
Donepezil Regulates LPS and Aβ-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling

Abstract
:1. Introduction
2. Results
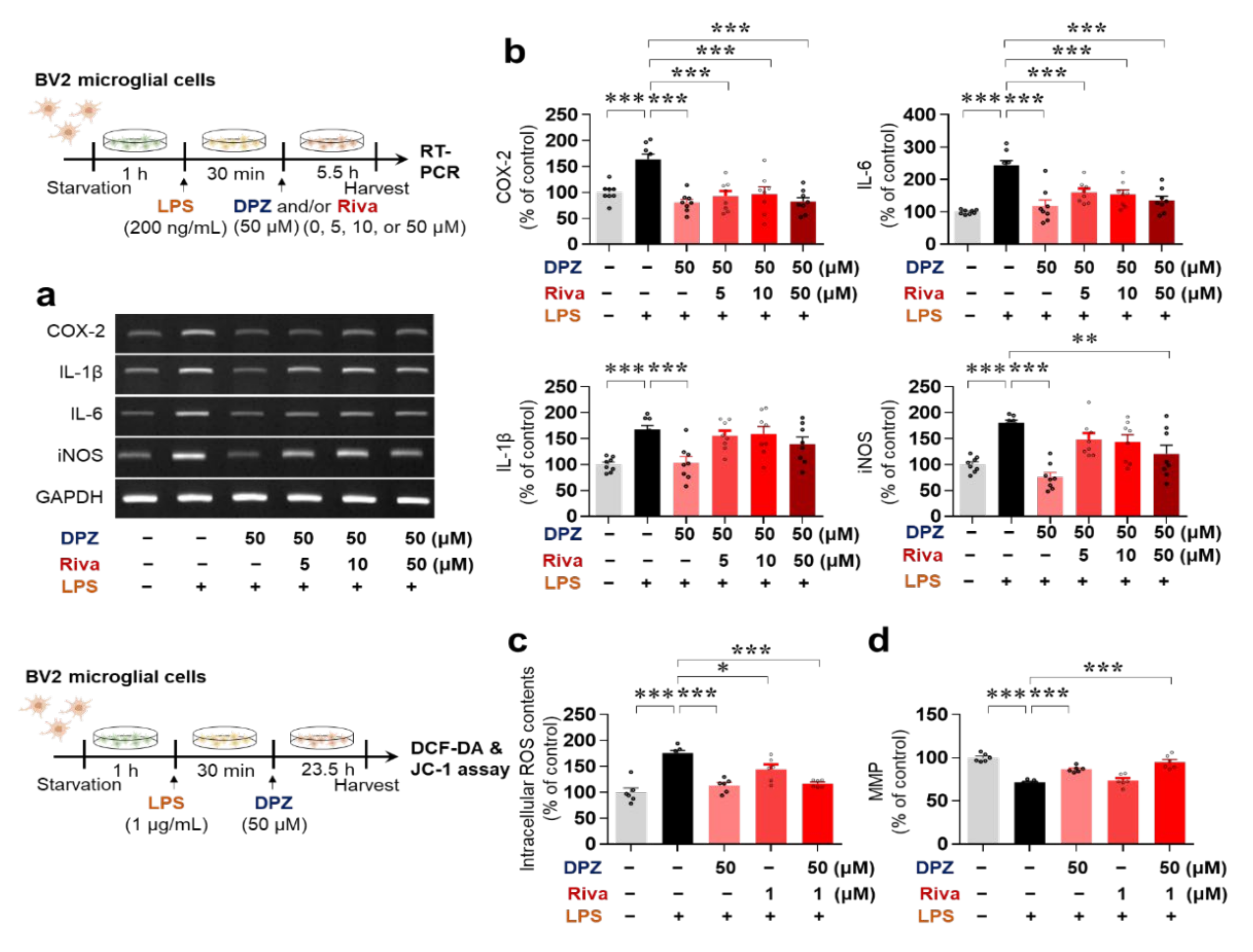
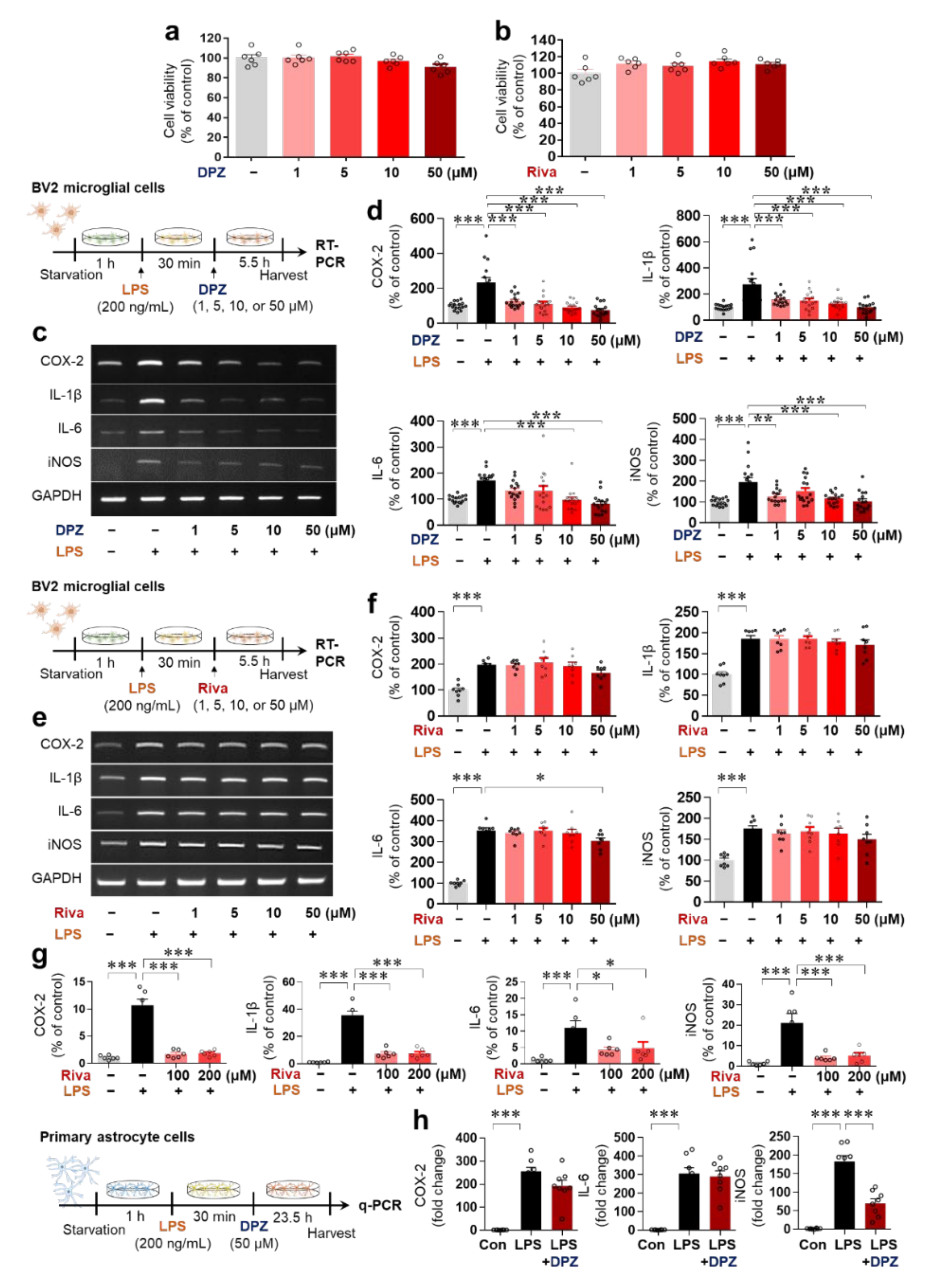
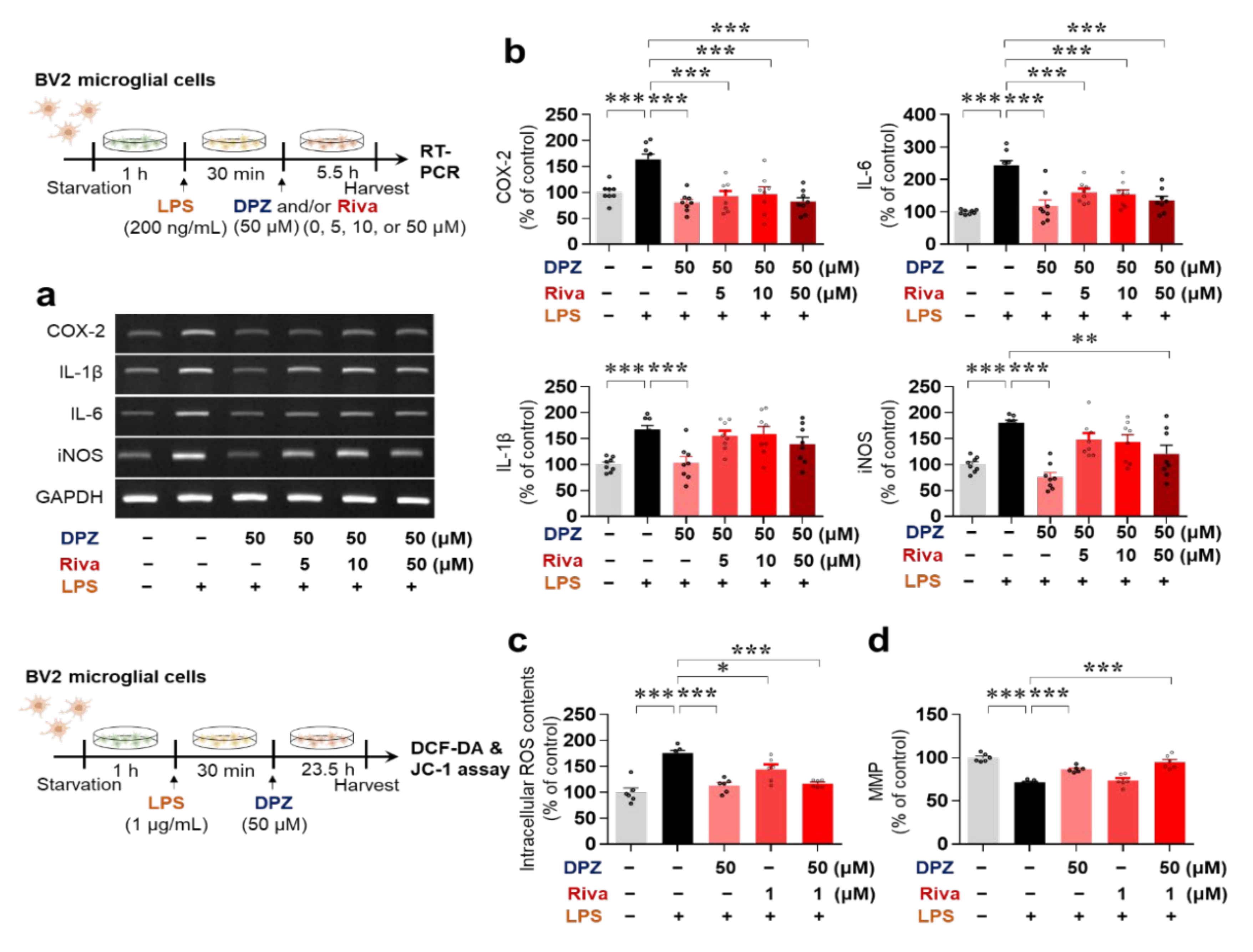
2.1. Donepezil Reduces LPS-Mediated Proinflammatory Cytokine mRNA Levels in BV2 Microglial Cells
2.2. Donepezil Suppresses Only LPS-Induced iNOS mRNA Levels in Primary Astrocytes
2.3. Donepezil and Rivastigmine Independently Modulate LPS-Induced Intracellular ROS Production and Mitochondrial Dysfunction
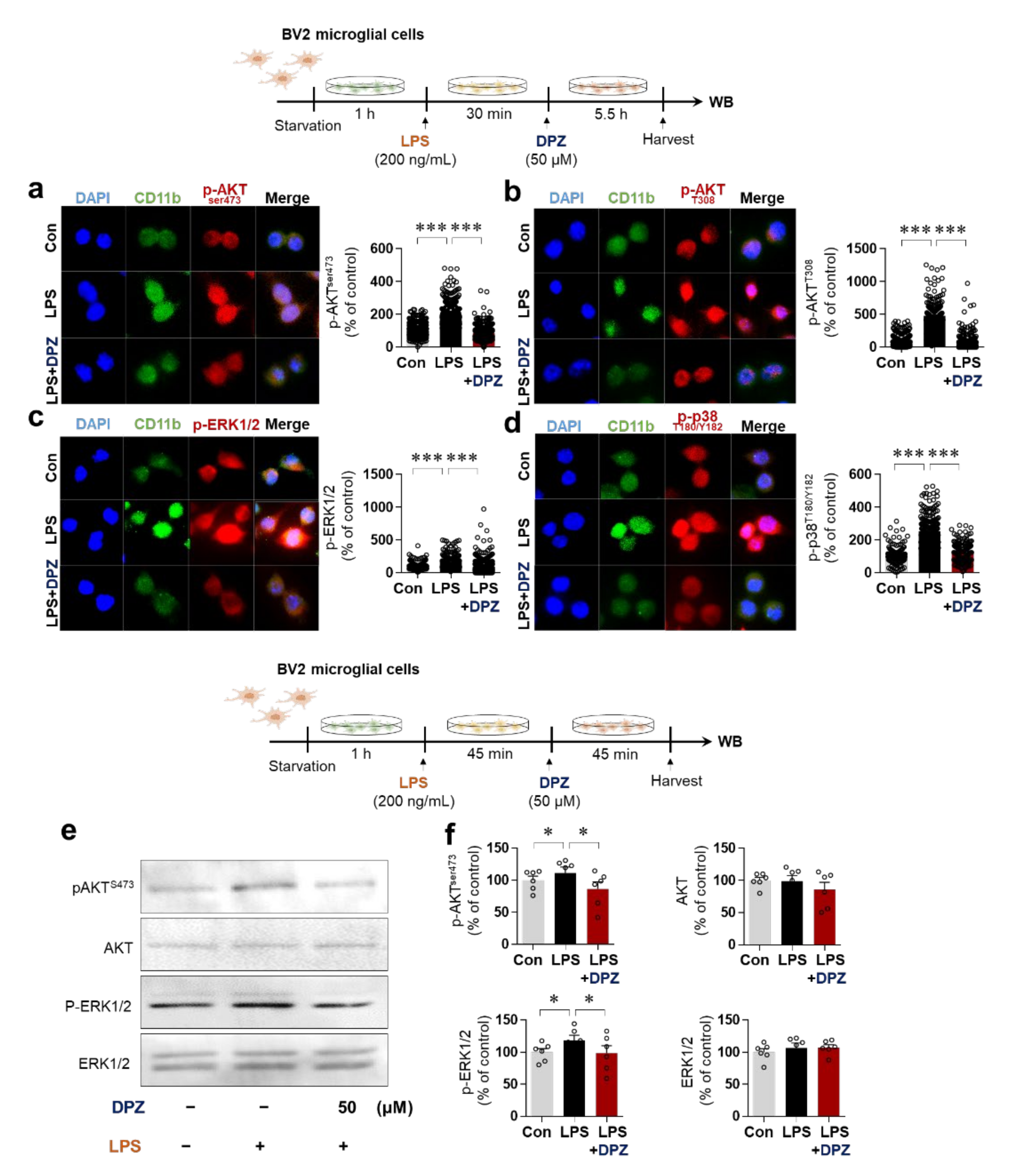
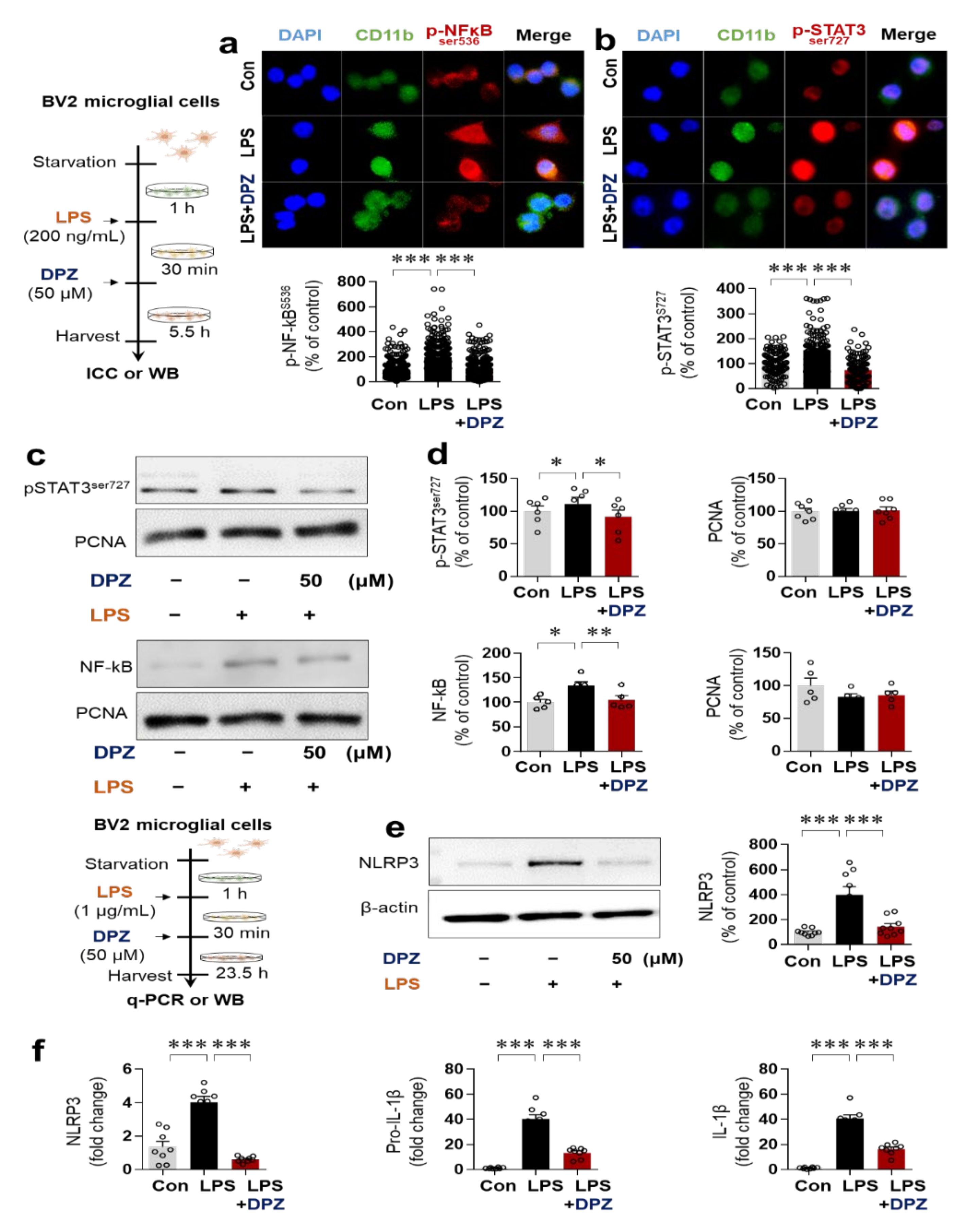
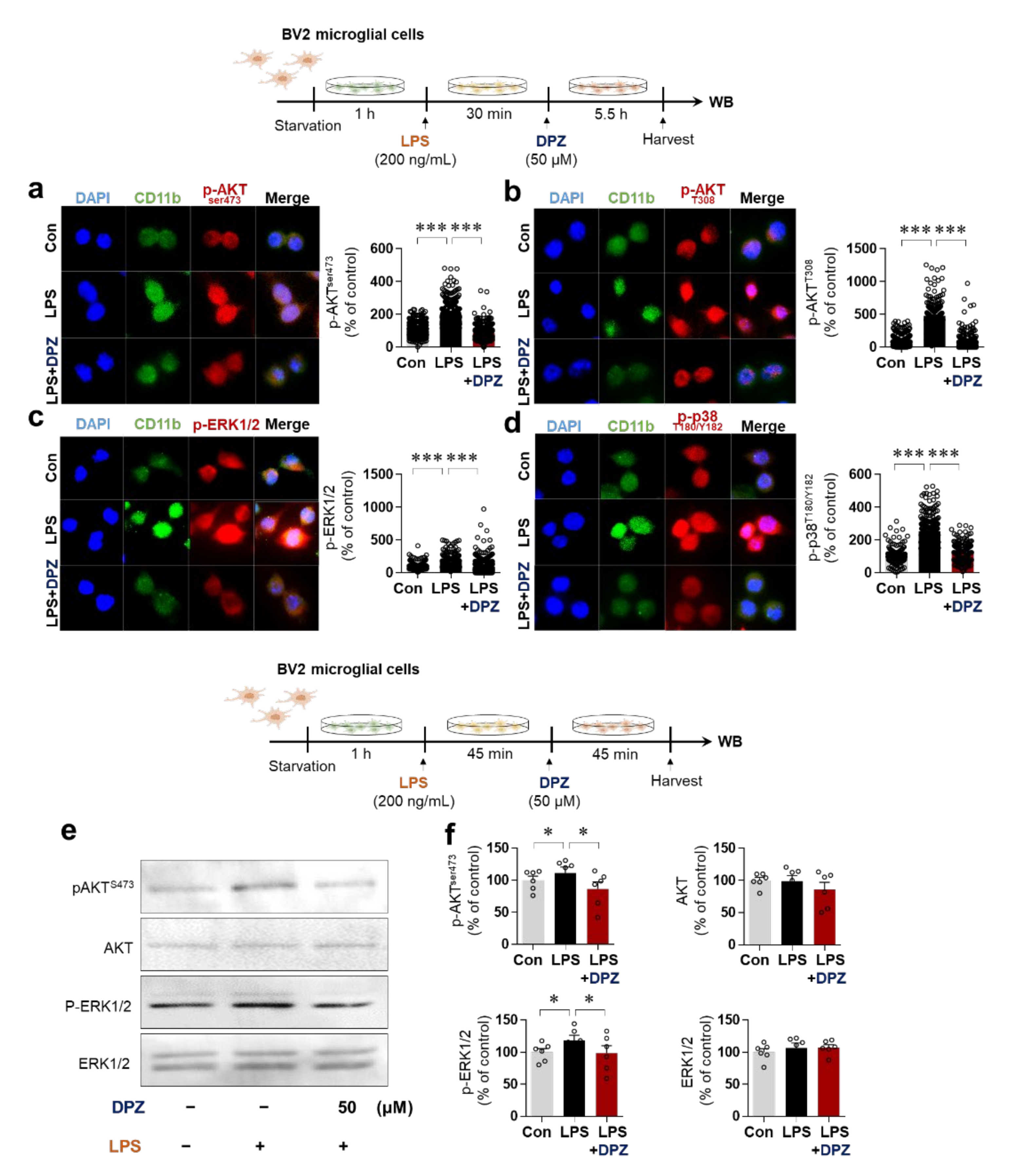
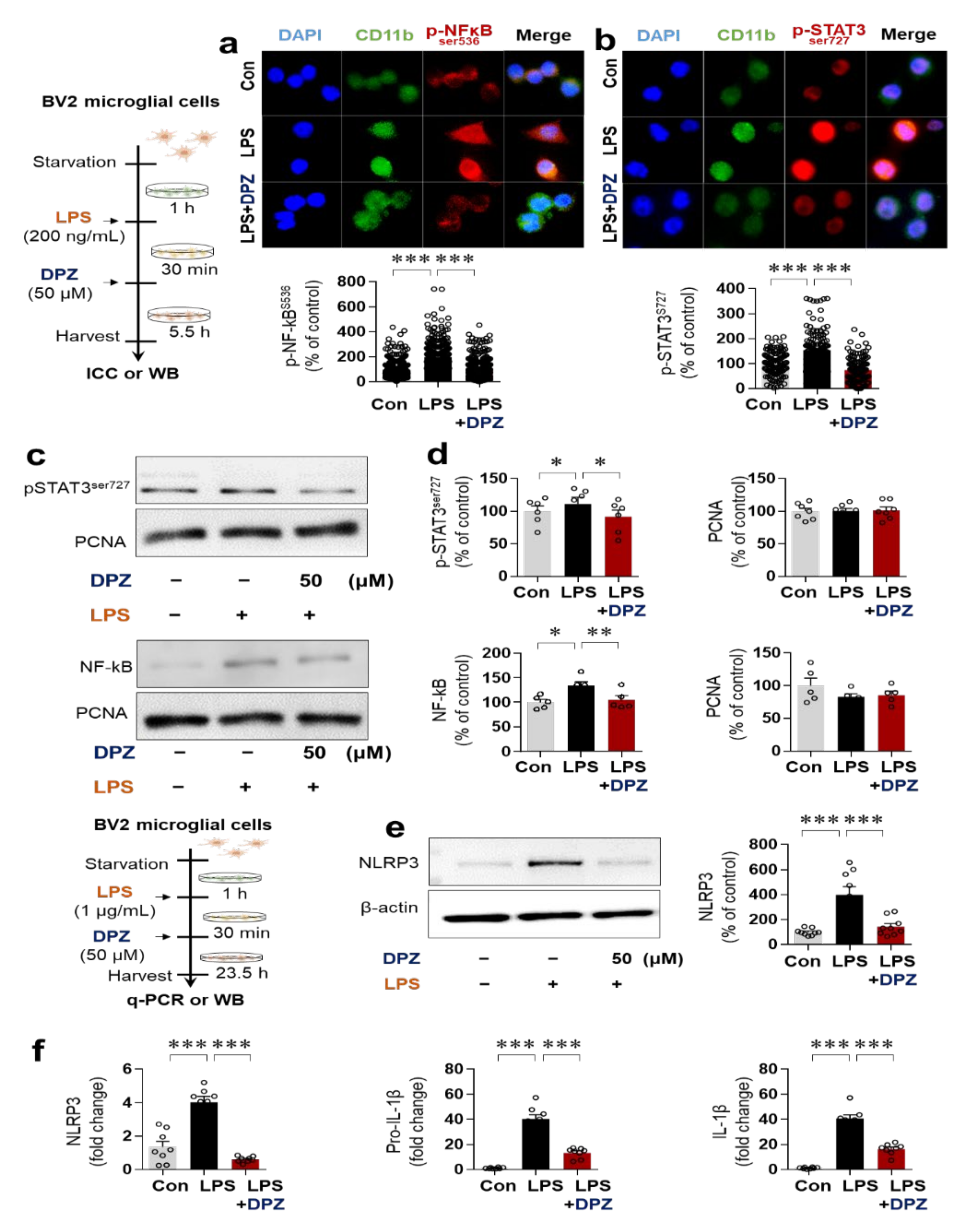
2.4. Donepezil Reduces LPS-Induced AKT/MAPK Signaling and Nuclear p-NF-κB/p-STAT3 Levels in BV2 Microglial Cells
2.5. Donepezil Ameliorates LPS-Induced NLRP3 Inflammasome Formation in BV2 Microglial Cells
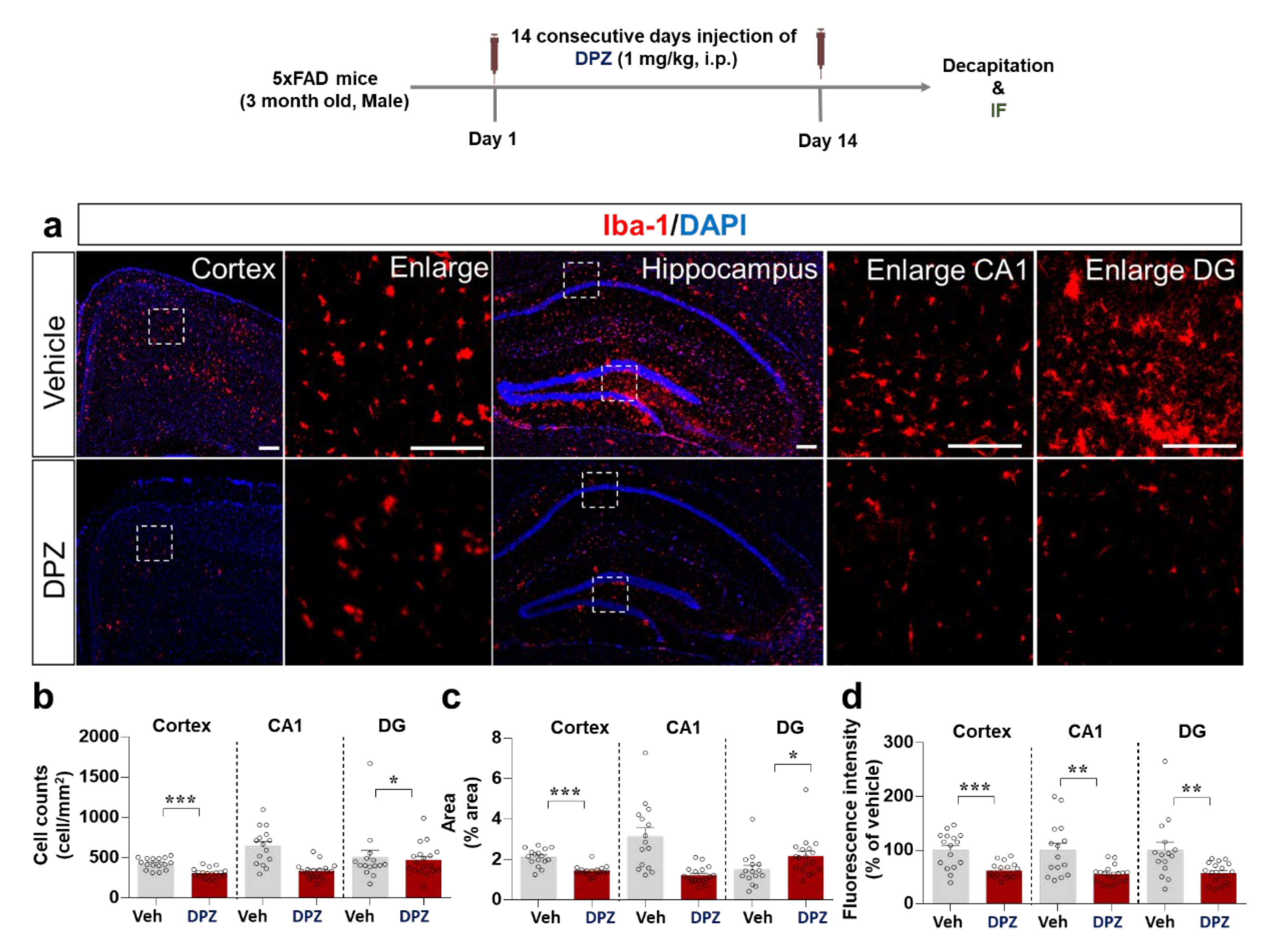
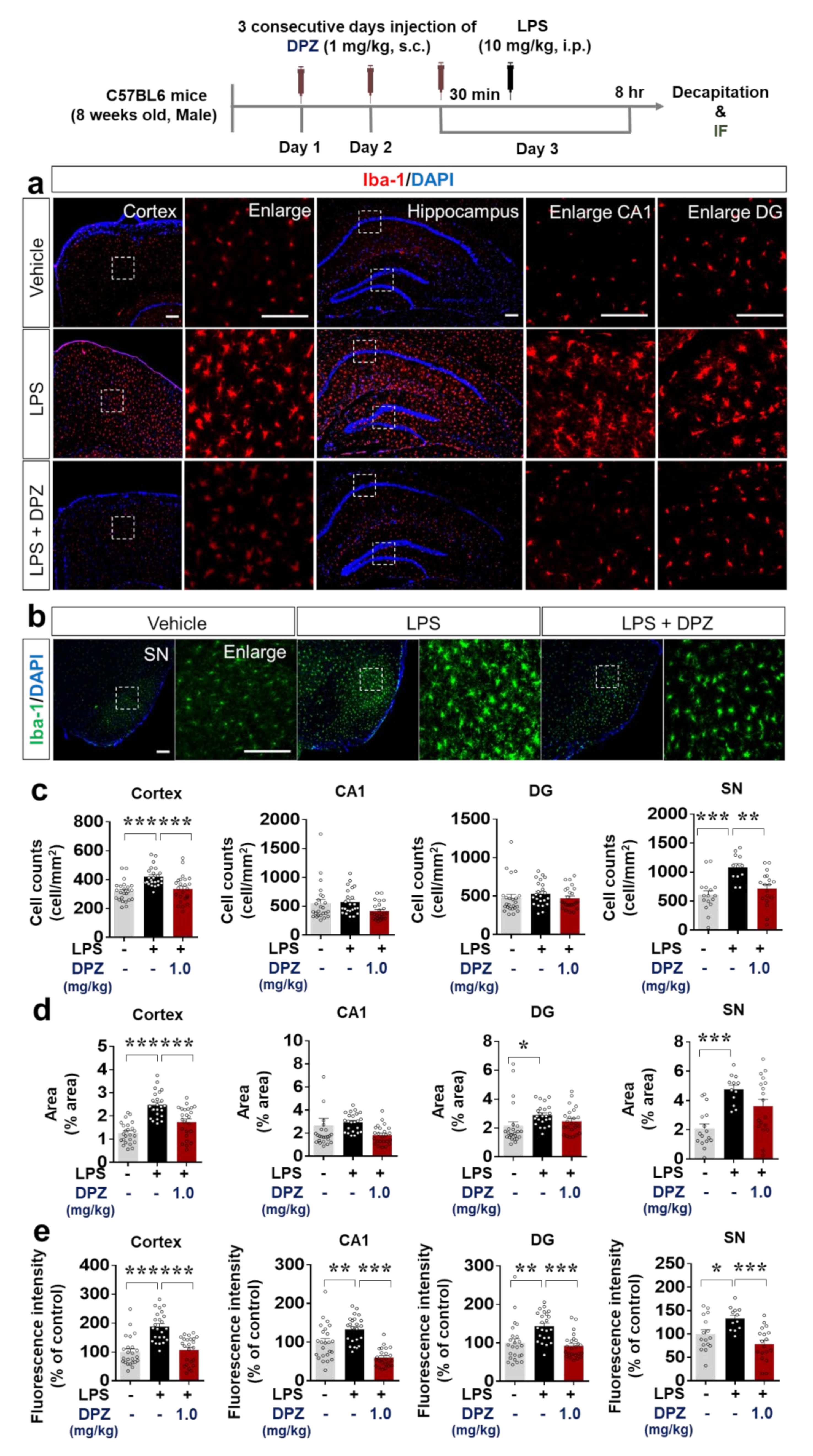
2.6. Donepezil Downregulates LPS-Mediated Microglial Density, Morphology and Activation in Wild-Type Mice
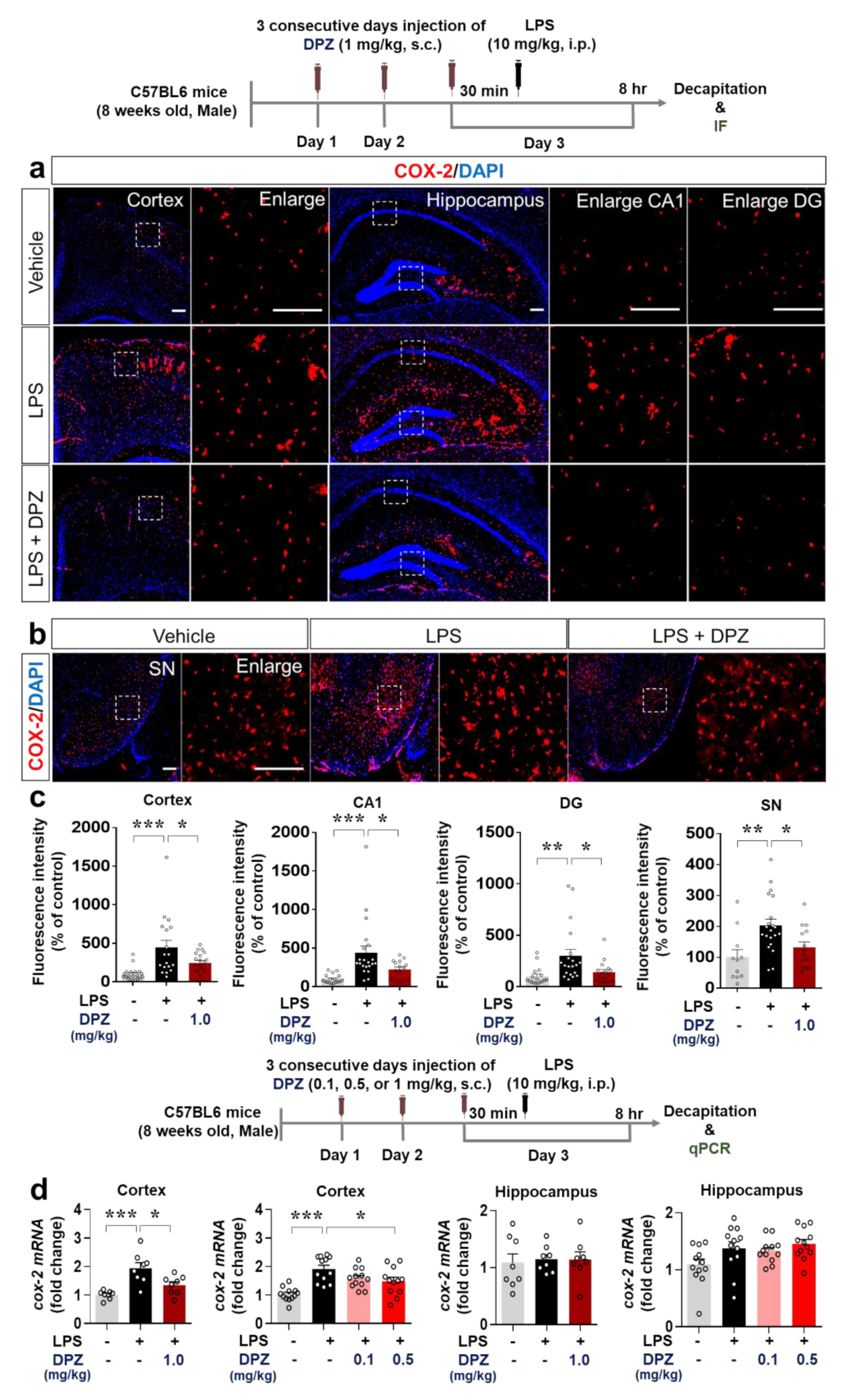
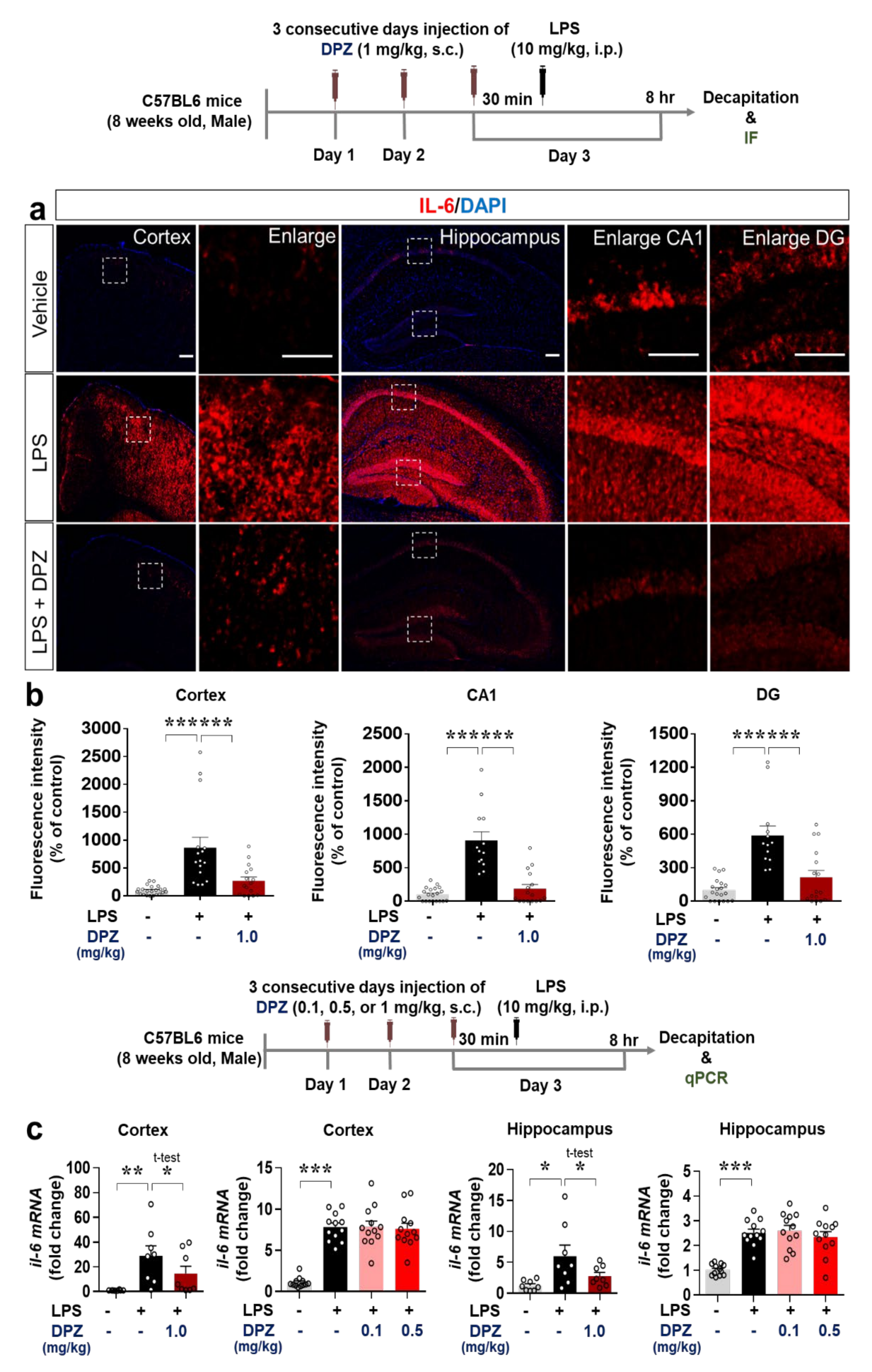
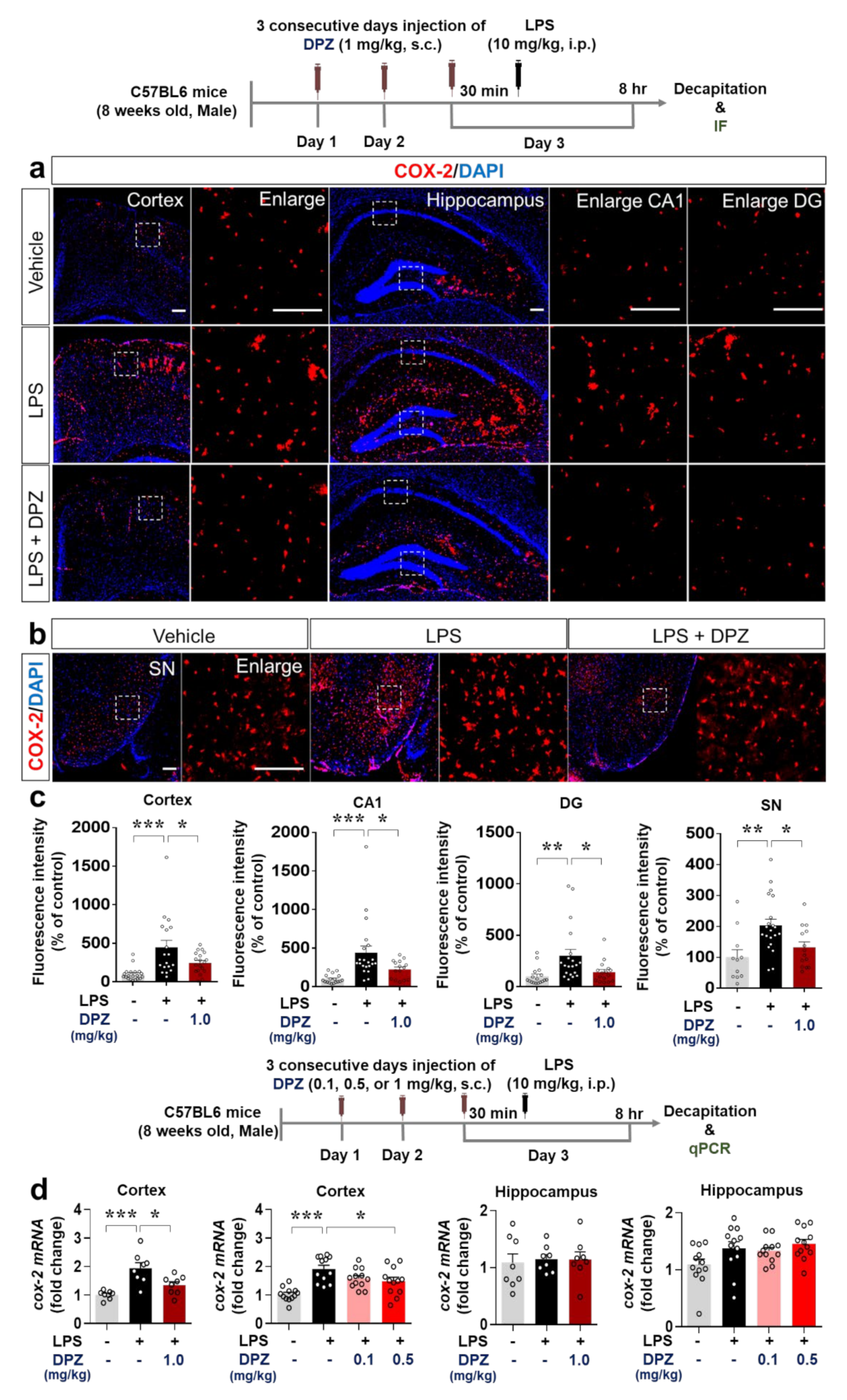
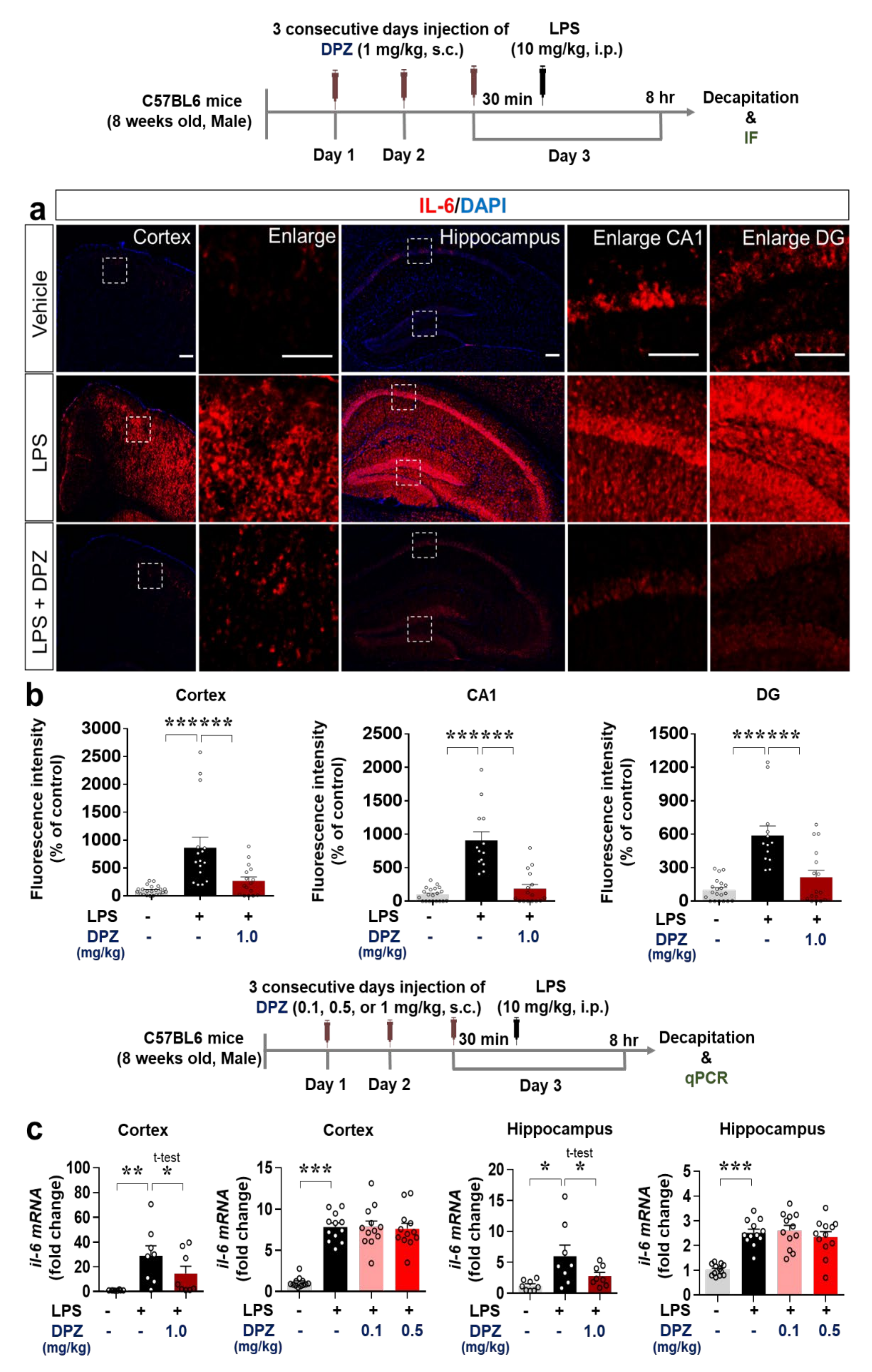
2.7. Donepezil Decreases the Induction of COX-2 and IL-6 Levels by LPS in Wild-Type Mice
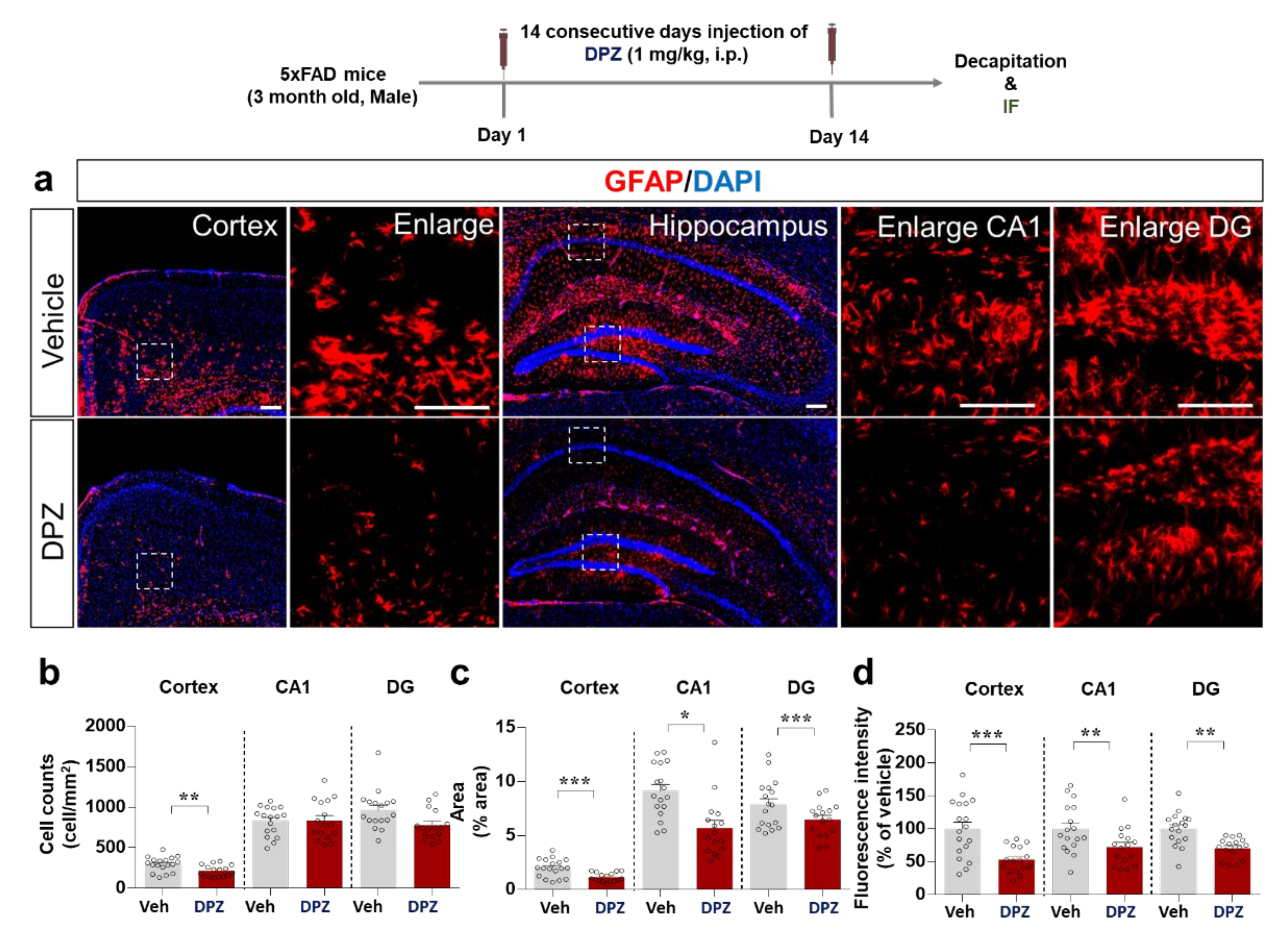
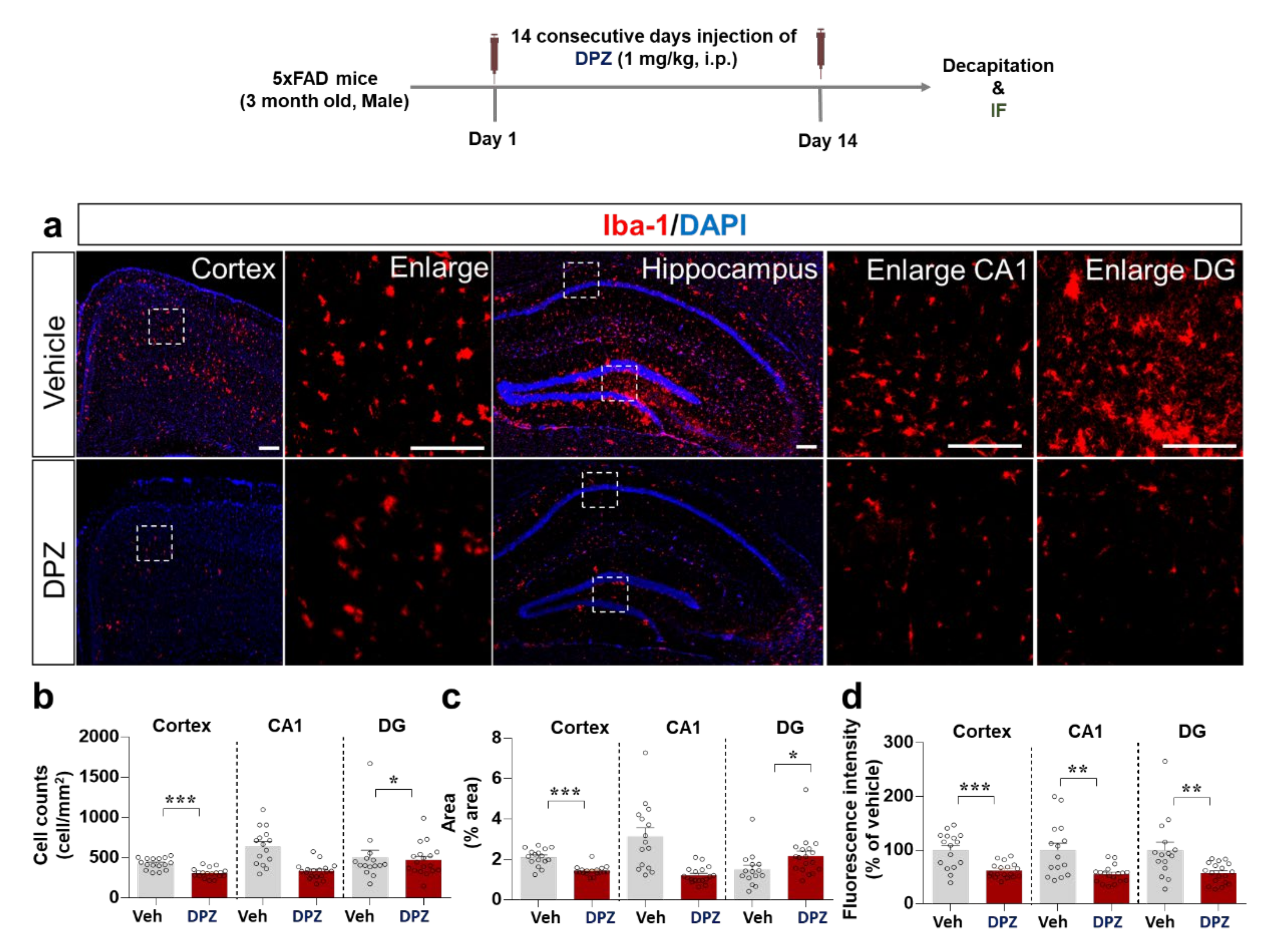
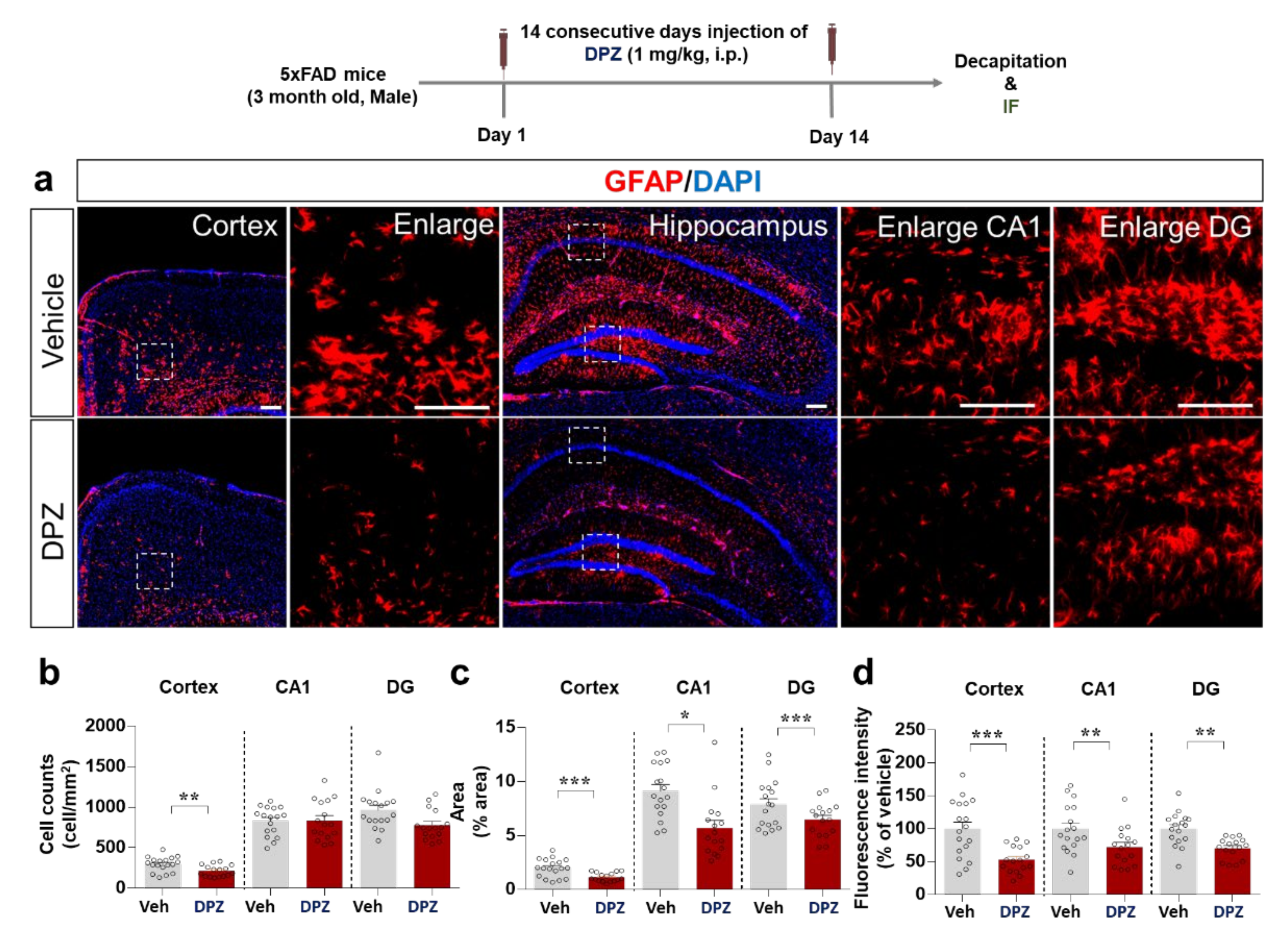
2.8. Donepezil Reduces Aβ-Mediated Neuroinflammation in a Mouse Model of AD
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. BV2 Cell Culture
4.3. Mouse Primary Astrocyte Cell Culture
4.4. MTT Assay
4.5. Reverse transcription PCR (RT-PCR)
4.6. Real-Time Polymerase Chain Reaction (Quantitative PCR, q-PCR)
4.7. Intracellular ROS Content
4.8. Mitochondrial Membrane Potential (MMP, ΔΨm)
4.9. Immunocytochemistry (ICC) Assay
4.10. Animals
4.11. LPS-Induced Neuroinflammation Mouse Model
4.12. Aβ-Induced Neuroinflammation Mouse Model (5xFAD Mice)
4.13. Western Blotting and Nuclear Fractionation
4.14. Nuclear Fractionation
4.15. Immunohistochemistry (IHC) Staining
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.-R.; Lee, B.; Joung, E.-J.; Gwon, W.-G.; Utsuki, T.; Kim, N.-G.; Kim, H.-R. 6, 6′-Bieckol suppresses inflammatory responses by down-regulating nuclear factor-κB activation via Akt, JNK, and p38 MAPK in LPS-stimulated microglial cells. Immunopharmacol. Immunotoxicol. 2016, 38, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, S.; Chen, L.; Liu, H.; Zhang, J.; Ge, H.; Zhang, Y.; Yu, B.; Kou, J. Tetrahydroberberrubine attenuates lipopolysaccharide-induced acute lung injury by down-regulating MAPK, AKT, and NF-κB signaling pathways. Biomed. Pharmacother. 2016, 82, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Guo, Y.-E.; Fang, J.-H.; Shi, C.-J.; Suo, N.; Zhang, R.; Xie, X. Donepezil, a drug for Alzheimer’s disease, promotes oligodendrocyte generation and remyelination. Acta Pharmacol. Sin. 2019, 40, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Sagal, J.P.; Colombres, M. Acetylcholinesterase interaction with Alzheimer amyloid β. Alzheimer’s Dis. 2005, 40, 299–317. [Google Scholar]
- Inestrosa, N.C.; Urra, S.; Colombres, M. Acetylcholinesterase (AChE)-amyloid-β-peptide complexes in Alzheimer’s disease. The Wnt signaling pathway. Curr. Alzheimer Res. 2004, 1, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Maroli, A.; Di Lascio, S.; Drufuca, L.; Cardani, S.; Setten, E.; Locati, M.; Fornasari, D.; Benfante, R. Effect of donepezil on the expression and responsiveness to LPS of CHRNA7 and CHRFAM7A in macrophages: A possible link to the cholinergic anti-inflammatory pathway. J. Neuroimmunol. 2019, 332, 155–166. [Google Scholar] [CrossRef]
- Kim, H.G.; Moon, M.; Choi, J.G.; Park, G.; Kim, A.J.; Hur, J.; Lee, K.T.; Oh, M.S. Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology 2014, 40, 23–32. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Chen, X.W.; Wei, W.; Liu, C. Activating alpha7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of beta-arrestin-1. CNS Neurosci. Ther. 2017, 23, 875–884. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Kwan, K.; Levine, Y.A.; Olofsson, P.S.; Yang, H.; Li, J.; Joshi, S.; Wang, H.; Andersson, U.; Chavan, S.S.; et al. alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 2014, 20, 350–358. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- De Jonge, W.J.; van der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.R.; Uematsu, S.; Akira, S.; van den Wijngaard, R.M.; et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.M.; Ochani, M.; Rosas-Ballina, M.; Liao, H.; Ochani, K.; Pavlov, V.A.; Gallowitsch-Puerta, M.; Ashok, M.; Czura, C.J.; Foxwell, B.; et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J. Exp. Med. 2006, 203, 1623–1628. [Google Scholar] [CrossRef] [Green Version]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S369–S379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, M.E.; Geurts, J.J.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion 2010, 10, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Han, M.H.; Park, C.; Jin, C.-Y.; Kim, G.-Y.; Choi, I.-W.; Kim, N.D.; Nam, T.-J.; Kwon, T.K.; Choi, Y.H. Anti-inflammatory effects of fucoidan through inhibition of NF-κB, MAPK and Akt activation in lipopolysaccharide-induced BV2 microglia cells. Food Chem. Toxicol. 2011, 49, 1745–1752. [Google Scholar] [CrossRef]
- Ryu, K.-Y.; Lee, H.-j.; Woo, H.; Kang, R.-J.; Han, K.-M.; Park, H.; Lee, S.M.; Lee, J.-Y.; Jeong, Y.J.; Nam, H.-W. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J. Neuroinflamm. 2019, 16, 1–36. [Google Scholar] [CrossRef]
- Dorey, E.; Chang, N.; Liu, Q.Y.; Yang, Z.; Zhang, W. Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharm. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Arikawa, M.; Kakinuma, Y.; Noguchi, T.; Todaka, H.; Sato, T. Donepezil, an acetylcholinesterase inhibitor, attenuates LPS-induced inflammatory response in murine macrophage cell line RAW 264.7 through inhibition of nuclear factor kappa B translocation. Eur. J. Pharmacol. 2016, 789, 17–26. [Google Scholar] [CrossRef]
- Hwang, J.; Hwang, H.; Lee, H.-W.; Suk, K. Microglia signaling as a target of donepezil. Neuropharmacology 2010, 58, 1122–1129. [Google Scholar] [CrossRef]
- Dasuri, K.; Zhang, L.; Kim, S.O.F.; Bruce-Keller, A.J.; Keller, J.N. Dietary and donepezil modulation of mTOR signaling and neuroinflammation in the brain. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 274–283. [Google Scholar] [CrossRef]
- Ongnok, B.; Khuanjing, T.; Chunchai, T.; Pantiya, P.; Kerdphoo, S.; Arunsak, B.; Nawara, W.; Jaiwongkam, T.; Apaijai, N.; Chattipakorn, N.; et al. Donepezil Protects Against Doxorubicin-Induced Chemobrain in Rats via Attenuation of Inflammation and Oxidative Stress Without Interfering With Doxorubicin Efficacy. Neurotherapeutics 2021. [Google Scholar] [CrossRef]
- Goschorska, M.; Baranowska-Bosiacka, I.; Gutowska, I.; Tarnowski, M.; Piotrowska, K.; Metryka, E.; Safranow, K.; Chlubek, D. Effect of acetylcholinesterase inhibitors donepezil and rivastigmine on the activity and expression of cyclooxygenases in a model of the inflammatory action of fluoride on macrophages obtained from THP-1 monocytes. Toxicology 2018, 406–407, 9–20. [Google Scholar] [CrossRef]
- Sreevathsa, M.R.; John, A.K. Double breasting technique for residual cavity in hepatic hydatid. J. R. Coll. Surg. Edinb. 1997, 42, 244–245. [Google Scholar] [PubMed]
- Polinsky, R.J. Clinical pharmacology of rivastigmine: A new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin. Ther. 1998, 20, 634–647. [Google Scholar] [CrossRef]
- Nam, H.Y.; Nam, J.H.; Yoon, G.; Lee, J.Y.; Nam, Y.; Kang, H.J.; Cho, H.J.; Kim, J.; Hoe, H.S. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J. Neuroinflamm. 2018, 15, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samavati, L.; Rastogi, R.; Du, W.; Huttemann, M.; Fite, A.; Franchi, L. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol. Immunol. 2009, 46, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Balic, J.J.; Albargy, H.; Luu, K.; Kirby, F.J.; Jayasekara, W.S.N.; Mansell, F.; Garama, D.J.; De Nardo, D.; Baschuk, N.; Louis, C.; et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1beta expression. Nat. Commun. 2020, 11, 3816. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, Y.; Gilboa, A.; Ben-Menachem, O.; Ben-Hur, T.; Soreq, H.; Yirmiya, R. Acetylcholinesterase inhibitors reduce brain and blood interleukin-1beta production. Ann. Neurol. 2005, 57, 741–745. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Relationships between mitochondria and neuroinflammation: Implications for Alzheimer’s disease. Curr. Top. Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef]
- Gubandru, M.; Margina, D.; Tsitsimpikou, C.; Goutzourelas, N.; Tsarouhas, K.; Ilie, M.; Tsatsakis, A.M.; Kouretas, D. Alzheimer’s disease treated patients showed different patterns for oxidative stress and inflammation markers. Food Chem. Toxicol. 2013, 61, 209–214. [Google Scholar] [CrossRef]
- Noh, H.; Jeon, J.; Seo, H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 2014, 69, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hawking, Z.L. Alzheimer’s disease: The role of mitochondrial dysfunction and potential new therapies. Biosci. Horiz. Int. J. Stud. Res. 2016, 9. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.Y.; Lei, Y.; Tang, X.C.; Zhang, H.Y. Donepezil attenuates Aβ-associated mitochondrial dysfunction and reduces mitochondrial Aβ accumulation in vivo and in vitro. Neuropharmacology 2015, 95, 29–36. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Tang, X.; Di, X.; Liu, Y. Protective effects of Donepezil against endothelial permeability. Eur. J. Pharmacol. 2017, 811, 60–65. [Google Scholar] [CrossRef]
- Liu, E.Y.; Xia, Y.; Kong, X.; Guo, M.S.; Anna, X.; Zheng, B.Z.; Mak, S.; Xu, M.L.; Tsim, K.W. Interacting with α7 nAChR is a new mechanism for AChE to enhance the inflammatory response in macrophages. Acta Pharm. Sin. B 2020, 10, 1926–1942. [Google Scholar] [CrossRef]
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The potential role of the NLRP3 inflammasome activation as a link between mitochondria ROS generation and neuroinflammation in postoperative cognitive dysfunction. Front. Cell. Neurosci. 2019, 13, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Wang, J.; Wang, P.; Ren, H.; Yue, Y.; Song, Z.; Fu, X. Donepezil protects glycerol-induced acute renal failure through the cholinergic anti-inflammatory and nitric oxide pathway in rats. Immunopharmacol. Immunotoxicol. 2020, 42, 625–631. [Google Scholar] [CrossRef] [PubMed]
- López, D.E.; Ballaz, S. The role of brain cyclooxygenase-2 (COX-2) beyond neuroinflammation: Neuronal homeostasis in memory and anxiety. Mol. Neurobiol. 2020, 57, 5167–5176. [Google Scholar] [CrossRef]
- Simon, L.S. Role and regulation of cyclooxygenase-2 during inflammation. Am. J. Med. 1999, 106, 37S–42S. [Google Scholar] [CrossRef]
- Cacquevel, M.; Lebeurrier, N.; Chéenne, S.; Vivien, D. Cytokines in neuroinflammation and Alzheimer’s disease. Curr. Drug Targets 2004, 5, 529–534. [Google Scholar] [CrossRef]
- Gambi, F.; Reale, M.; Iarlori, C.; Salone, A.; Toma, L.; Paladini, C.; De Luca, G.; Feliciani, C.; Salvatore, M.; Salerno, R.M. Alzheimer patients treated with an AchE inhibitor show higher IL-4 and lower IL-1β levels and expression in peripheral blood mononuclear cells. J. Clin. Psychopharmacol. 2004, 24, 314–321. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Hussain, M.D.; Yan, L.-J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Cheng, Y.; Wu, J.; Wang, C.; Wang, H.; Zhang, C.; Qiu, Z.; Xu, J. Donepezil improves learning and memory deficits in APP/PS1 mice by inhibition of microglial activation. Neurosci. Bull. 2015, 290, 530–542. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, R.; Wang, H.; Zheng, R. Effects of rivastigmine hydrogen tartrate and donepezil hydrochloride on the cognitive function and mental behavior of patients with Alzheimer’s disease. Exp. Ther. Med. 2020, 20, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Makitani, K.; Nakagawa, S.; Izumi, Y.; Akaike, A.; Kume, T. Inhibitory effect of donepezil on bradykinin-induced increase in the intracellular calcium concentration in cultured cortical astrocytes. J. Pharmacol. Sci. 2017, 134, 37–44. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′–3′) | |
|---|---|---|
| COX2 | Forward | GCC AGC AAA GCC TAG AGC AA |
| Reverse | GCC TTC TGC AGT CCA GGT TC | |
| IL-6 | Forward | GCC AGC AAA GCC TAG AGC AA |
| Reverse | GCC TTC TGC AGT CCA GGT TC | |
| IL-1β | Forward | AGC TGG AGA GTG TGG ATC CC |
| Reverse | CCT GTC TTG GCC GAG GAC TA | |
| iNOS | Forward | CCG GCA AAC CCA AGG TCT AC |
| Reverse | GCA TTT CGC TGT CTC CCC AA | |
| GAPDH | Forward | CAG GAG CGA GAC CCC ACT AA |
| Reverse | ATC ACG CCA CAG CTT TCC AG | |
| Gene | Primer Sequence (5′–3′) | |
|---|---|---|
| GAPDH | Forward | TGG GCT ACA CTG AGG ACC ACT |
| Reverse | GGG AGT GTC TGT TGA AGT CG | |
| iNOS | Forward | GGA TCT TCC CAG GCA ACC A |
| Reverse | TCC ACA ACT CGC TCC AAG ATT | |
| COX-2 | Forward | CCA CTT CAA GGG AGT CTG GA |
| Reverse | AGT CAT CTG CTA CGG GAG GA | |
| IL-6 | Forward | CCA CGG CCT TCC CTA CTT C |
| Reverse | TTG GGA GTG GTA TCC TCT GTG A | |
| IL-1β | Forward | TTG ACG GAC CCC AAA AGA TG |
| Reverse | AGG ACA GCC CAG GTC AAA G | |
| Pro-IL-1β | Forward | TCT TTG AAG TTG ACG GAC CC |
| Reverse | TGA GTG ATA CTG CCT GCC TG | |
| NLRP3 | Forward | TCC ACA ATT CTG ACC CAC AA |
| Reverse | ACC TCA CAG AGG GTC ACC AC | |
| Primary Antibodies | Host Species | Dilution | Manufacturer | Catalog No. |
| Anti-Iba-1 | Rabbit | 1:500 | Wako | 019-19741 |
| Anti-GFAP | Rabbit | 1:500 | Neuromics | RA22101 |
| Anti-IL-6 | Mouse | 1:50 | Santa Cruz | SC-57315 |
| Anti-COX-2 | Rabbit | 1:200 | Abcam | ab15191 |
| Secondary Antibodies | Dilution | Manufacturer | Catalog No. | |
| Goat anti-rabbit IgG, 555 | 1:200 | Invitrogen | A21428 | |
| Goat anti-rabbit IgG, 488 | 1:200 | Invitrogen | A11008 | |
| Goat anti-mouse IgG, 488 | 1:200 | Invitrogen | A11001 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Lee, H.-j.; Park, S.K.; Park, J.-H.; Jeong, H.-R.; Lee, S.; Lee, H.; Seol, E.; Hoe, H.-S. Donepezil Regulates LPS and Aβ-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling. Int. J. Mol. Sci. 2021, 22, 10637. https://doi.org/10.3390/ijms221910637
Kim J, Lee H-j, Park SK, Park J-H, Jeong H-R, Lee S, Lee H, Seol E, Hoe H-S. Donepezil Regulates LPS and Aβ-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling. International Journal of Molecular Sciences. 2021; 22(19):10637. https://doi.org/10.3390/ijms221910637
Chicago/Turabian StyleKim, Jieun, Hyun-ju Lee, Seon Kyeong Park, Jin-Hee Park, Ha-Ram Jeong, Soojung Lee, Heeyong Lee, Eunyoung Seol, and Hyang-Sook Hoe. 2021. "Donepezil Regulates LPS and Aβ-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling" International Journal of Molecular Sciences 22, no. 19: 10637. https://doi.org/10.3390/ijms221910637
APA StyleKim, J., Lee, H.-j., Park, S. K., Park, J.-H., Jeong, H.-R., Lee, S., Lee, H., Seol, E., & Hoe, H.-S. (2021). Donepezil Regulates LPS and Aβ-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling. International Journal of Molecular Sciences, 22(19), 10637. https://doi.org/10.3390/ijms221910637