
Hydroxylation of the Acetyltransferase NAA10 Trp38 Is Not an Enzyme-Switch in Human Cells

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
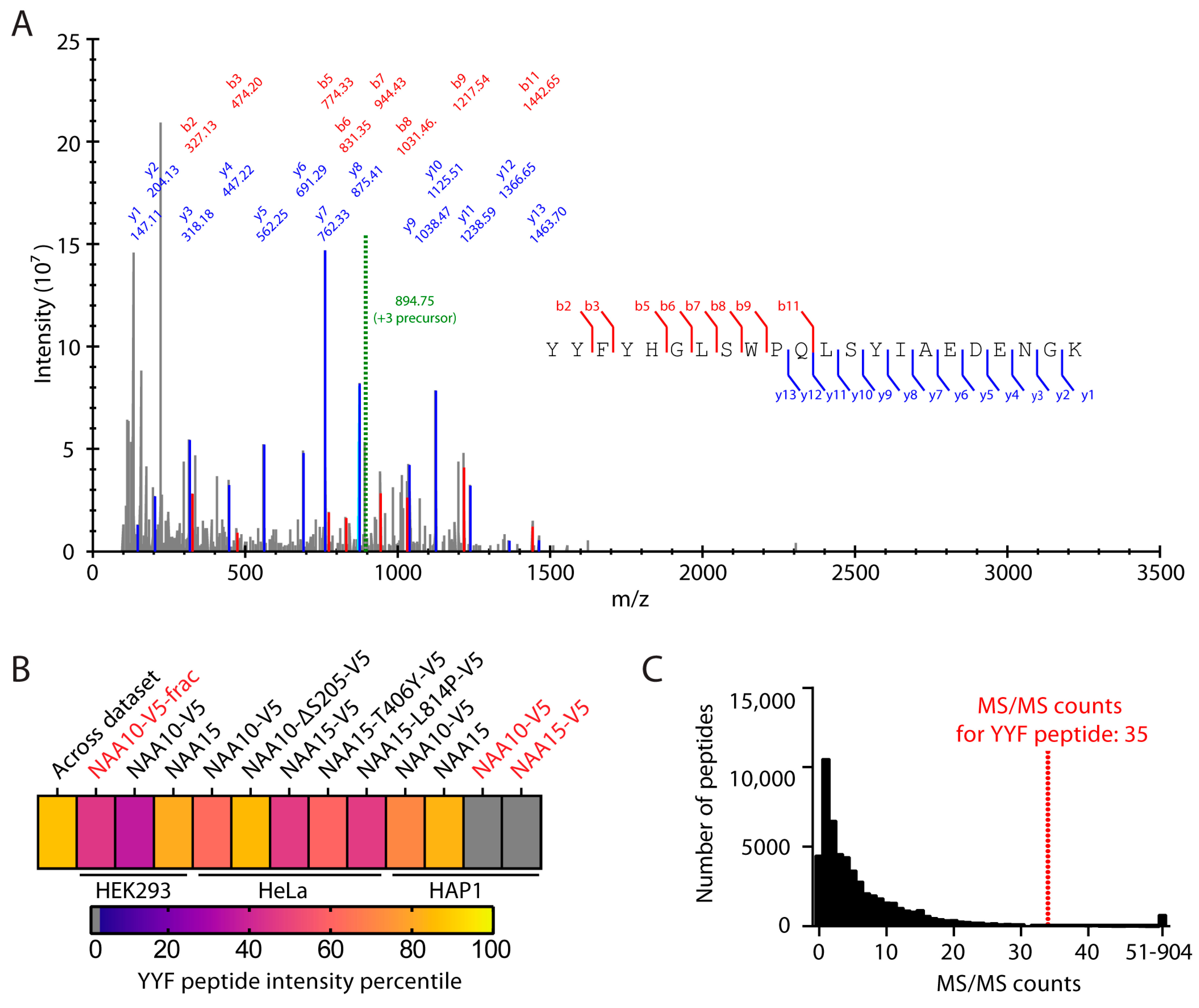
2.1. NAA10 Is Not Hydroxylated at Trp38
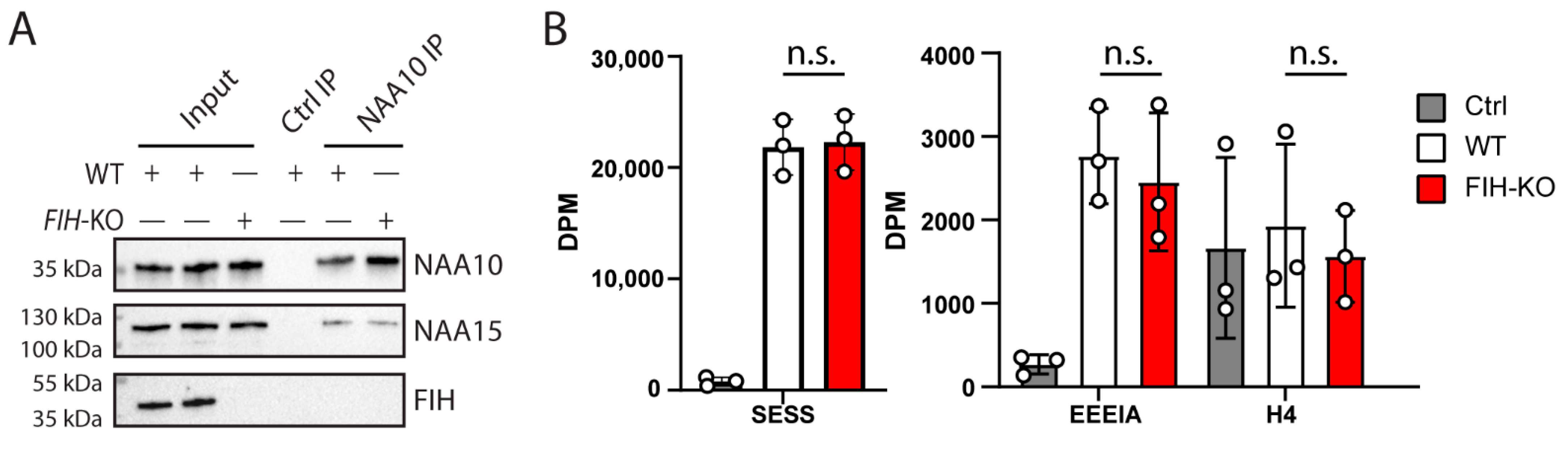
2.2. NAA10 Does Not Interact with FIH
2.3. FIH Does Not Influence the Activity of NAA10
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Culture and Transfection
4.3. Immunoprecipitation for Mass Spectrometry
4.4. Sample Preparation for Mass Spectrometry
4.5. Western Blotting
4.6. 14C Acetylation Assays
4.7. LC/MS
4.8. LC/MS Data Analysis and Processing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aksnes, H.; Ree, R.; Arnesen, T. Co-translational, Post-translational, and Non-catalytic Roles of N-Terminal Acetyltransferases. Mol. Cell 2019, 73, 1097–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trexler, A.J.; Rhoades, E. N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, B.; Tong, X.; Li, D.; Hu, Y.; He, W.; Zhao, C.; Hu, R.; Li, X.; Shao, Y.; Liu, C.; et al. N-Terminal Acetylation Preserves α-Synuclein from Oligomerization by Blocking Intermolecular Hydrogen Bonds. ACS Chem. Neurosci. 2017, 8, 2145–2151. [Google Scholar] [CrossRef]
- Arnesen, T.; Starheim, K.K.; Van Damme, P.; Evjenth, R.; Dinh, H.; Betts, M.J.; Ryningen, A.; Gevaert, K.; Anderson, D. The Chaperone-Like Protein HYPK Acts Together with NatA in Cotranslational N-Terminal Acetylation and Prevention of Huntingtin Aggregation. Mol. Cell. Biol. 2010, 30, 1898–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, W.M.; Mannakee, B.K.; Gutenkunst, R.N.; Serio, T.R. Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat. Commun. 2014, 5, 4383. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.-S.; Shemorry, A.; Varshavsky, A. N-Terminal Acetylation of Cellular Proteins Creates Specific Degradation Signals. Science 2010, 327, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Shemorry, A.; Hwang, C.; Varshavsky, A. Control of Protein Quality and Stoichiometries by N-Terminal Acetylation and the N-End Rule Pathway. Mol. Cell 2013, 50, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Goetze, S.; Qeli, E.; Mosimann, C.; Staes, A.; Gerrits, B.; Roschitzki, B.; Mohanty, S.; Niederer, E.M.; Laczko, E.; Timmerman, E.; et al. Identification and Functional Characterization of N- Terminally Acetylated Proteins in Drosophila melanogaster. PLoS Biol. 2009, 7, e1000236. [Google Scholar] [CrossRef] [Green Version]
- Myklebust, L.M.; Van Damme, P.; Støve, S.I.; Dörfel, M.J.; Abboud, A.; Kalvik, T.V.; Grauffel, C.; Jonckheere, V.; Wu, Y.; Swensen, J.; et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum. Mol. Genet. 2015, 24, 1956–1976. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-E.; Kim, J.-M.; Seok, O.-H.; Cho, H.; Wadas, B.; Kim, S.-Y.; Varshavsky, A.; Hwang, C.-S. Control of mammalian G protein signaling by N-terminal acetylation and the N-end rule pathway. Science 2015, 347, 1249–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.C.; Monda, J.K.; Bennett, E.J.; Harper, J.W.; Schulman, B.A. N-Terminal Acetylation Acts as an Avidity Enhancer Within an Interconnected Multiprotein Complex. Science 2011, 334, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monda, J.K.; Scott, D.C.; Miller, D.J.; Lydeard, J.; King, D.; Harper, J.W.; Bennett, E.J.; Schulman, B.A. Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure 2013, 21, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaudo, N.; Fernández, I.S.; Mclaughlin, S.H.; Peak-chew, S.Y.; Rhodes, D.; Martino, F. The N-terminal acetylation of Sir3 stabilizes its binding to the nucleosome core particle. Nat. Struct. Mol. Biol. 2013, 20, 1119–1121. [Google Scholar] [CrossRef]
- Yang, D.; Fang, Q.; Wang, M.; Ren, R.; Wang, H.; He, M.; Sun, Y.; Yang, N.; Xu, R.M. Nα-acetylated Sir3 stabilizes the conformation of a nucleosome-binding loop in the BAH domain. Nat. Struct. Mol. Biol. 2013, 20, 1116–1118. [Google Scholar] [CrossRef]
- Behnia, R.; Panic, B.; Whyte, J.R.C.; Munro, S. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 2004, 6, 405–413. [Google Scholar] [CrossRef]
- Setty, S.R.G.; Strochlic, T.I.; Tong, A.H.Y.; Boone, C.; Burd, C.G. Golgi targeting of Arf-like GTPase Arl3p requires its Nα-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 2004, 6, 414–419. [Google Scholar] [CrossRef]
- Forte, G.M.A.; Pool, M.R.; Stirling, C.J. N-Terminal Acetylation Inhibits Protein Targeting to the Endoplasmic Reticulum. PLoS Biol. 2011, 9, e1001073. [Google Scholar] [CrossRef] [Green Version]
- Ree, R.; Varland, S.; Arnesen, T. Spotlight on protein N-terminal acetylation. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.; Chen, A.; Deng, Y.; Liu, M.; Wang, Y.; Wang, Y.; Nie, M.; Wang, C.; Ding, H.; Yao, B.; et al. NatD promotes lung cancer progression by preventing histone H4 serine phosphorylation to activate Slug expression. Nat. Commun. 2017, 8, 928. [Google Scholar] [CrossRef] [PubMed]
- Schiza, V.; Molina-Serrano, D.; Kyriakou, D.; Hadjiantoniou, A.; Kirmizis, A. N-alpha-terminal Acetylation of Histone H4 Regulates Arginine Methylation and Ribosomal DNA Silencing. PLoS Genet. 2013, 9, e1003805. [Google Scholar] [CrossRef] [Green Version]
- Molina-Serrano, D.; Schiza, V.; Demosthenous, C.; Stavrou, E.; Oppelt, J.; Kyriakou, D.; Liu, W.; Zisser, G.; Bergler, H.; Dang, W.; et al. Loss of Nat4 and its associated histone H4 N-terminal acetylation mediates calorie restriction-induced longevity. EMBO Rep. 2016, 17, 1829–1843. [Google Scholar] [CrossRef]
- Gautschi, M.; Just, S.; Mun, A.; Ross, S.; Rücknagel, P.; Dubaquié, Y.; Ehrenhofer-murray, A.; Rospert, S.; Gautschi, M.; Mun, A.; et al. The Yeast Nα-Acetyltransferase NatA Is Quantitatively Anchored to the Ribosome and Interacts with Nascent Polypeptides. Mol. Cell. Biol. 2003, 23, 7403–7414. [Google Scholar] [CrossRef] [Green Version]
- Polevoda, B.; Brown, S.; Cardillo, T.S.; Rigby, S.; Sherman, F. Yeast Nα-Terminal Acetyltransferases Are Associated With Ribosomes. J. Cell. Biochem. 2008, 103, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Starheim, K.K.; Arnesen, T.; Gromyko, D.; Ryningen, A.; Varhaug, J.E.; Lillehaug, J.R. Identification of the human N α -acetyltransferase complex B (hNatB): A complex important for cell-cycle progression. Biochem. J. 2008, 415, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hole, K.; van Damme, P.; Dalva, M.; Aksnes, H.; Glomnes, N.; Varhaug, J.E.; Lillehaug, J.R.; Gevaert, K.; Arnesen, T. The human N-Alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS ONE 2011, 6, e24713. [Google Scholar] [CrossRef] [Green Version]
- Polevoda, B.; Hoskins, J.; Sherman, F. Properties of Nat4, an Nα-Acetyltransferase of Saccharomyces cerevisiae That Modifies N Termini of Histones H2A and H4. Mol. Cell. Biol. 2009, 29, 2913–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, T.V.; Bienvenut, W.V.; Linster, E.; Feldman-Salit, A.; Jung, V.A.; Meinnel, T.; Hell, R.; Giglione, C.; Wirtz, M. Molecular identification and functional characterization of the first N-alpha-acetyltransferase in plastids by global acetylome profiling. Proteomics 2015, 15, 2426–2435. [Google Scholar] [CrossRef] [Green Version]
- Bienvenut, W.V.; Brünje, A.; Boyer, J.; Mühlenbeck, J.S.; Bernal, G.; Lassowskat, I.; Dian, C.; Linster, E.; Dinh, T.V.; Koskela, M.M.; et al. Dual lysine and N-terminal acetyltransferases reveal the complexity underpinning protein acetylation. Mol. Syst. Biol. 2020, 16, e9464. [Google Scholar] [CrossRef] [PubMed]
- Aksnes, H.; Van Damme, P.; Goris, M.; Starheim, K.K.; Marie, M.; Støve, S.I.; Hoel, C.; Kalvik, T.V.; Hole, K.; Glomnes, N.; et al. An organellar nα-acetyltransferase, naa60, acetylates cytosolic N termini of transmembrane proteins and maintains golgi integrity. Cell Rep. 2015, 10, 1362–1374. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Hole, K.; Pimenta-Marques, A.; Helsens, K.; Vandekerckhove, J.; Martinho, R.G.; Gevaert, K.; Arnesen, T. NatF Contributes to an Evolutionary Shift in Protein N-Terminal Acetylation and Is Important for Normal Chromosome Segregation. PLoS Genet. 2011, 7, e1002169. [Google Scholar] [CrossRef] [Green Version]
- Drazic, A.; Aksnes, H.; Marie, M.; Boczkowska, M.; Varland, S.; Timmerman, E.; Foyn, H.; Glomnes, N.; Rebowski, G.; Impens, F.; et al. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc. Natl. Acad. Sci. USA 2018, 115, 4399–4404. [Google Scholar] [CrossRef] [Green Version]
- Wiame, E.; Tahay, G.; Tyteca, D.; Vertommen, D.; Stroobant, V.; Bommer, G.T.; Van Schaftingen, E. NAT6 acetylates the N-terminus of different forms of actin. FEBS J. 2018, 285, 3299–3316. [Google Scholar] [CrossRef] [Green Version]
- Goris, M.; Magin, R.S.; Foyn, H.; Myklebust, L.M.; Varland, S.; Ree, R.; Drazic, A.; Bhambra, P.; Støve, S.I.; Baumann, M.; et al. Structural determinants and cellular environment define processed actin as the sole substrate of the N-terminal acetyltransferase NAA80. Proc. Natl. Acad. Sci. USA 2018, 115, 4405–4410. [Google Scholar] [CrossRef] [Green Version]
- Rebowski, G.; Boczkowska, M.; Drazic, A.; Ree, R.; Goris, M.; Arnesen, T.; Dominguez, R. Mechanism of actin N-terminal acetylation. Sci. Adv. 2020, 6, eaay8793. [Google Scholar] [CrossRef] [Green Version]
- Ree, R.; Kind, L.; Kaziales, A.; Varland, S.; Dai, M.; Richter, K.; Drazic, A.; Arnesen, T. PFN2 and NAA80 cooperate to efficiently acetylate the N-terminus of actin. J. Biol. Chem. 2020, 295, 16713–16731. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [Green Version]
- Fluge, Ø.; Bruland, O.; Akslen, L.A.; Varhaug, J.E.; Lillehaug, J.R. NATH, a novel gene overexpressed in papillary thyroid carcinomas. Oncogene 2002, 21, 5056–5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnesen, T.; Anderson, D.; Baldersheim, C.; Lanotte, M.; Varhaug, J.E.; Lillehaug, J.R. Identification and characterization of the human ARD1–NATH protein acetyltransferase complex. Biochem. J. 2005, 386, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, J.R.; Kayne, P.S.; Moerschell, R.P.; Tsunasawa, S.; Gribskov, M.; Colavito-Shepanski, M.; Grunstein, M.; Sherman, F.; Sternglanz, R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989, 8, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, L.; Marmorstein, R. Structure of Human NatA and Its Regulation by the Huntingtin Interacting Protein HYPK. Structure 2018, 26, 925–935.e8. [Google Scholar] [CrossRef] [Green Version]
- Liszczak, G.; Goldberg, J.M.; Foyn, H.; Petersson, E.J.; Arnesen, T.; Marmorstein, R. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat. Struct. Mol. Biol. 2013, 20, 1098–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Damme, P.; Evjenth, R.; Foyn, H.; Demeyer, K.; De Bock, P.-J.; Lillehaug, J.R.; Arnesen, T.; Gevaert, K. Proteome-derived Peptide Libraries Allow Detailed Analysis of the Substrate Specificities of Nα-acetyltransferases and Point to hNaa10p as the Post-translational Actin Nα-acetyltransferase. Mol. Cell. Proteom. 2011, 10, M110.004580. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Peng, S.H.; Shen, L.; Lee, C.F.; Du, T.H.; Kang, M.L.; Xu, G.L.; Upadhyay, A.K.; Cheng, X.; Yan, Y.T.; et al. The Role of N-α-acetyltransferase 10 Protein in DNA Methylation and Genomic Imprinting. Mol. Cell 2017, 68, 89–103.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Ou, D.S.C.; Lee, S.B.; Chang, L.H.; Lin, R.K.; Li, Y.S.; Upadhyay, A.K.; Cheng, X.; Wang, Y.C.; Hsu, H.S.; et al. hNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J. Clin. Investig. 2010, 120, 2920–2930. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Park, J.W.; Chun, Y. Human Arrest Defective 1 Acetylates and Activates B -Catenin, Promoting Lung Cancer Cell Proliferation. Cancer Res. 2006, 66, 10677–10682. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.; Kim, H.-L.; Chun, Y.-S.; Shin, D.H.; Lee, K.-H.; Shin, C.S.; Lee, D.Y.; Kim, H.-H.; Lee, Z.H.; Ryoo, H.-M.; et al. NAA10 controls osteoblast differentiation and bone formation as a feedback regulator of Runx2. Nat. Commun. 2014, 5, 5176. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.H.; Yoon, H.; Chun, Y.S.; Shin, H.W.; Lee, M.N.; Oh, G.T.; Park, J.W. Arrest defective 1 regulates the oxidative stress response in human cells and mice by acetylating methionine sulfoxide reductase A. Cell Death Dis. 2014, 5, e1490. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Park, J.H.; Lee, E.J.; Vo, T.T.L.; Choi, H.; Kim, J.Y.; Jang, J.K.; Wee, H.J.; Lee, H.S.; Jang, S.H.; et al. ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat. Commun. 2016, 7, 12882. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Cha, J.H.; Park, J.H.; Jeong, C.H.; Park, Z.Y.; Lee, H.S.; Oh, S.H.; Kang, J.H.; Suh, S.W.; Kim, K.H.; et al. Arrest defective 1 autoacetylation is a critical step in its ability to stimulate cancer cell proliferation. Cancer Res. 2010, 70, 4422–4432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji Lee, E.; Hae Seo, J.; Park, J.; Thuy Lu Vo, T.; An, S.; Bae, S.-J.; Le, H.; Shin Lee, H.; Wee, H.; Lee, D.; et al. SAMHD1 acetylation enhances its deoxynucleotide triphosphohydrolase activity and promotes cancer cell proliferation. Oncotarget 2017, 8, 68517–68529. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Z.; Guo, J.; Li, Y.; Bavarva, J.H.; Qian, C.; Brahimi-Horn, M.C.; Tan, D.; Liu, W. Inactivation of androgen-induced regulator ARD1 inhibits androgen receptor acetylation and prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 3053–3058. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.H.; Chun, Y.S.; Lee, K.H.; Shin, H.W.; Park, J.W. Arrest defective-1 controls tumor cell behavior by acetylating myosin light chain kinase. PLoS ONE 2009, 4, e7451. [Google Scholar] [CrossRef] [Green Version]
- Iwai, K.; Yamanaka, K.; Kamura, T.; Minato, N.; Conaway, R.C.; Conaway, J.W.; Klausner, R.D.; Pause, A. Identification of the von Hippel-Lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 12436–12441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Lisztwan, J.; Imbert, G.; Wirbelauer, C.; Gstaiger, M.; Krek, W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999, 13, 1822–1833. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Bilton, R.; Mazure, N.; Trottier, E.; Hattab, M.; Déry, M.A.; Richard, D.E.; Pouysségur, J.; Brahimi-Horn, M.C. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF)-1α and is not induced by hypoxia or HIF. J. Biol. Chem. 2005, 280, 31132–31140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnesen, T.; Kong, X.; Evjenth, R.; Gromyko, D.; Varhaug, J.E.; Lin, Z.; Sang, N.; Caro, J.; Lillehaug, J.R. Interaction between HIF-1α (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1α. FEBS Lett. 2005, 579, 6428–6432. [Google Scholar] [CrossRef] [Green Version]
- Magin, R.S.; March, Z.M.; Marmorstein, R. The N-terminal Acetyltransferase Naa10 / ARD1 Does Not Acetylate Lysine Residues. J. Biol. Chem. 2016, 291, 5270–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, T.T.L.; Park, J.-H.; Lee, E.J.; Nguyen, Y.T.K.; Han, B.W.; Nguyen, H.T.T.; Mun, K.C.; Ha, E.; Kwon, T.K.; Kim, K.-W.; et al. Characterization of lysine acetyltransferase activity of recombinant human ARD1/NAA10. Molecules 2020, 25, 588. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Chun, Y.; Huh, J.; Park, J.W. FIH permits NAA10 to catalyze the oxygen-dependent lysyl-acetylation of HIF-1α. Redox Biol. 2018, 19, 364–374. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sørensen, K.D.; Høyer, S.; Ørntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, M.E.; Małecki, J.M.; Halabelian, L.; Nilges, B.S.; Pinto, R.; Kudithipudi, S.; Munk, S.; Davydova, E.; Zuhairi, F.R.; Arrowsmith, C.H.; et al. The dual methyltransferase METTL13 targets N terminus and Lys55 of eEF1A and modulates codon-specific translation rates. Nat. Commun. 2018, 9, 3411. [Google Scholar] [CrossRef] [PubMed]
- Schwanhüusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Mctiernan, N.; Wei, X.; Arnesen, T.; Marmorstein, R. Molecular basis for N-terminal acetylation by human NatE and its modulation by HYPK. Nat. Commun. 2020, 11, 818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drazic, A.; Arnesen, T. [14C]-Acetyl-Coenzyme A-Based In Vitro N-Terminal Acetylation Assay. In Protein Terminal Profiling. Methods in Molecular Biology; Schilling, O., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1574, ISBN 978-1-4939-6850-3. [Google Scholar]
- Rope, A.F.; Wang, K.; Evjenth, R.; Xing, J.; Johnston, J.J.; Swensen, J.J.; Johnson, W.E.; Moore, B.; Huff, C.D.; Bird, L.M.; et al. Using VAAST to Identify an X-Linked Disorder Resulting in Lethality in Male Infants Due to N-Terminal Acetyltransferase Deficiency. Am. J. Hum. Genet. 2011, 89, 28–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, J.P.; Støve, S.I.; McGorrian, C.; Galvin, J.; Blenski, M.; Dunne, A.; Ennis, S.; Brett, F.; King, M.D.; Arnesen, T.; et al. NAA10 mutation causing a novel intellectual disability syndrome with Long QT due to N-terminal acetyltransferase impairment. Sci. Rep. 2015, 5, 16022. [Google Scholar] [CrossRef] [PubMed]
- Støve, S.I.; Blenski, M.; Stray-Pedersen, A.; Wierenga, K.J.; Jhangiani, S.N.; Akdemir, Z.C.; Crawford, D.; McTiernan, N.; Myklebust, L.M.; Purcarin, G.; et al. A novel NAA10 variant with impaired acetyltransferase activity causes developmental delay, intellectual disability, and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2018, 26, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, B.; Støve, S.I.; Endele, S.; Myklebust, L.M.; Hoyer, J.; Sticht, H.; Azzarello-Burri, S.; Rauch, A.; Arnesen, T.; Reis, A. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur. J. Hum. Genet. 2015, 23, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunier, C.; Støve, S.I.; Popp, B.; Gérard, B.; Blenski, M.; AhMew, N.; de Bie, C.; Goldenberg, P.; Isidor, B.; Keren, B.; et al. Expanding the Phenotype Associated with NAA10-Related N-Terminal Acetylation Deficiency. Hum. Mutat. 2016, 37, 755–764. [Google Scholar] [CrossRef] [Green Version]
- McTiernan, N.; Støve, S.I.; Aukrust, I.; Mårli, M.T.; Myklebust, L.M.; Houge, G.; Arnesen, T. NAA10 dysfunction with normal NatA-complex activity in a girl with non-syndromic ID and a de novo NAA10 p.(V111G) variant—A case report. BMC Med. Genet. 2018, 19, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ree, R.; Geithus, A.S.; Tørring, P.M.; Sørensen, K.P.; Damkjær, M. A novel NAA10 p.(R83H) variant with impaired acetyltransferase activity identified in two boys with ID and microcephaly. BMC Med. Genet. 2019, 20, 101. [Google Scholar] [CrossRef]
- Cheng, H.; Gottlieb, L.; Marchi, E.; Kleyner, R.; Bhardwaj, P.; Rope, A.F.; Rosenheck, S.; Moutton, S.; Philippe, C.; Eyaid, W.; et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 2019, 28, 2900–2919. [Google Scholar] [CrossRef]
- Johnston, J.J.; Williamson, K.A.; Chou, C.M.; Sapp, J.C.; Ansari, M.; Chapman, H.M.; Cooper, D.N.; Dabir, T.; Dudley, J.N.; Holt, R.J.; et al. NAA10 polyadenylation signal variants cause syndromic microphthalmia. J. Med. Genet. 2019, 56, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Esmailpour, T.; Riazifar, H.; Liu, L.; Donkervoort, S.; Huang, V.H.; Madaan, S.; Shoucri, B.M.; Busch, A.; Wu, J.; Towbin, A.; et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J. Med. Genet. 2014, 51, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, I.; McTiernan, N.; Darbakk, C.; Boltshauser, E.; Ree, R.; Ebner, S.; Mayr, J.A.; Arnesen, T. Severe syndromic ID and skewed X-inactivation in a girl with NAA10 dysfunction and a novel heterozygous de novo NAA10 p.(His16Pro) variant—A case report. BMC Med. Genet. 2020, 21, 153. [Google Scholar] [CrossRef]
- McTiernan, N.; Gill, H.; Prada, C.E.; Pachajoa, H.; Lores, J.; Arnesen, T. NAA10 p.(N101K) disrupts N-terminal acetyltransferase complex NatA and is associated with developmental delay and hemihypertrophy. Eur. J. Hum. Genet. 2021, 29, 280–288. [Google Scholar] [CrossRef]
- Wu, Y.; Lyon, G.J. NAA10-related syndrome. Exp. Mol. Med. 2018, 50, 85. [Google Scholar] [CrossRef]
- Ree, R.; Myklebust, L.M.; Thiel, P.; Foyn, H.; Fladmark, K.E.; Arnesen, T. The N-terminal acetyltransferase Naa10 is essential for zebrafish development. Biosci. Rep. 2015, 35, e00249. [Google Scholar] [CrossRef]
- Wang, Y.; Mijares, M.; Gall, M.D.; Turan, T.; Javier, A.; Bornemann, D.J.; Manage, K.; Warrior, R. Drosophila variable nurse cells Encodes Arrest Defective 1 ( ARD1 ), the Catalytic Subunit of the Major N-Terminal Acetyltransferase Complex. Dev. Dyn. 2010, 239, 2813–2827. [Google Scholar] [CrossRef]
- Ingram, A.K.; Cross, G.A.M.; Horn, D. Genetic manipulation indicates that ARD1 is an essential Nα-acetyltransferase in Trypanosoma brucei. Mol. Biochem. Parasitol. 2000, 111, 309–317. [Google Scholar] [CrossRef]
- Sönnichsen, B.; Koski, L.B.; Walsh, A.; Marschall, P.; Neumann, B.; Brehm, M.; Alleaume, A.M.; Artelt, J.; Bettencourt, P.; Cassin, E.; et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature 2005, 434, 462–469. [Google Scholar] [CrossRef]
- Linster, E.; Stephan, I.; Bienvenut, W.V.; Maple-Grødem, J.; Myklebust, L.M.; Huber, M.; Reichelt, M.; Sticht, C.; Møller, S.G.; Meinnel, T.; et al. Downregulation of N-terminal acetylation triggers ABA-mediated drought responses in Arabidopsis. Nat. Commun. 2015, 6, 7640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromyko, D.; Arnesen, T.; Ryningen, A.; Varhaug, J.E.; Lillehaug, J.R. Depletion of the human Nα-terminal acetyltransferase A induces p53-dependent apoptosis and p53-independent growth inhibition. Int. J. Cancer 2010, 127, 2777–2789. [Google Scholar] [CrossRef]
- Arnesen, T. HIF1α and ARD1: Enemies, friends or neither? Nat. Rev. Cancer 2006, 6, 8–9. [Google Scholar] [CrossRef] [Green Version]
- Klont, F.; Bras, L.; Wolters, J.C.; Ongay, S.; Bischoff, R.; Halmos, G.B.; Horvatovich, P. Assessment of Sample Preparation Bias in Mass Spectrometry-Based Proteomics. Anal. Chem. 2018, 90, 5405–5413. [Google Scholar] [CrossRef] [Green Version]
- Froelich, J.M.; Reid, G.E. The origin and control of ex vivo oxidative peptide modifications prior to mass spectrometry analysis. Proteomics 2008, 8, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Kappock, T.J.; Caradonna, J.P. Pterin-dependent amino acid hydroxylases. Chem. Rev. 1996, 96, 2659–2756. [Google Scholar] [CrossRef]
- Jakobsson, M.E.; Moen, A.; Falnes, P.Ø. Correspondence: On the enzymology and significance of HSPA1 lysine methylation. Nat. Commun. 2016, 7, 11464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockman, M.E.; Lippl, K.; Tian, Y.M.; Pegg, H.B.; Figg, W.D.; Abboud, M.I.; Heilig, R.; Fischer, R.; Myllyharju, J.; Schofield, C.J.; et al. Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates. Elife 2019, 8, e46490. [Google Scholar] [CrossRef] [PubMed]
- Murray-Rust, T.A.; Oldham, N.J.; Hewitson, K.S.; Schofield, C.J. Purified recombinant hARD1 does not catalyse acetylation of Lys 532 of HIF-1α fragments in vitro. FEBS Lett. 2006, 580, 1911–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, C.C.; Cavadas, M.A.S.; Tambuwala, M.M.; Hams, E.; Rodríguez, J.; Von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.S.; Crifo, B.; Halligan, D.N.; et al. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Ge, W.; Chowdhury, R.; Claridge, T.D.W.; Kramer, H.B.; Schmierer, B.; McDonough, M.A.; Gong, L.; Kessler, B.M.; Ratcliffe, P.J.; et al. Asparagine and aspartate hydroxylation of the cytoskeletal ankyrin family is catalyzed by factor-inhibiting hypoxia-inducible factor. J. Biol. Chem. 2011, 286, 7648–7660. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Chowdhury, R.; Ge, W.; Hamed, R.B.; McDonough, M.A.; Claridge, T.D.W.; Kessler, B.M.; Cockman, M.E.; Ratcliffe, P.J.; Schofield, C.J. Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. FEBS J. 2011, 278, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockman, M.E.; Webb, J.D.; Ratcliffe, P.J. FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Ann. N. Y. Acad. Sci. 2009, 1177, 9–18. [Google Scholar] [CrossRef]
- Cockman, M.E.; Webb, J.D.; Kramer, H.B.; Kessler, B.M.; Ratcliffe, P.J. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteom. 2009, 8, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Bludau, I.; Aebersold, R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat. Rev. Mol. Cell Biol. 2020, 21, 327–340. [Google Scholar] [CrossRef]
- Rodriguez, J.; Pilkington, R.; Munoz, A.G.; Nguyen, L.K.; Rauch, N.; Kennedy, S.; Monsefi, N.; Herrero, A.; Taylor, C.T.; Kriegsheim, A. Von Substrate-Trapped Interactors of PHD3 and FIH Cluster in Distinct Signaling Pathways. Cell Rep. 2016, 14, 2745–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigl, T.B.; Kjosås, I.; Seljeseth, E.; Glomnes, N.; Aksnes, H. Efficient and crucial quality control of HAP1 cell ploidy status. Biol. Open 2020, 9, bio057174. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Kelstrup, C.D.; Bekker-Jensen, D.B.; Arrey, T.N.; Hogrebe, A.; Harder, A.; Olsen, J. V Performance Evaluation of the Q Exactive HF-X for Shotgun Proteomics. J. Proteome Res. 2018, 17, 727–738. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; MacLean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ree, R.; Krogstad, K.; McTiernan, N.; Jakobsson, M.E.; Arnesen, T. Hydroxylation of the Acetyltransferase NAA10 Trp38 Is Not an Enzyme-Switch in Human Cells. Int. J. Mol. Sci. 2021, 22, 11805. https://doi.org/10.3390/ijms222111805
Ree R, Krogstad K, McTiernan N, Jakobsson ME, Arnesen T. Hydroxylation of the Acetyltransferase NAA10 Trp38 Is Not an Enzyme-Switch in Human Cells. International Journal of Molecular Sciences. 2021; 22(21):11805. https://doi.org/10.3390/ijms222111805
Chicago/Turabian StyleRee, Rasmus, Karoline Krogstad, Nina McTiernan, Magnus E. Jakobsson, and Thomas Arnesen. 2021. "Hydroxylation of the Acetyltransferase NAA10 Trp38 Is Not an Enzyme-Switch in Human Cells" International Journal of Molecular Sciences 22, no. 21: 11805. https://doi.org/10.3390/ijms222111805
APA StyleRee, R., Krogstad, K., McTiernan, N., Jakobsson, M. E., & Arnesen, T. (2021). Hydroxylation of the Acetyltransferase NAA10 Trp38 Is Not an Enzyme-Switch in Human Cells. International Journal of Molecular Sciences, 22(21), 11805. https://doi.org/10.3390/ijms222111805