
Construction of IL-13 Receptor α2-Targeting Resveratrol Nanoparticles against Glioblastoma Cells: Therapeutic Efficacy and Molecular Effects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. IL-13Rα2 Expression in C6 Cells
2.2. Sustained Release of Res from Pep-PP@Res
2.3. Nontoxicity of the Drug Carrier to C6 Cells
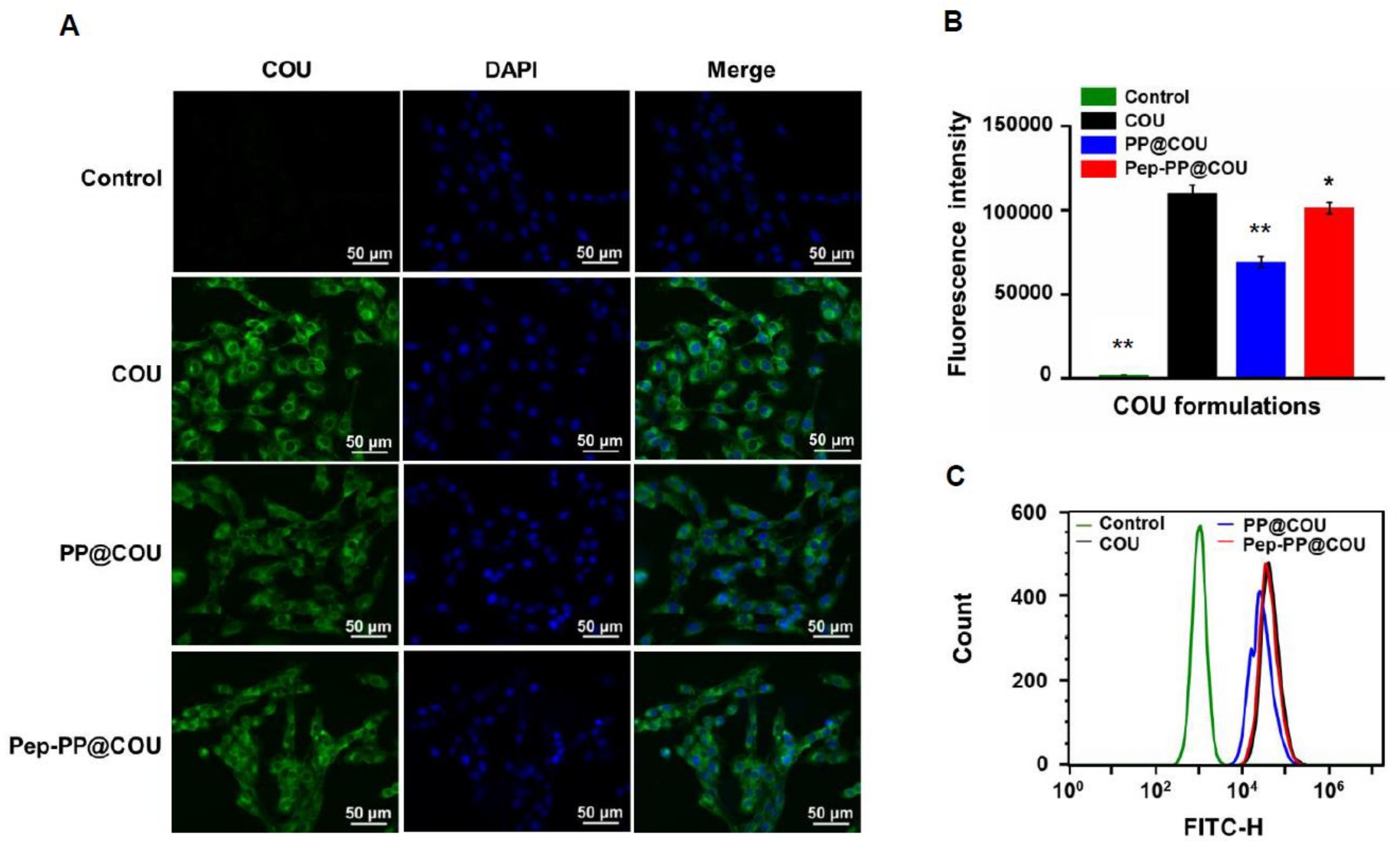
2.4. Pep-1 Modification Enhanced Internalization of Nanoparticles
2.5. Better Anti-Glioblastoma Efficacy of Pep-PP@Res
2.6. Safety of Pep-PP@Res towards Normal Rat Brain Cells
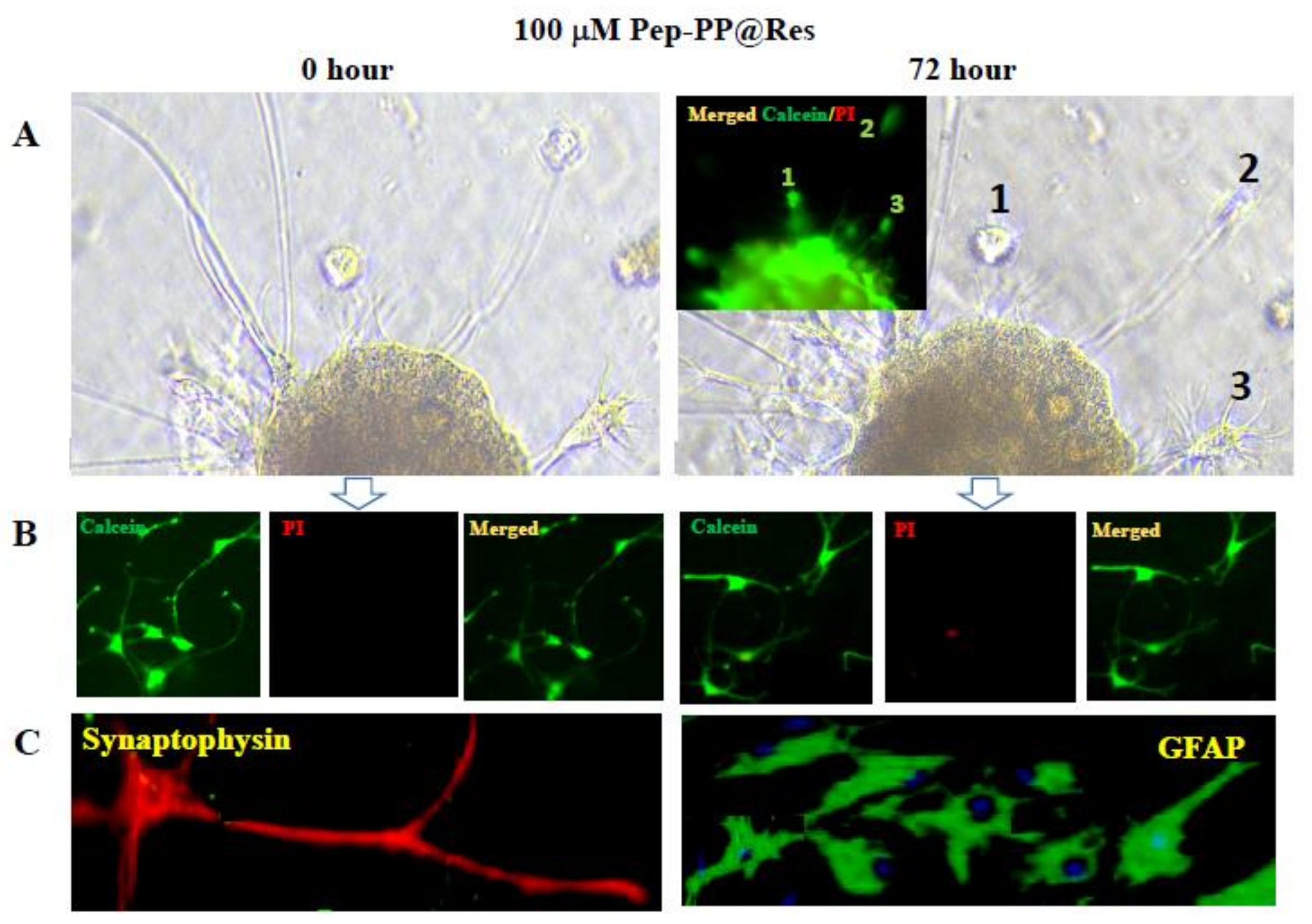
2.7. Prolonged Res Retention Time in Pep-PP@Res-Treated Cells
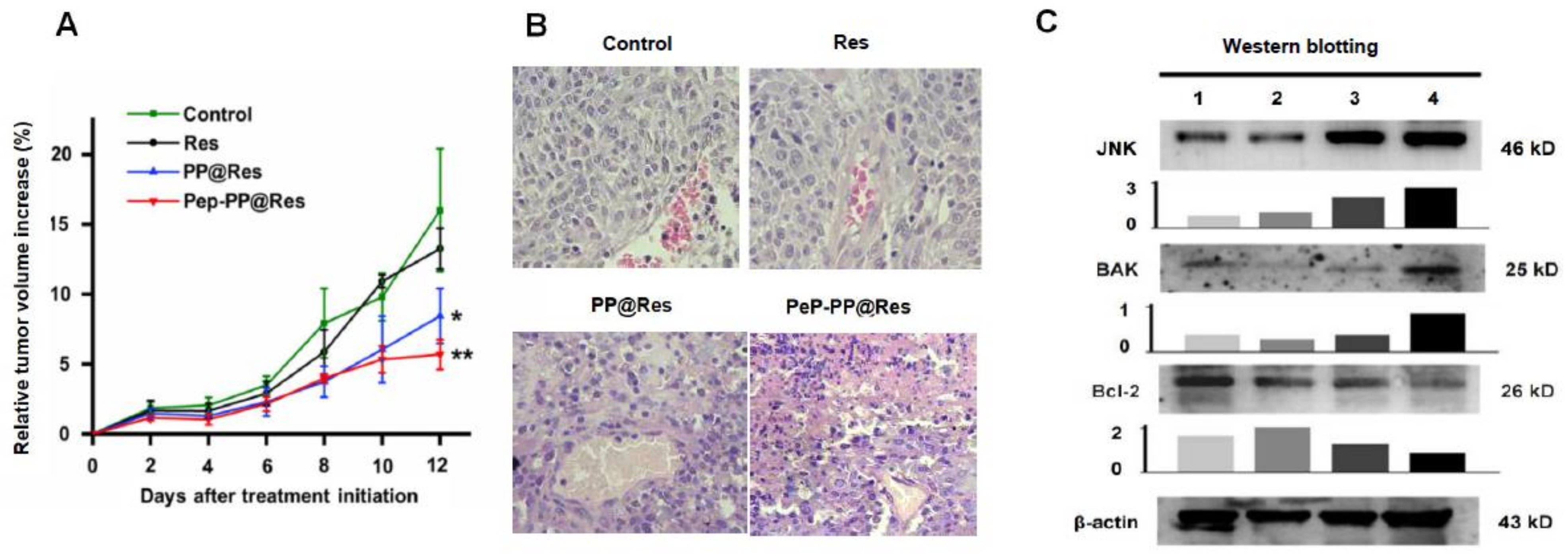
2.8. Improved Inhibitory Effects of Pep-PP@Res on the C6 Xenograft in Nude Mice
2.9. Activated JNK Signaling in the Res Formulation-Treated C6 Tumors
3. Discussion
4. Materials and Methods
4.1. Experimental Materials
4.2. GBM Cell Line
4.3. Immunocytochemical and Immunofluorescent Staining
4.4. Synthesis of Pep-PEG3.5K-b-PCL4K
4.5. Preparation of Nanoparticle Formulations
4.6. Characterization of the Res-Loaded Formulations
4.7. In Vitro Drug Release Analysis
4.8. Drug Carrier Safety Study
4.9. Internalization of Pep-PP@COU into GBM Cells
4.10. Cell Proliferation Assay
4.11. Safety of Pep-PP@Res on Normal Rat Brain Cells
4.12. Intracellular Res Retention Time and Distribution Pattern
4.13. Evaluation of In Vivo Tumor-Suppressive Effects
4.14. Protein Preparation and Western Blotting
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, v1–v86. [Google Scholar] [CrossRef]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.; Lightner, D.D.; Barnholtz-Sloan, J.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Brandes, A.; Dittrich, C.; Fumoleau, P.; Coudert, B.; Clement, P.M.; Frenay, M.; Rampling, R.; Stupp, R.; Kros, J.M.; et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: A european organisation for research and treatment of cancer brain tumor group study. J. Clin. Oncol. 2008, 26, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Hottinger, A.F.; Pacheco, P.; Stupp, R. Tumor treating fields: A novel treatment modality and its use in brain tumors. Neuro Oncol. 2016, 18, 1338–1349. [Google Scholar] [CrossRef]
- Liu, Q.; Xue, Y.; Chen, Q.; Chen, H.; Zhang, X.; Wang, L.; Han, C.; Que, S.; Lou, M.; Lan, J. PomGnT1 enhances temozolomide resistance by activating epithelial-mesenchymal transition signaling in glioblastoma. Oncol. Rep. 2017, 38, 2911–2918. [Google Scholar] [CrossRef]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef]
- Robich, M.P.; Chu, L.M.; Chaudray, M.; Nezafat, R.; Han, Y.; Clements, R.T.; Laham, R.J.; Manning, W.J.; Coady, M.A.; Sellke, F.W. Anti-angiogenic effect of high-dose resveratrol in a swine model of metabolic syndrome. Surgery 2010, 148, 453–462. [Google Scholar] [CrossRef]
- Song, X.; Shu, X.H.; Sha, L.; Bian, J.; Wang, L.L.; Gu, J.Y.; Shi, S.; Li, P.-N.; Wu, M.-L.; Wang, Q.; et al. Lumbar puncture-administered resveratrol inhibits STAT3 activation, enhancing autophagy and apoptosis in orthotopic rat glioblastomas. Oncotarget 2016, 7, 75790–75799. [Google Scholar]
- Shu, X.H.; Li, H.; Sun, X.X.; Wang, Q.; Sun, Z.; Wu, M.L.; Chen, X.Y.; Li, C.; Kong, Q.Y.; Liu, J. Metabolic patterns and biotransformation activities of resveratrol in human glioblastoma cells: Relevance with therapeutic efficacies. PLoS ONE 2011, 6, e27484. [Google Scholar] [CrossRef] [PubMed]
- Bonferoni, M.C.; Rossi, S.; Sandri, G.; Ferrari, F. Nanoparticle formulations to enhance tumor targeting of poorly soluble polyphenols with potential anticancer properties. Semin. Cancer Biol. 2017, 46, 205–214. [Google Scholar] [CrossRef]
- Du, Y.-Z.; Situ, J.-Q.; Ye, Y.-Q.; Zhu, X.-L.; Yu, R.-S.; You, J.; Yuan, H.; Hu, F.-Q. Specific targeting of A54 homing peptide-functionalized dextran-g-poly(lactic-co-glycolic acid) micelles to tumor cells. Int. J. Nanomed. 2015, 10, 665–675. [Google Scholar] [CrossRef][Green Version]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Bu, L.; Gan, L.-C.; Guo, X.-Q.; Chen, F.-Z.; Song, Q.; Zhao, Q.-; Gou, X.-J.; Hou, S.-X.; Yao, Q. Trans-resveratrol loaded chitosan nanoparticles modified with biotin and avidin to target hepatic carcinoma. Int. J. Pharm. 2013, 452, 355–362. [Google Scholar] [CrossRef]
- Regerat, F.; Texier, O.; Barthomeuf, C.; Pourrat, H. Antienzymatic activities of two Leguminosae: Bean pods and bean bulls. Antiamylase activity. Ann. Pharm. Fr. 1989, 47, 74–79. [Google Scholar]
- Figueiró, F.; Bernardi, A.; Frozza, R.L.; Terroso, T.; Zanotto-Filho, A.; Jandrey, E.H.F.; Moreira, J.C.F.; Salbego, C.G.; Edelweiss, M.I.; Pohlmann, A.; et al. Resveratrol-loaded lipid-core nanocapsules treatment reduces in vitro and in vivo glioma growth. J. Biomed. Nanotechnol. 2013, 9, 516–526. [Google Scholar] [CrossRef]
- Akiyama, Y.; Komiyama, M.; Miyata, H.; Yagoto, M.; Ashizawa, T.; Iizuka, A.; Oshita, C.; Kume, A.; Nogami, M.; Ito, I.; et al. Novel cancer-testis antigen expression on glioma cell lines derived from high-grade glioma patients. Oncol. Rep. 2014, 31, 1683–1690. [Google Scholar] [CrossRef]
- Kawakami, K.; Taguchi, J.; Murata, T.; Puri, R.K. The interleukin-13 receptor alpha2 chain: An essential component for binding and internalization but not for interleukin-13-induced signal transduction through the STAT6 pathway. Blood 2001, 97, 2673–2679. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M.; Hulet, S.W.; Connor, J.R.; Gillespie, G.Y. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin. Cancer Res. 1999, 5, 985–990. [Google Scholar] [PubMed]
- Jiang, Y.; Wang, X.; Liu, X.; Lv, W.; Zhang, H.; Zhang, M.; Li, X.; Xin, H.; Xu, Q. Enhanced antiglioma efficacy of ultrahigh loading capacity paclitaxel prodrug conjugate self-assembled targeted nanoparticles. ACS Appl. Mater. Interfaces 2017, 9, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pandya, H.; Gibo, D.M.; Garg, S.; Kridel, S.; Debinski, W. An interleukin 13 receptor α 2–specific peptide homes to human glioblastoma multiforme xenografts. Neuro Oncol. 2011, 14, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Lançon, A.; Delma, D.; Osman, H.; Thénot, J.-P.; Jannin, B.; Latruffe, N. Human hepatic cell uptake of resveratrol: Involvement of both passive diffusion and carrier-mediated process. Biochem. Biophys. Res. Commun. 2004, 316, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Lee, S.-L.-O.; Son, A.-R.; Ahn, J.; Song, J.-Y. Niclosamide enhances ROS-mediated cell death through c-Jun activation. Biomed. Pharmacother. 2014, 68, 619–624. [Google Scholar] [CrossRef]
- Inoshita, S.; Takeda, K.; Hatai, T.; Terada, Y.; Sano, M.; Hata, J.; Umezawa, A.; Ichijo, H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J. Biol. Chem. 2002, 277, 43730–43734. [Google Scholar] [CrossRef]
- Shu, X.-H.; Wang, L.-L.; Li, H.; Song, X.; Shi, S.; Gu, J.-Y.; Wu, M.-L.; Chen, X.-Y.; Kong, Q.-Y.; Liu, J. Diffusion efficiency and bioavailability of resveratrol administered to rat brain by different routes: Therapeutic implications. Neurotherapeutics 2015, 12, 491–501. [Google Scholar] [CrossRef]
- Gagliano, N.; Aldini, G.; Colombo, G.; Rossi, R.; Colombo, R.; Gioia, M.; Milzani, A.D.G.; Dalle-Donne, I. The potential of resveratrol against human gliomas. Anti Cancer Drugs 2010, 21, 140–150. [Google Scholar] [CrossRef]
- Asensi, M.; Medina, I.; Ortega, A.; Carretero, J.; Bañó, C.; Obrador, E.; Estrela, J.M. Inhibition of cancer growth by resveratrol is related to its low bioavailability. Free Radic. Biol. Med. 2002, 33, 387–398. [Google Scholar] [CrossRef]
- Jose, S.; Anju, S.S.; Cinu, T.A.; Aleykutty, N.A.; Thomas, S.; Souto, E.B. In vivo pharmacokinetics and biodistribution of resveratrol-loaded solid lipid nanoparticles for brain delivery. Int. J. Pharm. 2014, 474, 6–13. [Google Scholar] [CrossRef]
- Muthu, M.S.; Singh, S. Targeted nanomedicines: Effective treatment modalities for cancer, AIDS and brain disorders. Nanomedicine 2009, 4, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Knight, F.C.; Gilchuk, P.; Kumar, A.; Becker, K.W.; Sevimli, S.; Jacobson, M.E.; Suryadevara, N.; Wang-Bishop, L.; Boyd, K.L.; Crowe, J.E., Jr.; et al. Mucosal immunization with a pH-responsive nanoparticle vaccine induces protective CD8(+) lung-resident memory T cells. ACS Nano 2019, 13, 10939–10960. [Google Scholar] [CrossRef]
- Gao, F.; Wu, J.; Niu, S.; Sun, T.; Li, F.; Bai, Y.; Jin, L.; Lin, L.; Shi, Q.; Zhu, L.M.; et al. Biodegradable, pH-sensitive hollow mesoporous organosilica nanoparticle (HMON) with controlled release of pirfenidone and ultrasound-target-microbubble-destruction (UTMD) for pancreatic cancer treatment. Theranostics 2019, 9, 6002–6018. [Google Scholar] [CrossRef] [PubMed]
- Maier-Salamon, A.; Böhmdorfer, M.; Riha, J.; Thalhammer, T.; Szekeres, T.; Jaeger, W. Interplay between metabolism and transport of resveratrol. Ann. N. Y. Acad. Sci. 2013, 1290, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-L.; Yi, L.; Jin, X.; Xie, Q.; Zhang, T.; Zhou, X.; Chang, H.; Fu, Y.-J.; Zhu, J.-D.; Zhang, Q.-Y.; et al. Absorption of resveratrol by vascular endothelial cells through passive diffusion and an SGLT1-mediated pathway. J. Nutr. Biochem. 2013, 24, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, Q.; Peng, F.; Liu, L.; Gong, C. Strategies of polymeric nanoparticles for enhanced internalization in cancer therapy. Colloids Surf. B Biointerfaces 2015, 135, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Li, A.; Jia, Z.; Yuan, Y.; Dai, H.; Li, H. Transferrin modified PEG-PLA-resveratrol conjugates: In vitro and in vivo studies for glioma. Eur. J. Pharmacol. 2013, 718, 41–47. [Google Scholar] [CrossRef]
- Delmas, D.; Limagne, E.; Ghiringhelli, F.; Aires, V. Immune Th17 lymphocytes play a critical role in the multiple beneficial properties of resveratrol. Food Chem. Toxicol. 2020, 137, 111091. [Google Scholar] [CrossRef]
- Vlahopoulos, S.; Zoumpourlis, V.C. JNK: A key modulator of intracellular signaling. Biochemistry 2004, 69, 844–854. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, J.; Sun, W.; Zhang, T.; Zuo, N.; Wang, H.; Wang, G.; Xu, J.; Yin, F.; Mao, M.; et al. Antitumor activity of Raddeanin A is mediated by Jun amino-terminal kinase activation and signal transducer and activator of transcription 3 inhibition in human osteosarcoma. Cancer Sci. 2019, 110, 1746–1759. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Zheng, X.; Wu, M.-L.; Tian, X.-T.; Song, X.; Liu, Y.-N.; Li, P.-N.; Liu, J. Increased reactive oxygen species and distinct oxidative damage in resveratrol-suppressed glioblastoma cells. J. Cancer 2021, 12, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, J.; Weng, C.; Chen, R.; Zheng, Y.; Chen, Q.; Tang, H. Identification of the protein–protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem. Biophys. Res. Commun. 2003, 305, 989–996. [Google Scholar] [CrossRef]
- Weng, C.; Li, Y.; Xu, D.; Shi, Y.; Tang, H. Specific cleavage of Mcl-1 by Caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J. Biol. Chem. 2005, 280, 10491–10500. [Google Scholar] [CrossRef]
- Shuai, X.; Merdan, T.; Unger, F.; Wittmar, A.M.; Kissel, T. Novel biodegradable ternary copolymers hy-PEI-g-PCL-b-PEG: Synthesis, characterization, and potential as efficient nonviral gene delivery vectors. Macromolecules 2003, 36, 5751–5759. [Google Scholar] [CrossRef]
- Zhang, P.; Hu, L.; Yin, Q.; Zhang, Z.; Feng, L.; Li, Y. Transferrin-conjugated polyphosphoester hybrid micelle loading paclitaxel for brain-targeting delivery: Synthesis, preparation and in vivo evaluation. J. Control. Release 2012, 159, 429–434. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, Q.; Zhang, J.; Lu, Z.; Wang, A.; Fei, X.; Dai, X.; Wu, J.; Wang, Z.; Zhao, Y.; et al. Advantages of a dual-color fluorescence-tracing glioma orthotopic implantation model: Detecting tumor location, angiogenesis, cellular fusion and the tumor microenvironment. Exp. Ther. Med. 2015, 10, 2047–2054. [Google Scholar] [CrossRef][Green Version]
- Xiong, L.; Lin, X.M.; Nie, J.H.; Ye, H.S.; Liu, J. Resveratrol and its nanoparticle suppress doxorubicin/docetaxel-resistant anaplastic thyroid cancer cells in vitro and in vivo. Nanotheranostics 2021, 5, 143–154. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.-M.; Shi, X.-X.; Xiong, L.; Nie, J.-H.; Ye, H.-S.; Du, J.-Z.; Liu, J. Construction of IL-13 Receptor α2-Targeting Resveratrol Nanoparticles against Glioblastoma Cells: Therapeutic Efficacy and Molecular Effects. Int. J. Mol. Sci. 2021, 22, 10622. https://doi.org/10.3390/ijms221910622
Lin X-M, Shi X-X, Xiong L, Nie J-H, Ye H-S, Du J-Z, Liu J. Construction of IL-13 Receptor α2-Targeting Resveratrol Nanoparticles against Glioblastoma Cells: Therapeutic Efficacy and Molecular Effects. International Journal of Molecular Sciences. 2021; 22(19):10622. https://doi.org/10.3390/ijms221910622
Chicago/Turabian StyleLin, Xiao-Min, Xiao-Xiao Shi, Le Xiong, Jun-Hua Nie, Hai-Shan Ye, Jin-Zi Du, and Jia Liu. 2021. "Construction of IL-13 Receptor α2-Targeting Resveratrol Nanoparticles against Glioblastoma Cells: Therapeutic Efficacy and Molecular Effects" International Journal of Molecular Sciences 22, no. 19: 10622. https://doi.org/10.3390/ijms221910622
APA StyleLin, X.-M., Shi, X.-X., Xiong, L., Nie, J.-H., Ye, H.-S., Du, J.-Z., & Liu, J. (2021). Construction of IL-13 Receptor α2-Targeting Resveratrol Nanoparticles against Glioblastoma Cells: Therapeutic Efficacy and Molecular Effects. International Journal of Molecular Sciences, 22(19), 10622. https://doi.org/10.3390/ijms221910622