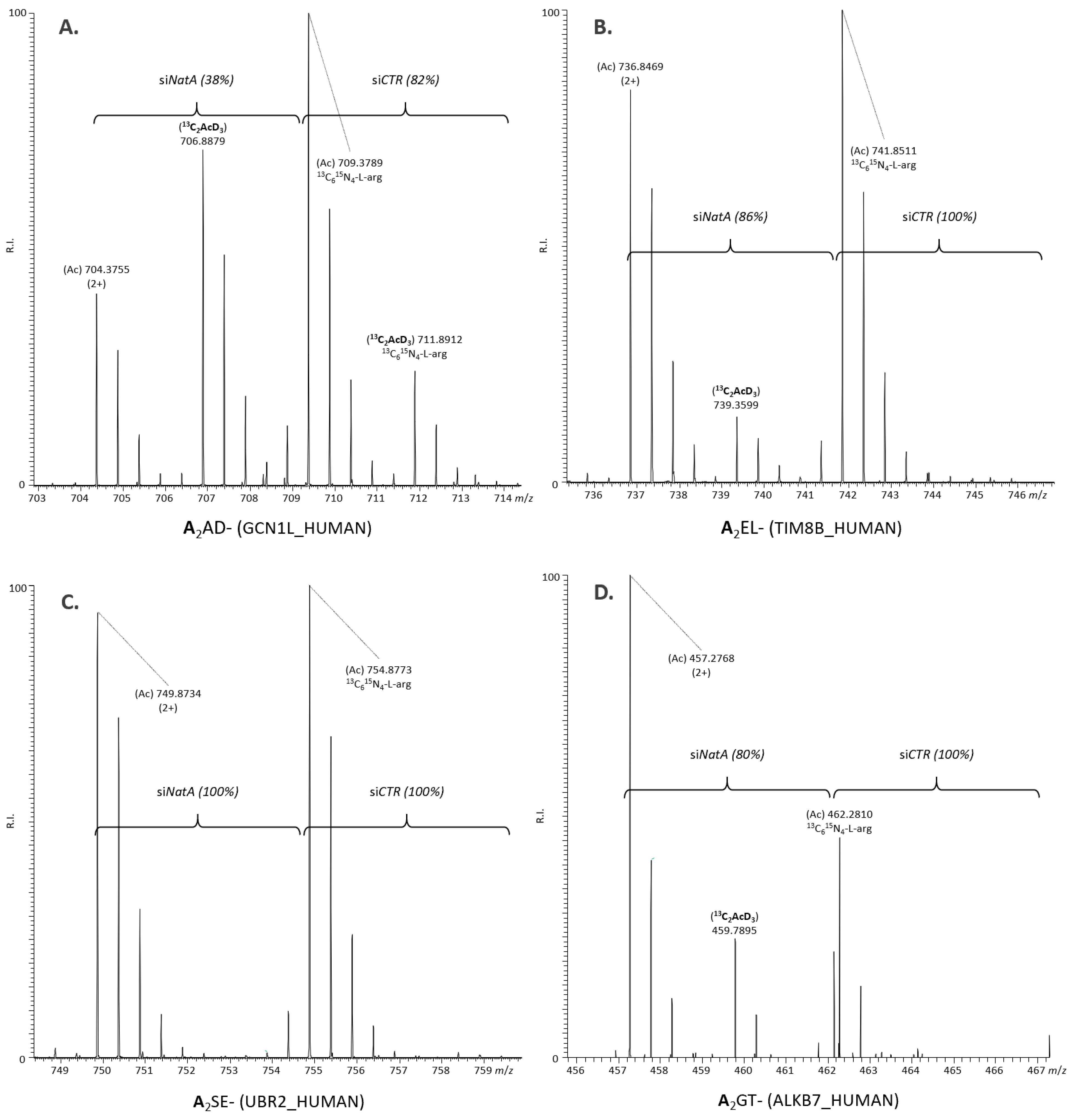
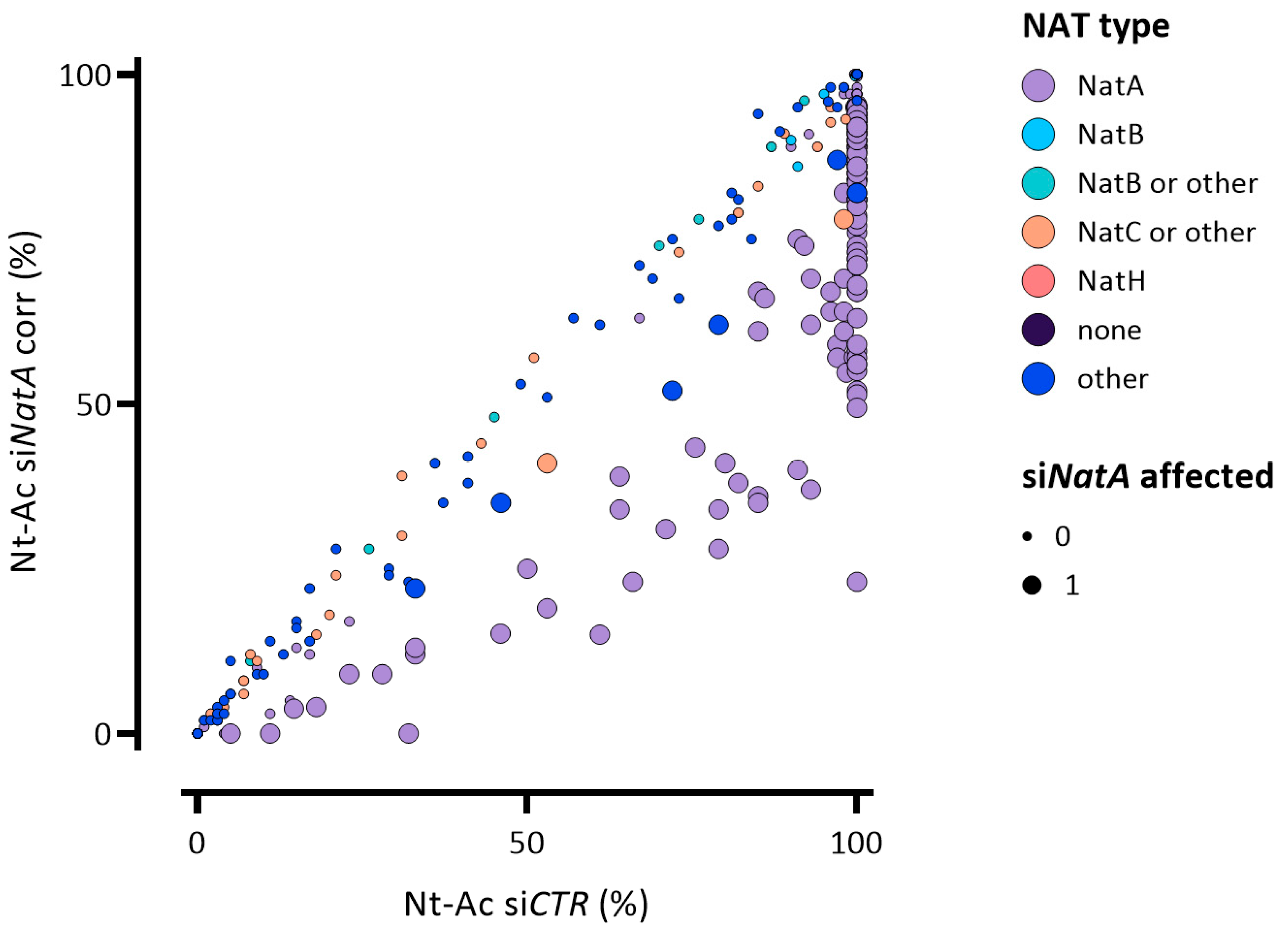
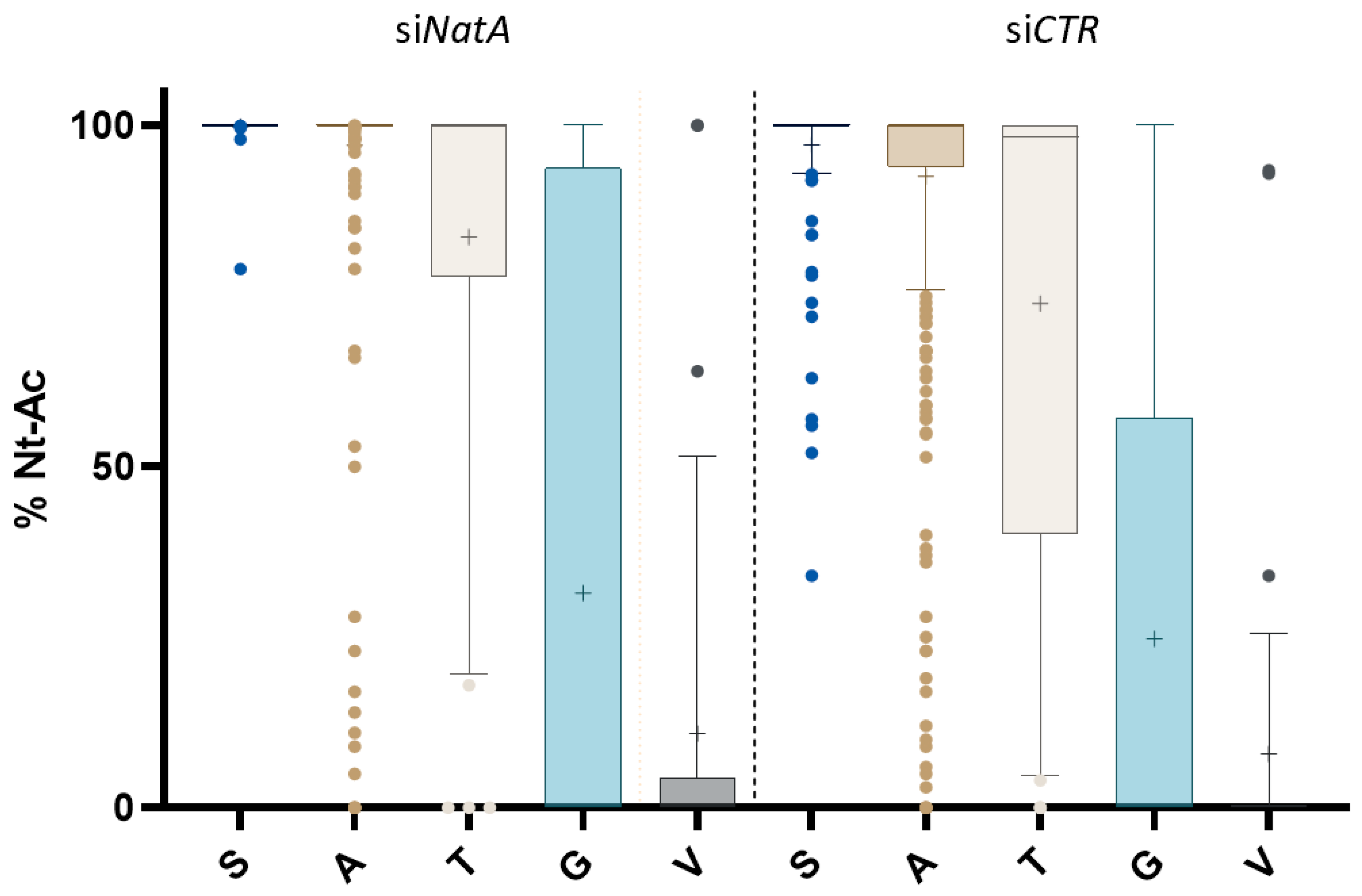
2.1. N-Terminal Proteomics to Explore the Human Nt-Acetylome upon Pronounced NatA Knockdown
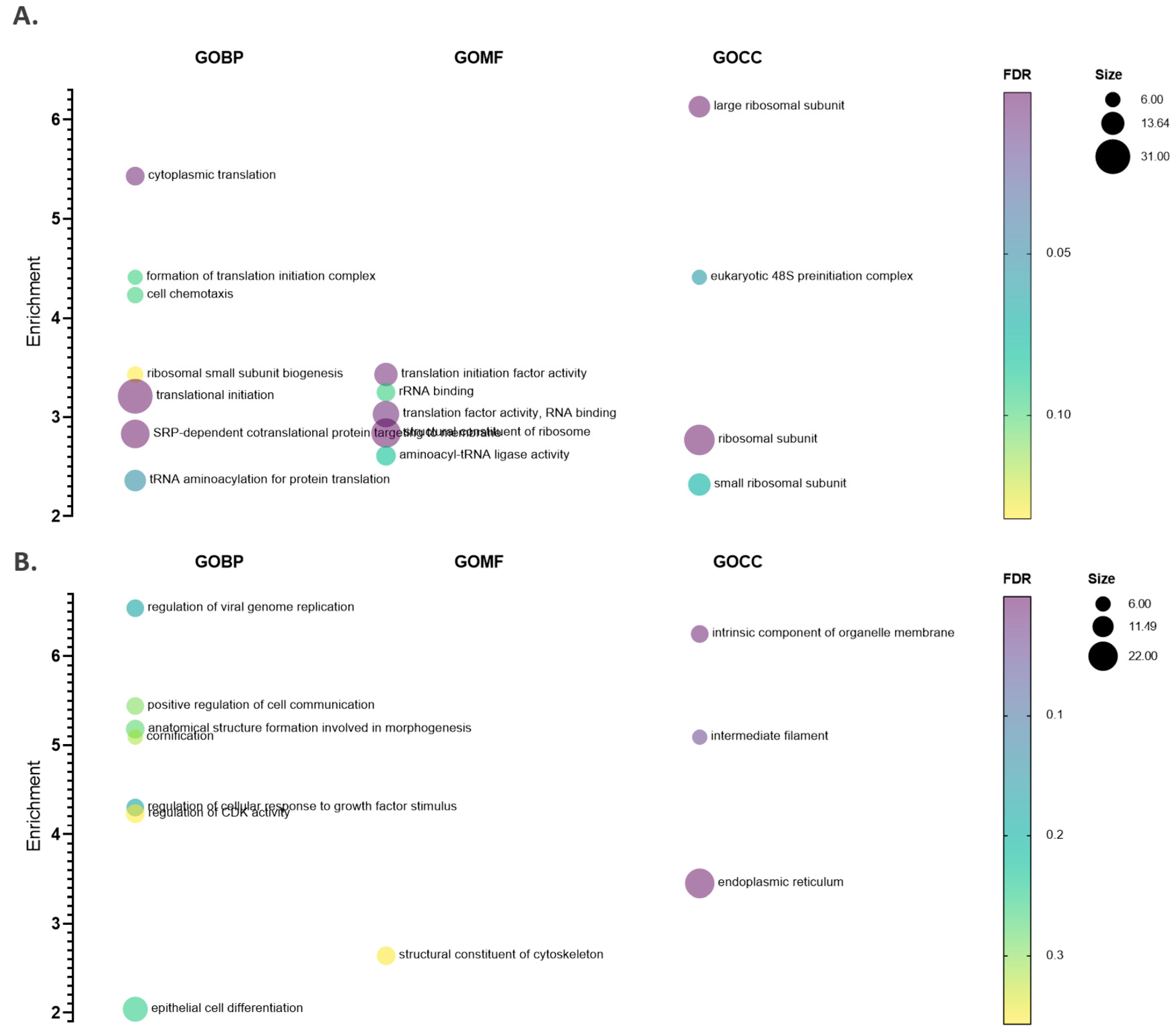
2.2. hNatA Affects Translation, Lipid Metabolism and Regulation of Cyclin-Dependent Protein Kinase Activity
3. Discussion
4. Materials and Methods
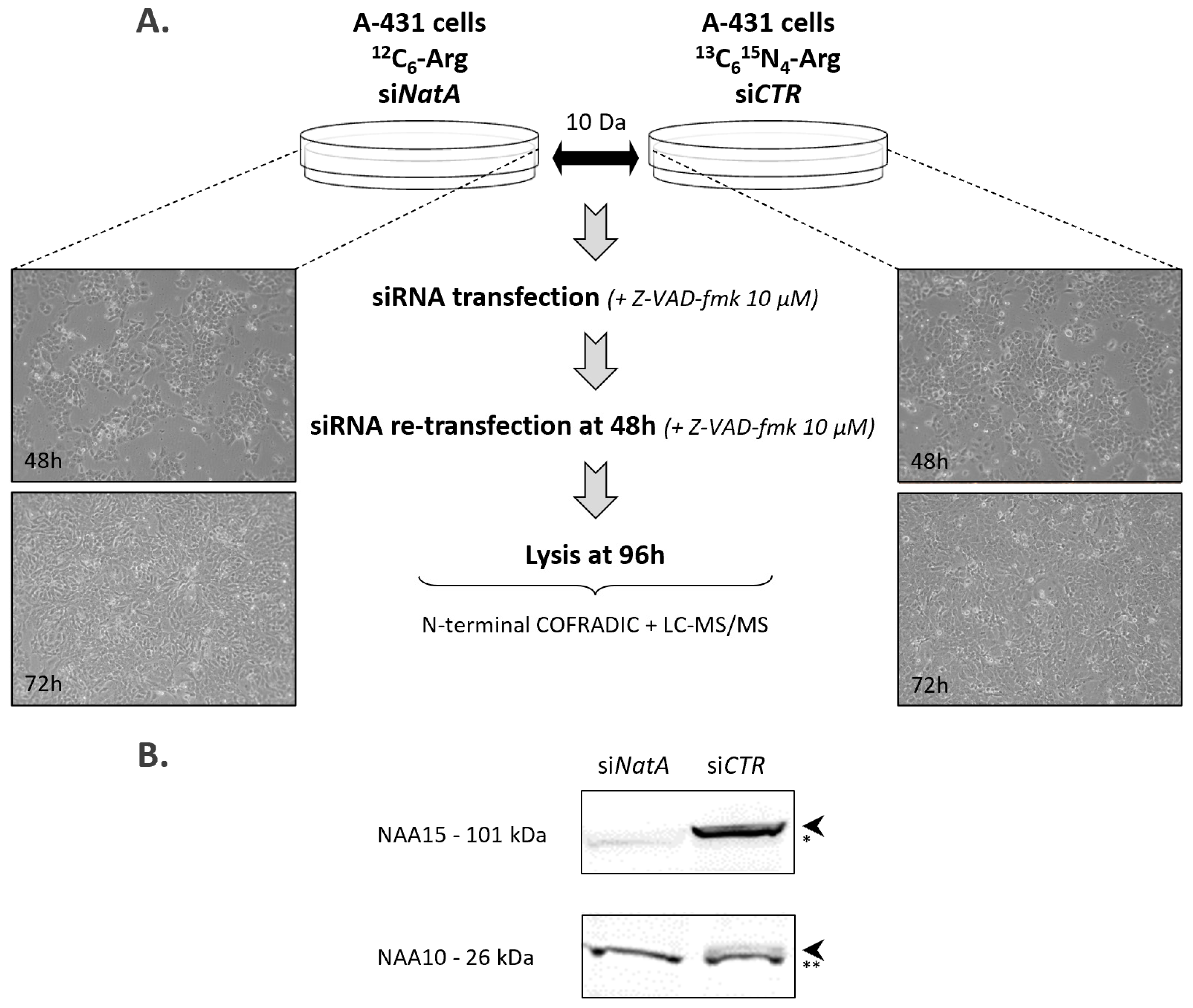
4.1. Cell Culture
4.2. siRNA Transfection
4.3. N-Terminal Proteomics
4.4. LC-MS/MS Analysis and Data Analysis
4.5. SDS-PAGE and Immunoblotting
Supplementary Materials
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Damme, P.; Hole, K.; Pimenta-Marques, A.; Helsens, K.; Vandekerckhove, J.; Martinho, R.G.; Gevaert, K.; Arnesen, T. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 2011, 7, e1002169. [Google Scholar] [CrossRef] [Green Version]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawron, D.; Ndah, E.; Gevaert, K.; Van Damme, P. Positional proteomics reveals differences in N-terminal proteoform stability. Mol. Syst. Biol. 2016, 12, 858. [Google Scholar] [CrossRef]
- Deng, S.; Marmorstein, R. Protein N-Terminal Acetylation: Structural Basis, Mechanism, Versatility, and Regulation. Trends. Biochem. Sci. 2021, 46, 15–27. [Google Scholar] [CrossRef]
- Aksnes, H.; Ree, R.; Arnesen, T. Co-translational, Post-translational, and Non-catalytic Roles of N-Terminal Acetyltransferases. Mol. Cell 2019, 73, 1097–1114. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Gottlieb, L.; Pan, B.; Supplee, J.; Wei, X.; Petersson, E.J.; Marmorstein, R. Molecular mechanism of N-terminal acetylation by the ternary NatC complex. Structure 2021. [Google Scholar] [CrossRef]
- Jonckheere, V.; Van Damme, P. N-Terminal Acetyltransferase Naa40p Whereabouts Put into N-Terminal Proteoform Perspective. Int. J. Mol. Sci. 2021, 22, 3690. [Google Scholar] [CrossRef]
- Hollebeke, J.; Van Damme, P.; Gevaert, K. N-terminal acetylation and other functions of Nalpha-acetyltransferases. Biol. Chem. 2012, 393, 291–298. [Google Scholar] [CrossRef]
- Dorfel, M.J.; Lyon, G.J. The biological functions of Naa10—From amino-terminal acetylation to human disease. Gene 2015, 567, 103–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linster, E.; Wirtz, M. N-terminal acetylation: An essential protein modification emerges as an important regulator of stress responses. J. Exp. Bot. 2018, 69, 4555–4568. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.N.; Kweon, H.Y.; Oh, G.T. N-alpha-acetyltransferase 10 (NAA10) in development: The role of NAA10. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [PubMed]
- Giglione, C.; Boularot, A.; Meinnel, T. Protein N-terminal methionine excision. Cell. Mol. Life Sci. 2004, 61, 1455–1474. [Google Scholar] [CrossRef] [PubMed]
- Giglione, C.; Fieulaine, S.; Meinnel, T. N-terminal protein modifications: Bringing back into play the ribosome. Biochimie 2015, 114, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Hole, K.; Gevaert, K.; Arnesen, T. N-terminal acetylome analysis reveals the specificity of Naa50 (Nat5) and suggests a kinetic competition between N-terminal acetyltransferases and methionine aminopeptidases. Proteomics 2015, 15, 2436–2446. [Google Scholar] [CrossRef]
- Polevoda, B.; Sherman, F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 2003, 325, 595–622. [Google Scholar] [CrossRef]
- Park, E.C.; Szostak, J.W. ARD1 and NAT1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO J. 1992, 11, 2087–2093. [Google Scholar] [CrossRef]
- Mullen, J.R.; Kayne, P.S.; Moerschell, R.P.; Tsunasawa, S.; Gribskov, M.; Colavito-Shepanski, M.; Grunstein, M.; Sherman, F.; Sternglanz, R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989, 8, 2067–2075. [Google Scholar] [CrossRef]
- Arnesen, T.; Anderson, D.; Baldersheim, C.; Lanotte, M.; Varhaug, J.E.; Lillehaug, J.R. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 2005, 386, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Knorr, A.G.; Schmidt, C.; Tesina, P.; Berninghausen, O.; Becker, T.; Beatrix, B.; Beckmann, R. Ribosome-NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat. Struct. Mol. Biol. 2019, 26, 35–39. [Google Scholar] [CrossRef]
- Magin, R.S.; Deng, S.; Zhang, H.; Cooperman, B.; Marmorstein, R. Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLoS ONE 2017, 12, e0186278. [Google Scholar] [CrossRef] [Green Version]
- Liszczak, G.; Goldberg, J.M.; Foyn, H.; Petersson, E.J.; Arnesen, T.; Marmorstein, R. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat. Struct. Mol. Biol. 2013, 20, 1098–1105. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; McTiernan, N.; Wei, X.; Arnesen, T.; Marmorstein, R. Molecular basis for N-terminal acetylation by human NatE and its modulation by HYPK. Nat. Commun. 2020, 11, 818. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, L.; Marmorstein, R. Structure of Human NatA and Its Regulation by the Huntingtin Interacting Protein HYPK. Structure 2018, 26, 925–935.e928. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Evjenth, R.; Foyn, H.; Demeyer, K.; De Bock, P.J.; Lillehaug, J.R.; Vandekerckhove, J.; Arnesen, T.; Gevaert, K. Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of N(alpha)-acetyltransferases and point to hNaa10p as the post-translational actin N(alpha)-acetyltransferase. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.; Chu, C.W.; Kong, X.; Yokomori, K.; Zou, H. The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. J. Cell Biol. 2007, 177, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Liszczak, G.; Marmorstein, R. Implications for the evolution of eukaryotic amino-terminal acetyltransferase (NAT) enzymes from the structure of an archaeal ortholog. Proc. Natl. Acad. Sci. USA 2013, 110, 14652–14657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.; Magin, R.S.; Wei, X.; Pan, B.; Petersson, E.J.; Marmorstein, R. Structure and Mechanism of Acetylation by the N-Terminal Dual Enzyme NatA/Naa50 Complex. Structure 2019, 27, 1057–1070.e1054. [Google Scholar] [CrossRef] [PubMed]
- Rathore, O.S.; Faustino, A.; Prudencio, P.; Van Damme, P.; Cox, C.J.; Martinho, R.G. Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Sci. Rep. 2016, 6, 21304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evjenth, R.; Hole, K.; Karlsen, O.A.; Ziegler, M.; Arnesen, T.; Lillehaug, J.R. Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J. Biol. Chem. 2009, 284, 31122–31129. [Google Scholar] [CrossRef] [Green Version]
- Arnesen, T.; Starheim, K.K.; Van Damme, P.; Evjenth, R.; Dinh, H.; Betts, M.J.; Ryningen, A.; Vandekerckhove, J.; Gevaert, K.; Anderson, D. The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol. Cell. Biol. 2010, 30, 1898–1909. [Google Scholar] [CrossRef] [Green Version]
- Rope, A.F.; Wang, K.; Evjenth, R.; Xing, J.; Johnston, J.J.; Swensen, J.J.; Johnson, W.E.; Moore, B.; Huff, C.D.; Bird, L.M.; et al. Using VAAST to Identify an X-Linked Disorder Resulting in Lethality in Male Infants Due to N-Terminal Acetyltransferase Deficiency. Am. J. Hum. Genet. 2011. [Google Scholar] [CrossRef] [Green Version]
- Myklebust, L.M.; Van Damme, P.; Stove, S.I.; Dorfel, M.J.; Abboud, A.; Kalvik, T.V.; Grauffel, C.; Jonckheere, V.; Wu, Y.; Swensen, J.; et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum. Mol. Genet. 2015, 24, 1956–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Damme, P.; Stove, S.I.; Glomnes, N.; Gevaert, K.; Arnesen, T. A Saccharomyces cerevisiae model reveals in vivo functional impairment of the Ogden syndrome N-terminal acetyltransferase Naa10S37P mutant. Mol. Cell. Proteom. 2014. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Lyon, G.J. NAA10-related syndrome. Exp. Mol. Med. 2018, 50, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Betts, M.J.; Pendino, F.; Liberles, D.A.; Anderson, D.; Caro, J.; Kong, X.; Varhaug, J.E.; Lillehaug, J.R. Characterization of hARD2, a processed hARD1 gene duplicate, encoding a human protein N-alpha-acetyltransferase. BMC Biochem. 2006, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Kweon, H.Y.; Lee, M.N.; Dorfel, M.; Seo, S.; Gottlieb, L.; PaPazyan, T.; McTiernan, N.; Ree, R.; Bolton, D.; Garcia, A.; et al. Naa12 compensates for Naa10 in mice in the amino-terminal acetylation pathway. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.; Tai, W.; Morton, S.; Impens, F.; Van Damme, P.; Van Haver, D.; Timmerman, E.; Venturini, G.; Zhang, K.; Jang, M.Y.; et al. Mechanisms of Congenital Heart Disease Caused by NAA15 Haploinsufficiency. Circ. Res. 2021, 128, 1156–1169. [Google Scholar] [CrossRef]
- Arnesen, T.; Gromyko, D.; Kagabo, D.; Betts, M.J.; Starheim, K.K.; Varhaug, J.E.; Anderson, D.; Lillehaug, J.R. A novel human NatA Nalpha-terminal acetyltransferase complex: hNaa16p-hNaa10p (hNat2-hArd1). BMC Biochem. 2009, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Arnesen, T.; Gromyko, D.; Pendino, F.; Ryningen, A.; Varhaug, J.E.; Lillehaug, J.R. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-alpha-acetyltransferase complex. Oncogene 2006, 25, 4350–4360. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Arnesen, T.; Gevaert, K. Protein alpha-N-acetylation studied by N-terminomics. FEBS J. 2011, 278, 3822–3834. [Google Scholar] [CrossRef]
- Van Damme, P.; Arnesen, T.; Ruttens, B.; Gevaert, K. In-gel N-acetylation for the quantification of the degree of protein In Vivo N-terminal acetylation. Methods Mol. Biol. 2013, 981, 115–126. [Google Scholar]
- Van Damme, P.; Lasa, M.; Polevoda, B.; Gazquez, C.; Elosegui-Artola, A.; Kim, D.S.; De Juan-Pardo, E.; Demeyer, K.; Hole, K.; Larrea, E.; et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl. Acad. Sci. USA 2012, 109, 12449–12454. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Kalvik, T.V.; Starheim, K.K.; Jonckheere, V.; Myklebust, L.M.; Menschaert, G.; Varhaug, J.E.; Gevaert, K.; Arnesen, T. A Role for Human N-alpha Acetyltransferase 30 (Naa30) in Maintaining Mitochondrial Integrity. Mol. Cell. Proteom. 2016, 15, 3361–3372. [Google Scholar] [CrossRef] [Green Version]
- Frottin, F.; Martinez, A.; Peynot, P.; Mitra, S.; Holz, R.C.; Giglione, C.; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteom. 2006, 5, 2336–2349. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, V.; Fijalkowska, D.; Van Damme, P. Omics Assisted N-terminal Proteoform and Protein Expression Profiling On Methionine Aminopeptidase 1 (MetAP1) Deletion. Mol. Cell. Proteom. 2018, 17, 694–708. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Gawron, D.; Van Criekinge, W.; Menschaert, G. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Mol. Cell. Proteom. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetze, S.; Qeli, E.; Mosimann, C.; Staes, A.; Gerrits, B.; Roschitzki, B.; Mohanty, S.; Niederer, E.M.; Laczko, E.; Timmerman, E.; et al. Identification and functional characterization of N-terminally acetylated proteins in Drosophila melanogaster. PLoS Biol. 2009, 7, e1000236. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, R.A.; Brickey, W.W.; Walker, K.W. N-terminal processing: The methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci. 1998, 23, 263–267. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomen, V.A.; Majek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Staes, A.; Impens, F.; Van Damme, P.; Ruttens, B.; Goethals, M.; Demol, H.; Timmerman, E.; Vandekerckhove, J.; Gevaert, K. Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat. Protoc. 2011, 6, 1130–1141. [Google Scholar] [CrossRef]
- Van Damme, P.; Van Damme, J.; Demol, H.; Staes, A.; Vandekerckhove, J.; Gevaert, K. A review of COFRADIC techniques targeting protein N-terminal acetylation. BMC Proc. 2009, 3 (Suppl. S6), S6. [Google Scholar] [CrossRef] [Green Version]
- Willems, P.; Ndah, E.; Jonckheere, V.; Stael, S.; Sticker, A.; Martens, L.; Van Breusegem, F.; Gevaert, K.; Van Damme, P. N-terminal Proteomics Assisted Profiling of the Unexplored Translation Initiation Landscape in Arabidopsis thaliana. Mol. Cell. Proteom. 2017, 16, 1064–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drazic, A.; Aksnes, H.; Marie, M.; Boczkowska, M.; Varland, S.; Timmerman, E.; Foyn, H.; Glomnes, N.; Rebowski, G.; Impens, F.; et al. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc. Natl. Acad. Sci. USA 2018, 115, 4399–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Damme, P.; Martens, L.; Van Damme, J.; Hugelier, K.; Staes, A.; Vandekerckhove, J.; Gevaert, K. Caspase-specific and nonspecific In Vivo protein processing during Fas-induced apoptosis. Nat. Methods 2005, 2, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Helsens, K.; Colaert, N.; Barsnes, H.; Muth, T.; Flikka, K.; Staes, A.; Timmerman, E.; Wortelkamp, S.; Sickmann, A.; Vandekerckhove, J.; et al. ms_lims, a simple yet powerful open source laboratory information management system for MS-driven proteomics. Proteomics 2010, 10, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Anderson, D.; Torsvik, J.; Halseth, H.B.; Varhaug, J.E.; Lillehaug, J.R. Cloning and characterization of hNAT5/hSAN: An evolutionarily conserved component of the NatA protein N-alpha-acetyltransferase complex. Gene 2006, 371, 291–295. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NatA Substrate Type (Excluding XP- N-Termini) 1 | Identified | NTA Determined | Full + Partial NTA (%) | siNatA Affected (% of Total/Putative Substrates) |
|---|---|---|---|---|
| A- | 479 | 458 | 454 (99%) | 116 (25%, 26%) |
| C- | 1 | 1 | 1 (100%) | 0 (0%) |
| G- | 43 | 38 | 14 (37%) | 8 (21%, 57%) |
| S- | 191 | 172 | 172 (100%) | 26 (15%) |
| T- | 45 | 40 | 37 (93%) | 16 (40%, 43%) |
| V- | 37 | 33 | 9 (27%) | 7 (21%, 78%) |
| Total | 796 | 742 | 687 (80%) | 173 (23%,25%) |
| Setup | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| siNatA | siCTR | |||||||||
| NAT Type | M-Starting | P1’ (P2’) | Free | Partial NTA | Fully NTA | Total | Free | Partial NTA | Fully NTA | Total |
| NatA | 0 | A | 4 | 134 | 320 | 458 | 4 | 25 | 429 | 458 |
| C | 1 | 1 | 1 | 1 | ||||||
| G | 26 | 6 | 6 | 38 | 24 | 6 | 8 | 38 | ||
| S | 30 | 142 | 172 | 1 | 171 | 172 | ||||
| T | 3 | 17 | 20 | 40 | 3 | 9 | 28 | 40 | ||
| V | 26 | 7 | 0 | 33 | 24 | 7 | 2 | 33 | ||
| Total | 59 | 194 | 489 | 742 | 55 | 48 | 639 | 742 | ||
| Other (iMet-retained NatA-type N-termini) | 1 | A | 2 | 10 | 12 | 2 | 10 | 12 | ||
| G | 2 | 2 | 2 | 6 | 2 | 2 | 2 | 6 | ||
| S | 1 | 1 | 3 | 5 | 1 | 1 | 3 | 5 | ||
| T | 1 | 10 | 16 | 27 | 1 | 8 | 18 | 27 | ||
| V | 2 | 12 | 11 | 25 | 2 | 12 | 11 | 25 | ||
| Total | 6 | 27 | 42 | 75 | 6 | 25 | 44 | 75 | ||
| NatB | 1 | D | 1 | 101 | 102 | 1 | 101 | 102 | ||
| E | 3 | 186 | 189 | 3 | 186 | 189 | ||||
| N | 3 | 44 | 47 | 3 | 44 | 47 | ||||
| Total | 7 | 331 | 338 | 7 | 331 | 338 | ||||
| NatB or other | 1 | Q | 1 | 4 | 14 | 19 | 5 | 14 | 19 | |
| Total | 1 | 4 | 14 | 19 | 0 | 5 | 14 | 19 | ||
| NatC or other | 1 | F | 2 | 2 | 14 | 18 | 3 | 1 | 14 | 18 |
| I | 2 | 6 | 5 | 13 | 2 | 6 | 5 | 13 | ||
| L | 4 | 15 | 25 | 44 | 3 | 14 | 27 | 44 | ||
| Y | 1 | 3 | 6 | 10 | 1 | 3 | 6 | 10 | ||
| Total | 9 | 26 | 50 | 85 | 9 | 24 | 52 | 85 | ||
| NatF or other | 1 | K | 19 | 20 | 8 | 47 | 18 | 21 | 8 | 47 |
| M | 1 | 12 | 13 | 1 | 12 | 13 | ||||
| Total | 19 | 21 | 20 | 60 | 18 | 22 | 20 | 60 | ||
| None | 0 | P | 62 | 62 | 62 | 62 | ||||
| 0 | X(P) | 15 | 15 | 15 | 15 | |||||
| 1 | P | 4 | 4 | 4 | 4 | |||||
| Total | 81 | 0 | 0 | 81 | 81 | 0 | 0 | 81 | ||
| NatH | 1 | D | 1 | 1 | 1 | 1 | ||||
| Total | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | ||
| Grand Total | 175 | 279 | 947 | 1401 | 169 | 131 | 1101 | 1401 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Damme, P. Charting the N-Terminal Acetylome: A Comprehensive Map of Human NatA Substrates. Int. J. Mol. Sci. 2021, 22, 10692. https://doi.org/10.3390/ijms221910692
Van Damme P. Charting the N-Terminal Acetylome: A Comprehensive Map of Human NatA Substrates. International Journal of Molecular Sciences. 2021; 22(19):10692. https://doi.org/10.3390/ijms221910692
Chicago/Turabian StyleVan Damme, Petra. 2021. "Charting the N-Terminal Acetylome: A Comprehensive Map of Human NatA Substrates" International Journal of Molecular Sciences 22, no. 19: 10692. https://doi.org/10.3390/ijms221910692
APA StyleVan Damme, P. (2021). Charting the N-Terminal Acetylome: A Comprehensive Map of Human NatA Substrates. International Journal of Molecular Sciences, 22(19), 10692. https://doi.org/10.3390/ijms221910692