
Monocarboxylate Transporter-2 Expression Restricts Tumor Growth in a Murine Model of Lung Cancer: A Multi-Omic Analysis

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. MCT2 Immunoreactivity
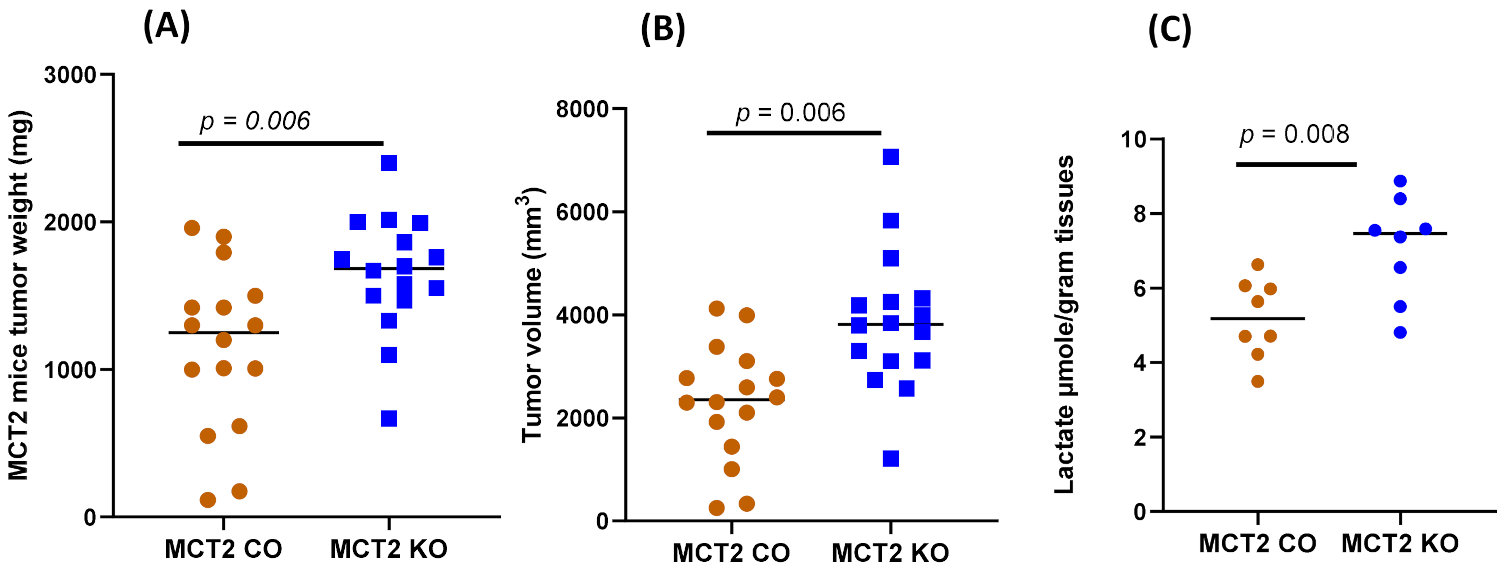
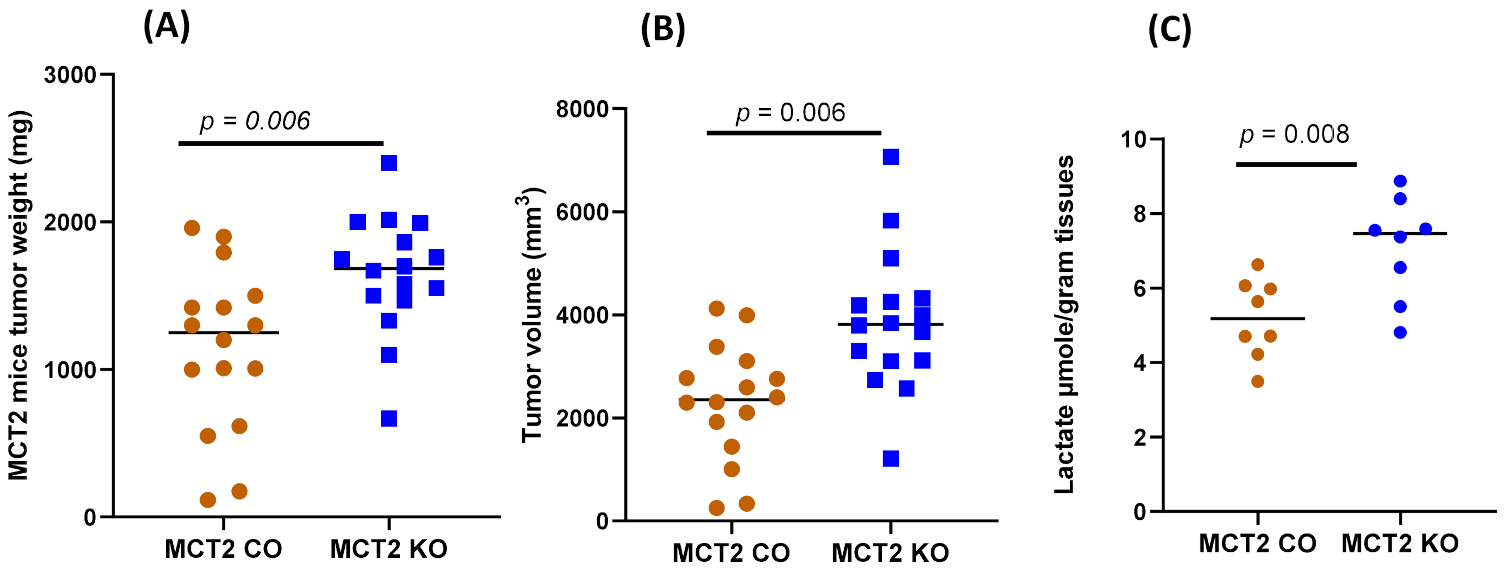
2.2. Tumor Growth and Local Invasiveness
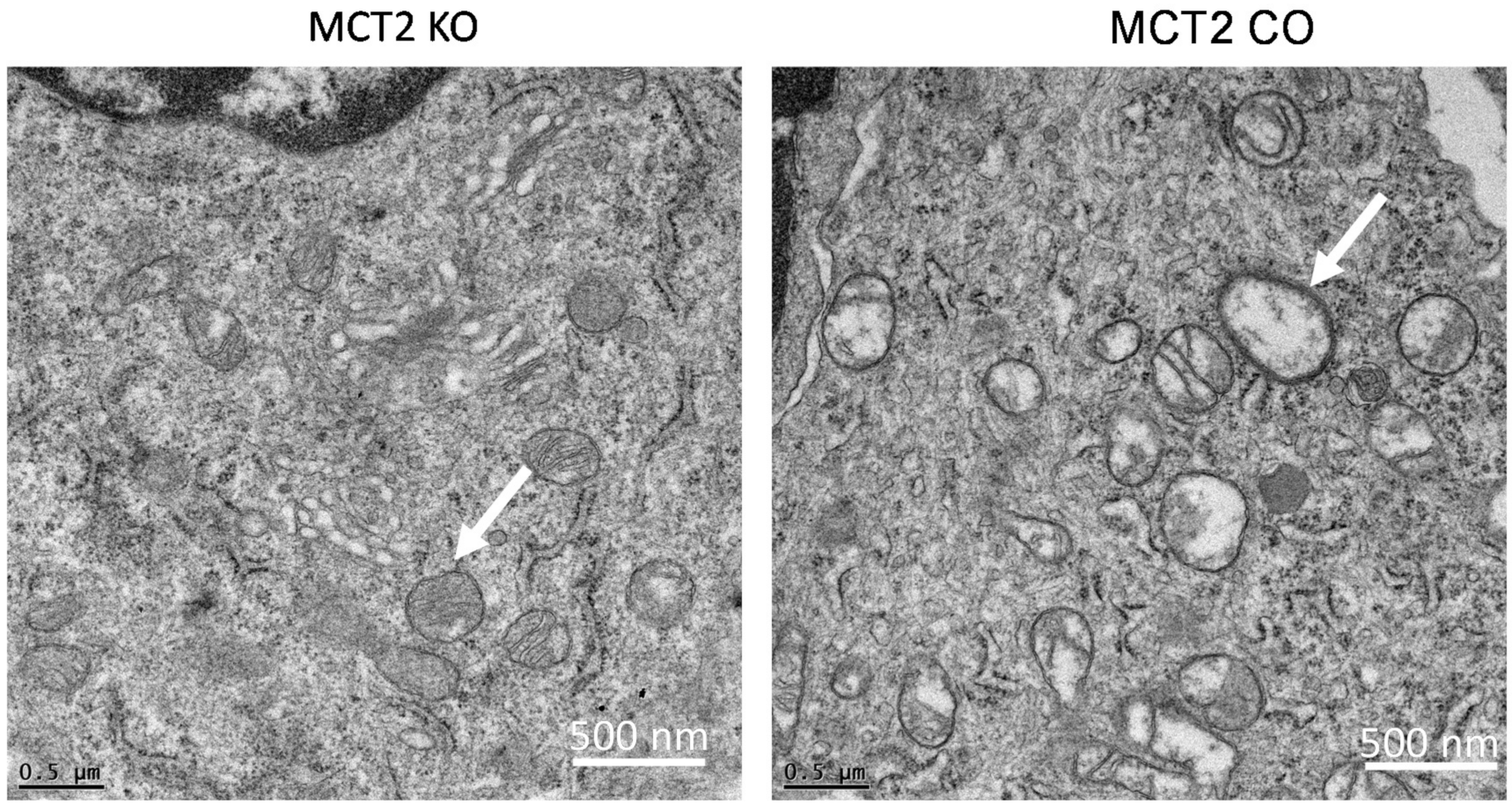
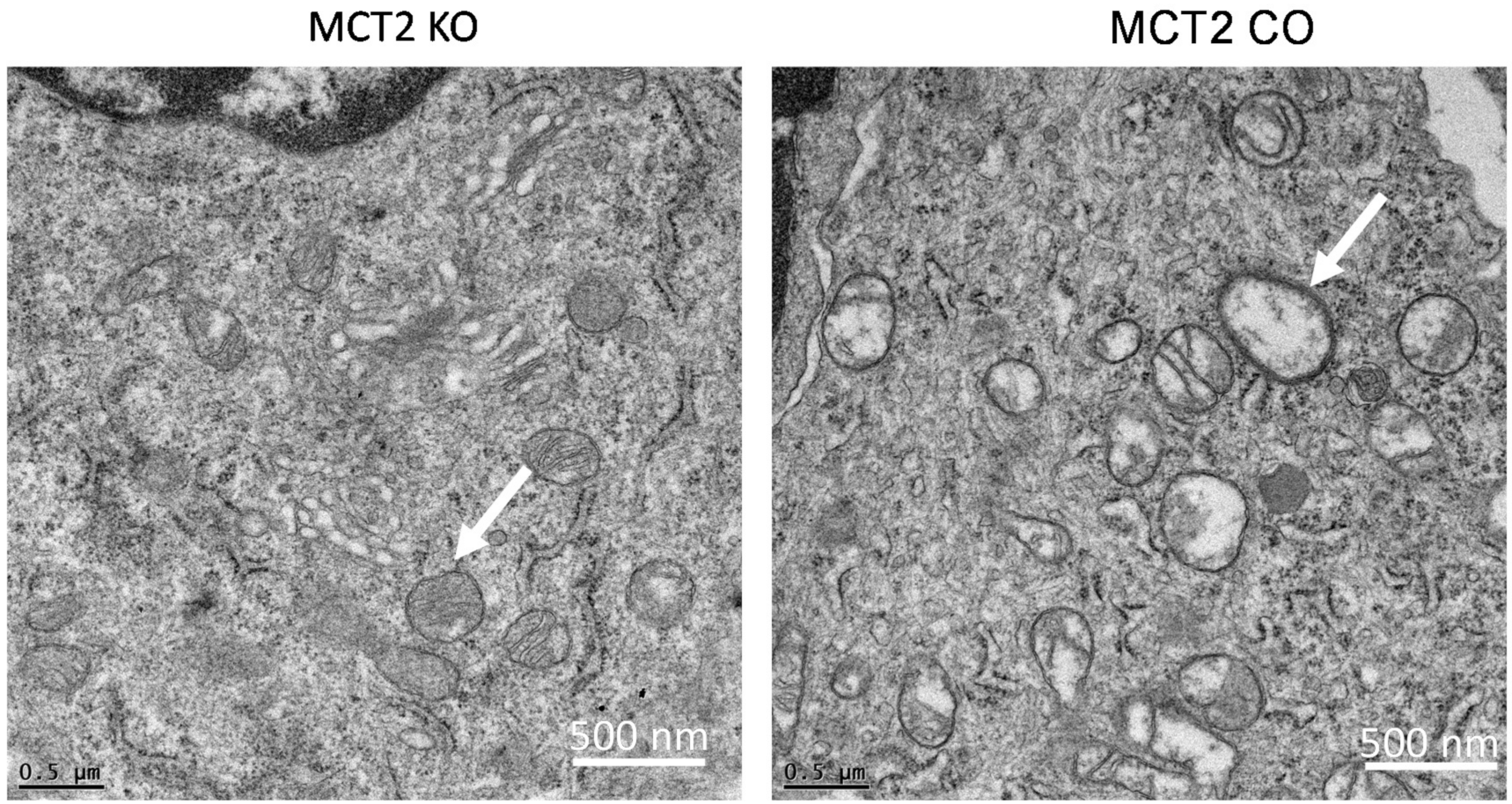
2.3. Electron Microscope
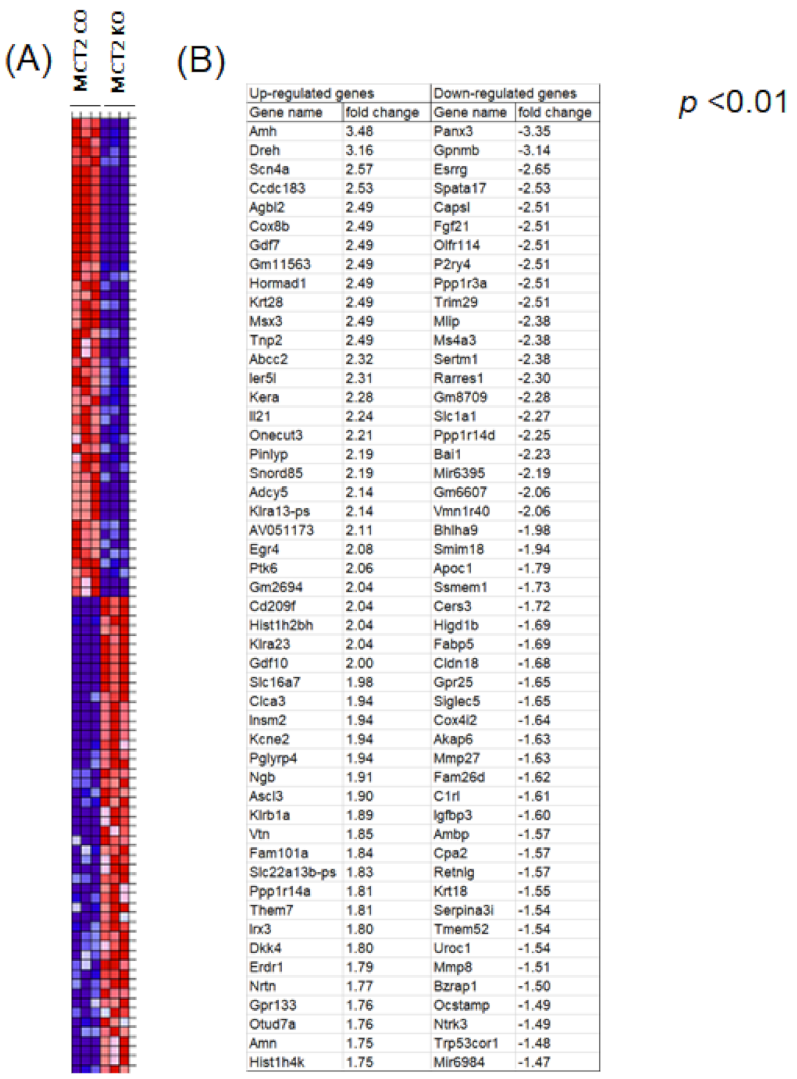
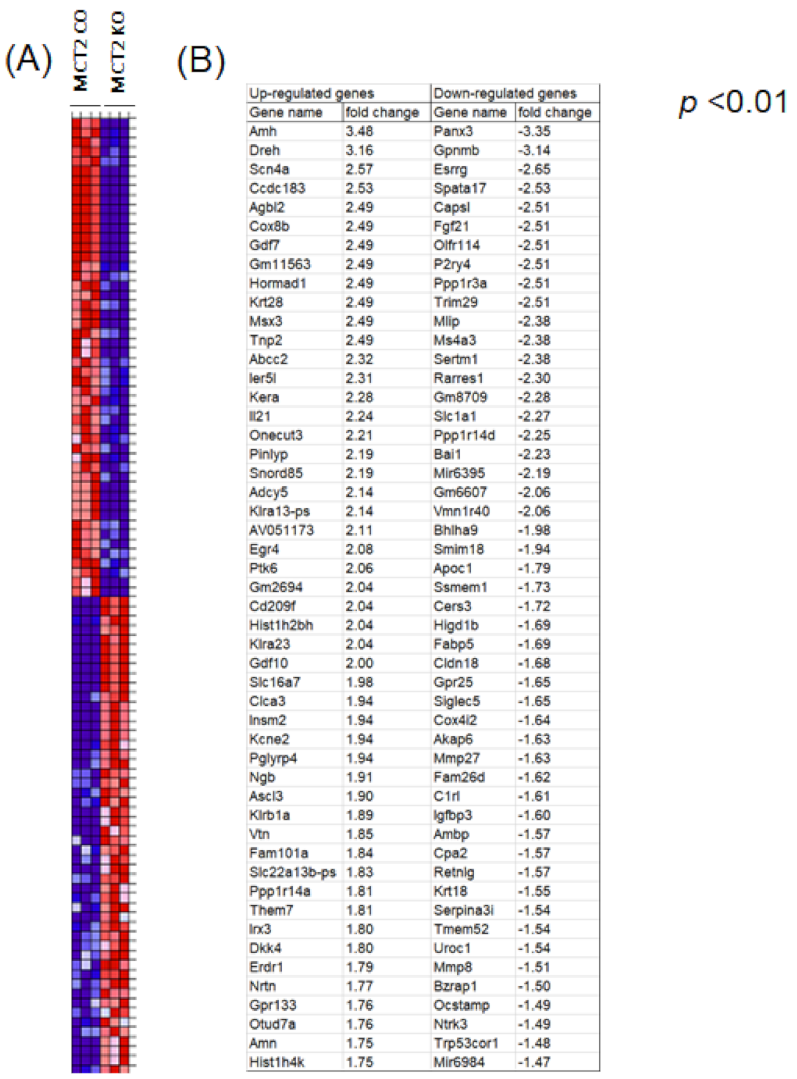
2.4. RNA-seq in Tumor-Associated Macrophages
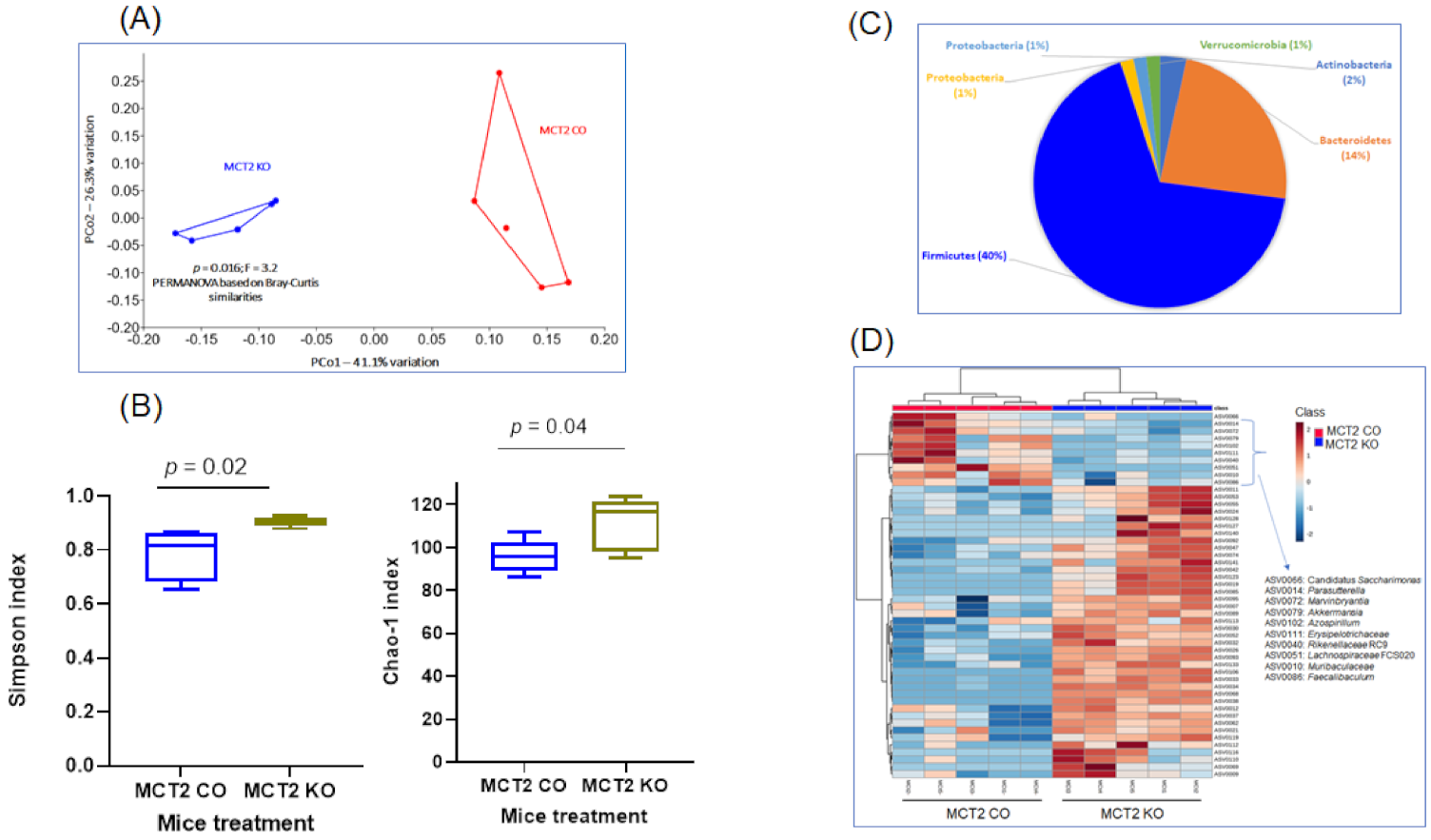
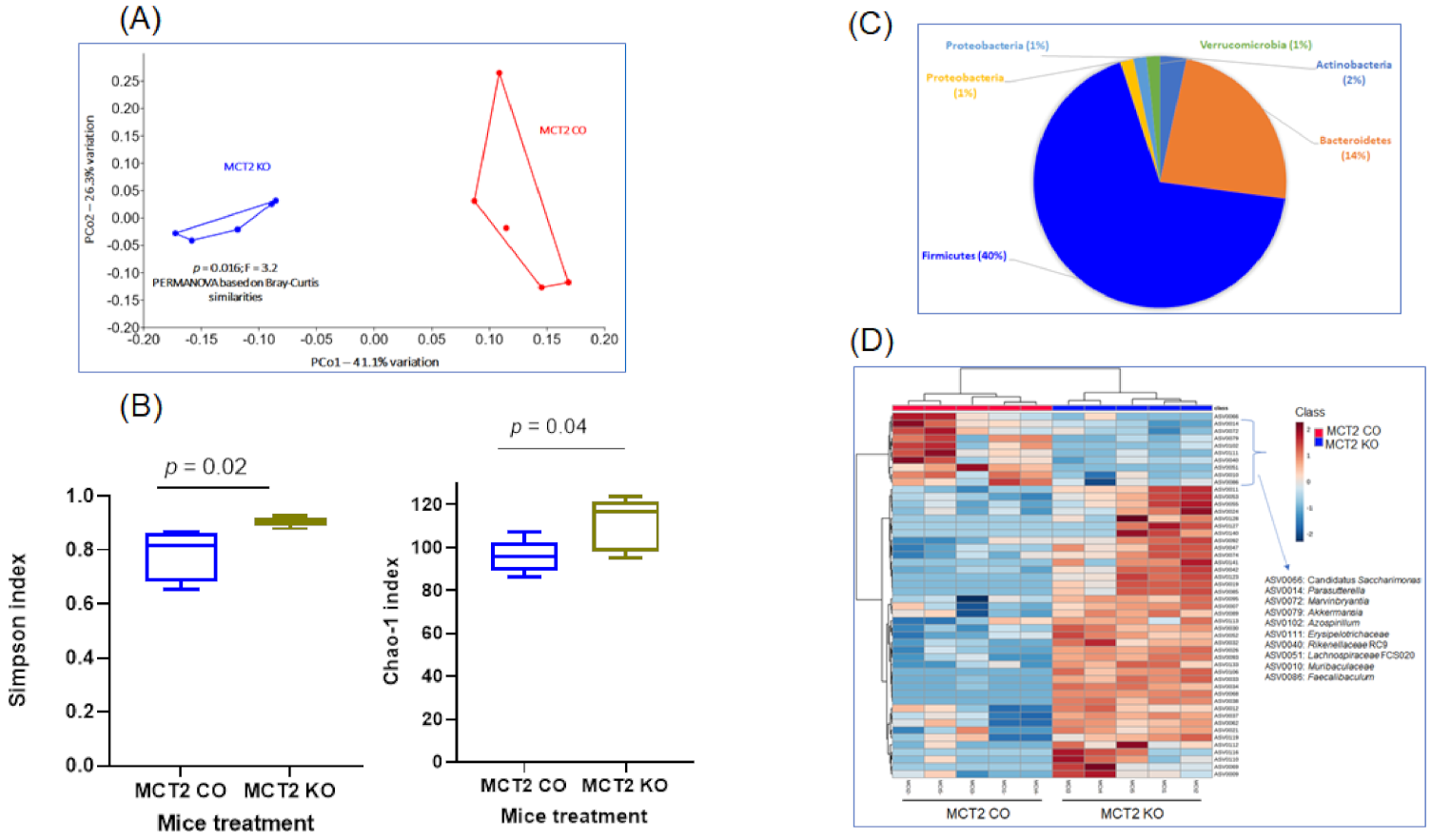
2.5. Fecal Microbiota
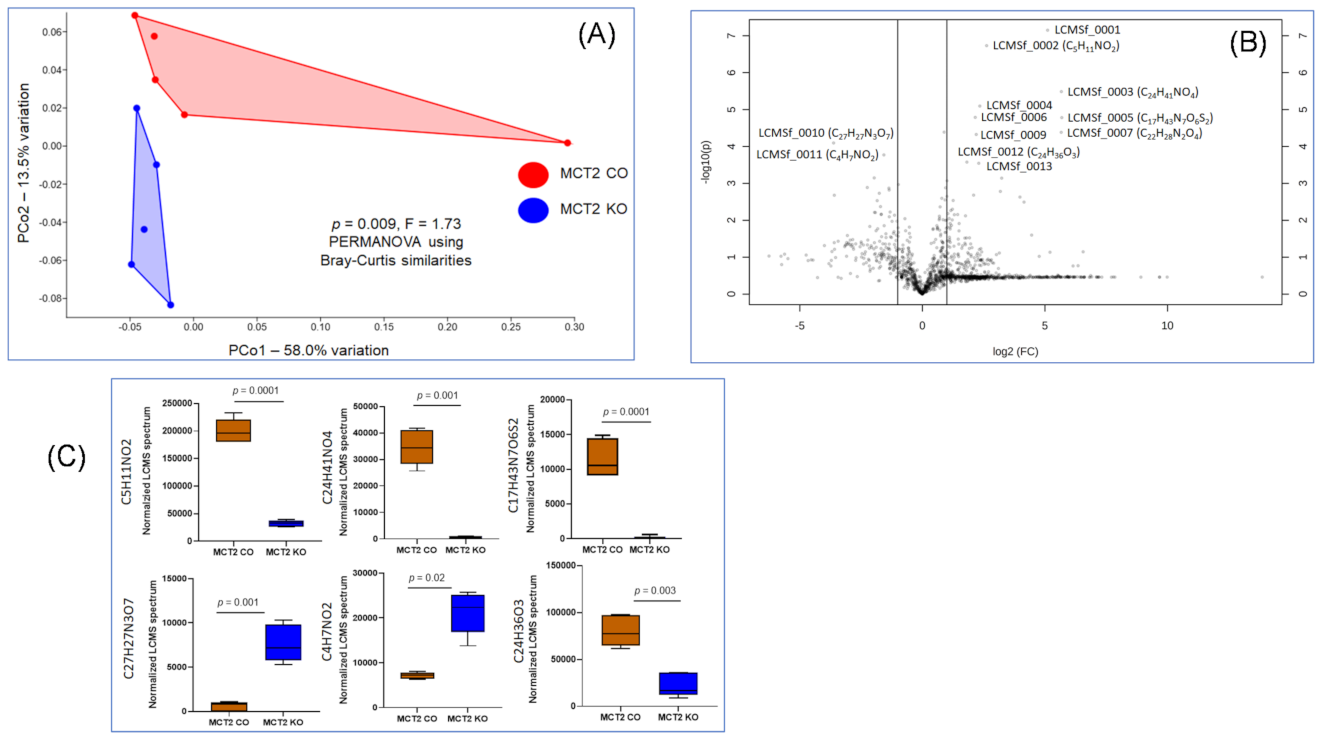
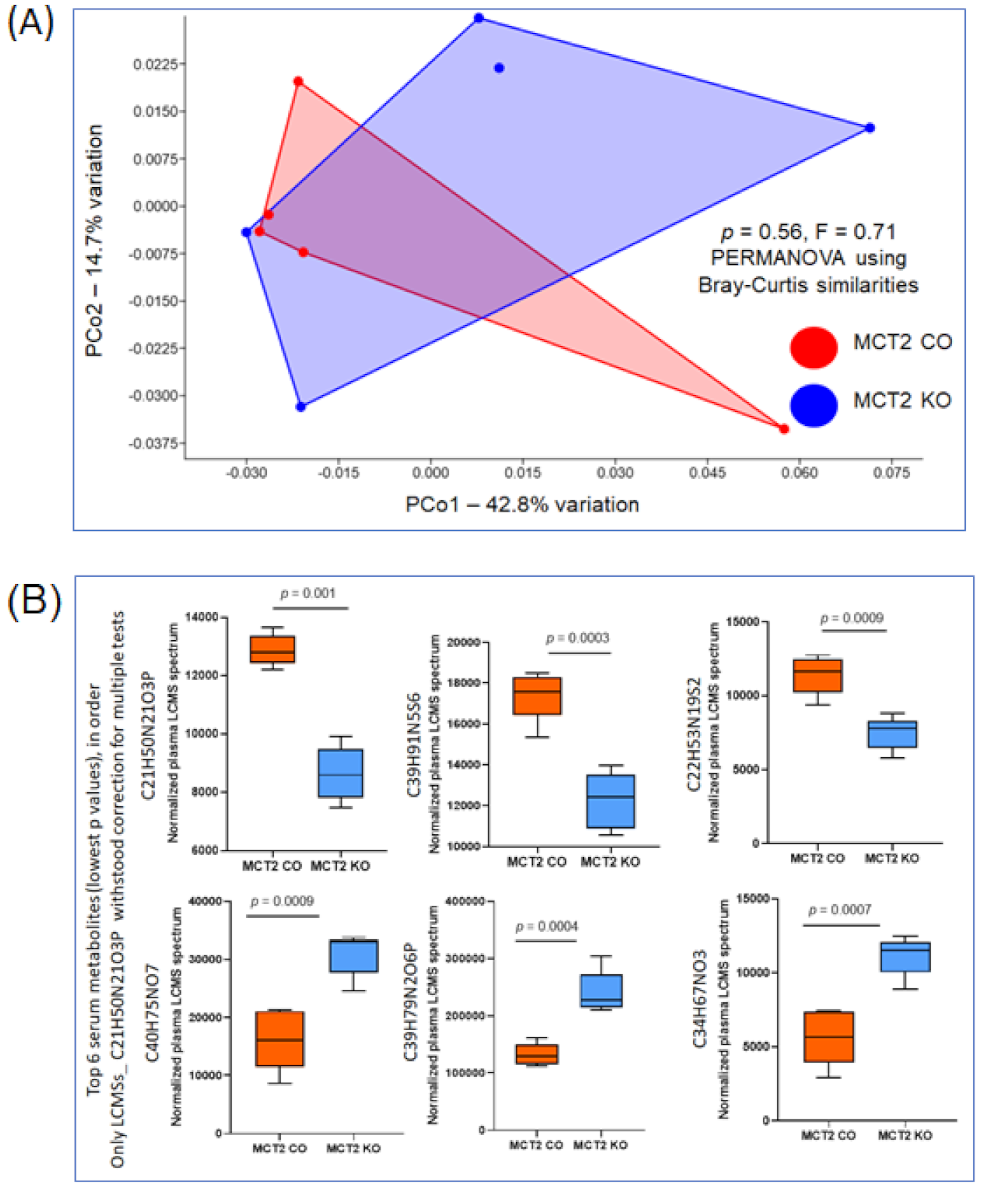
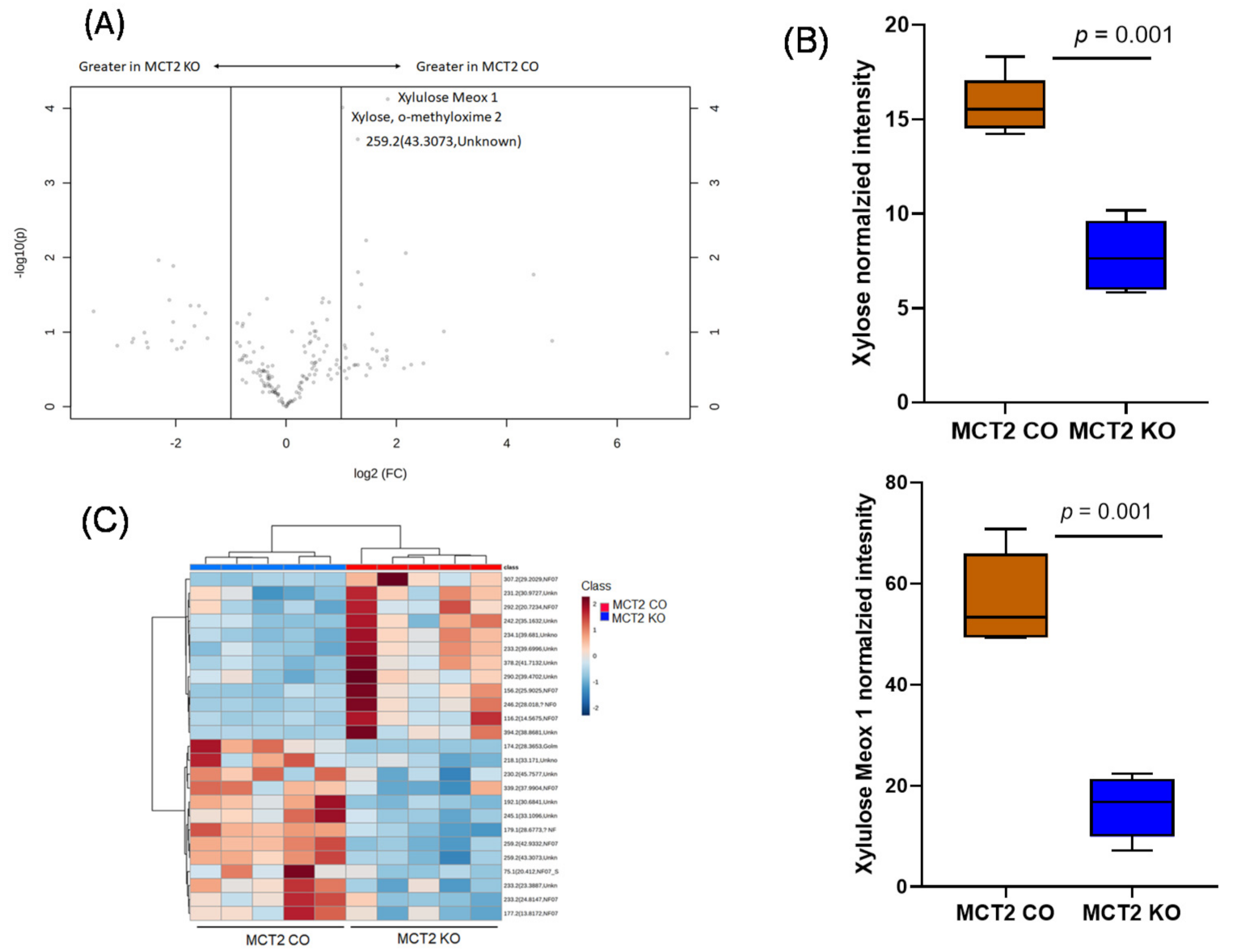
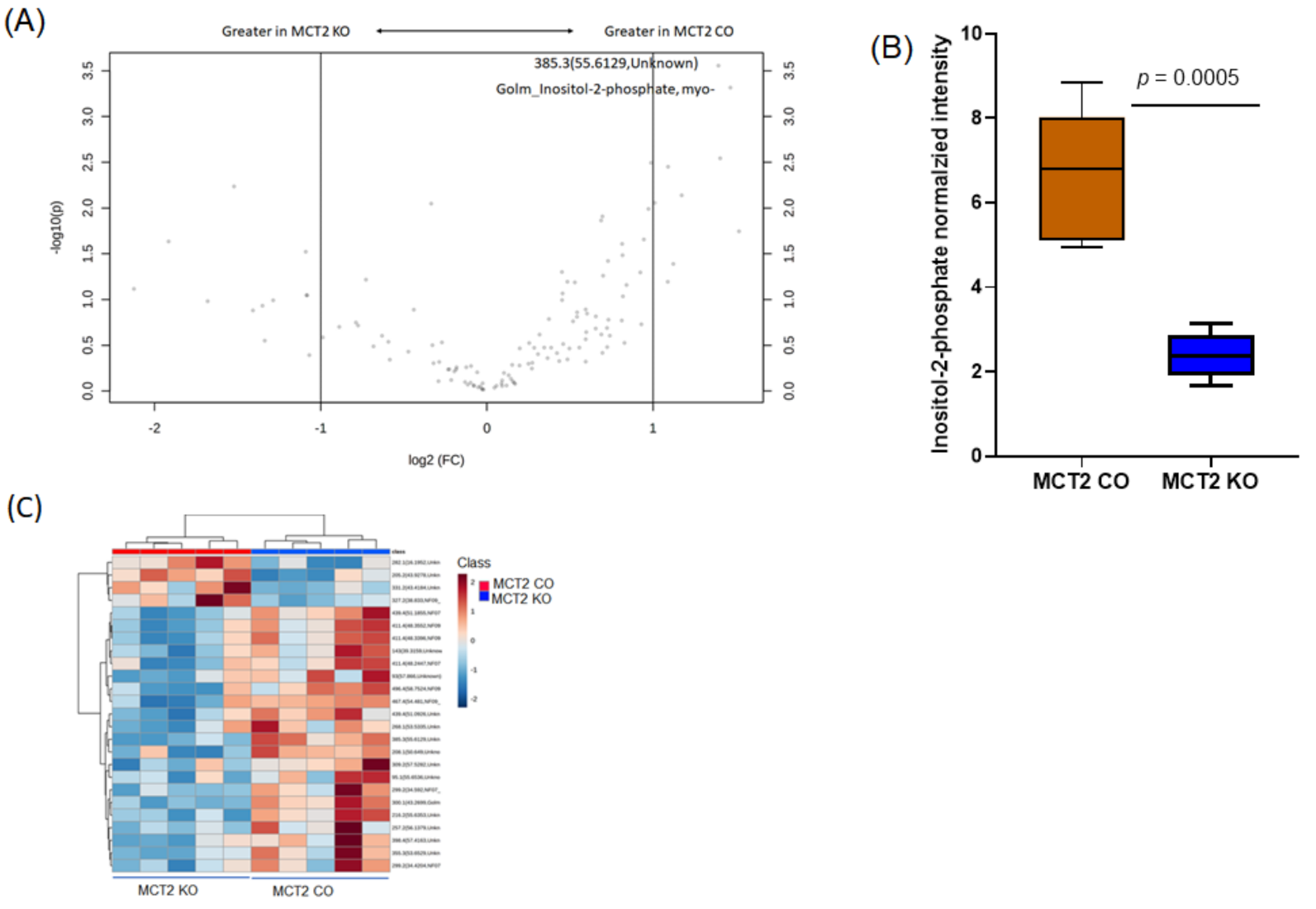
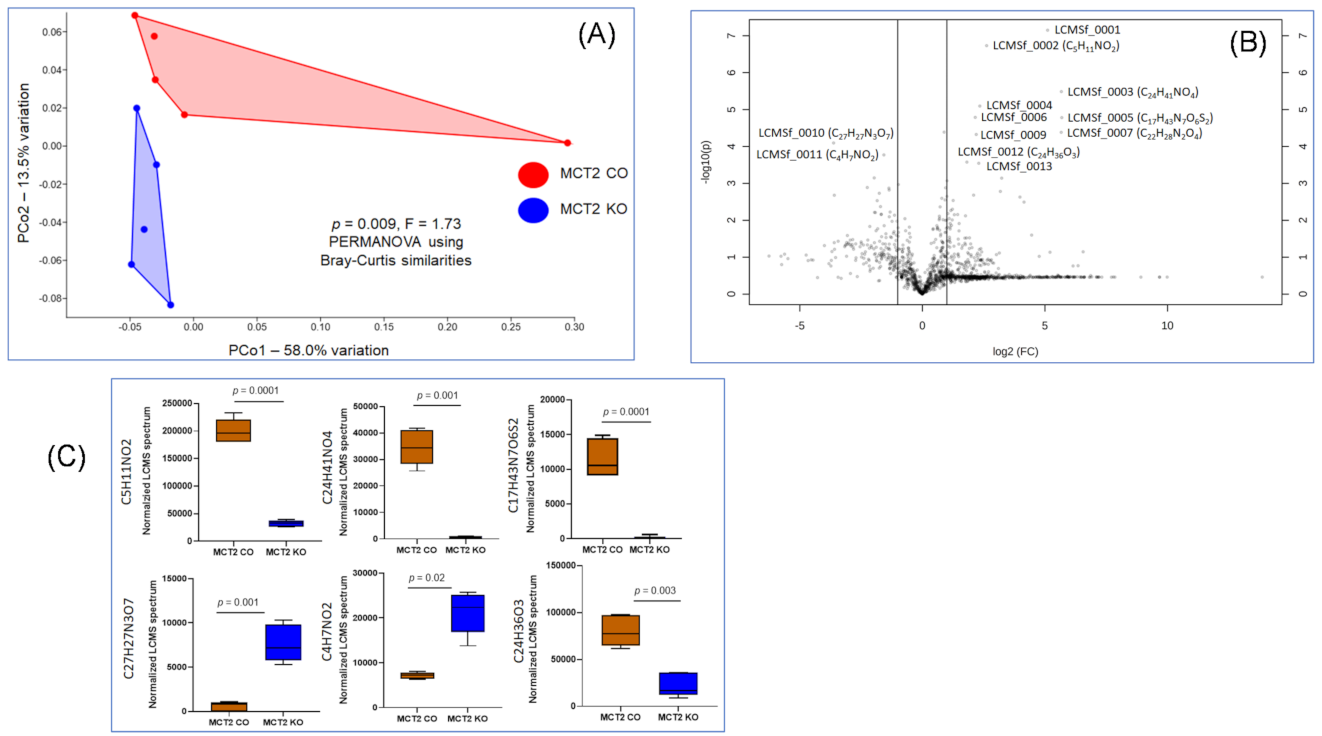
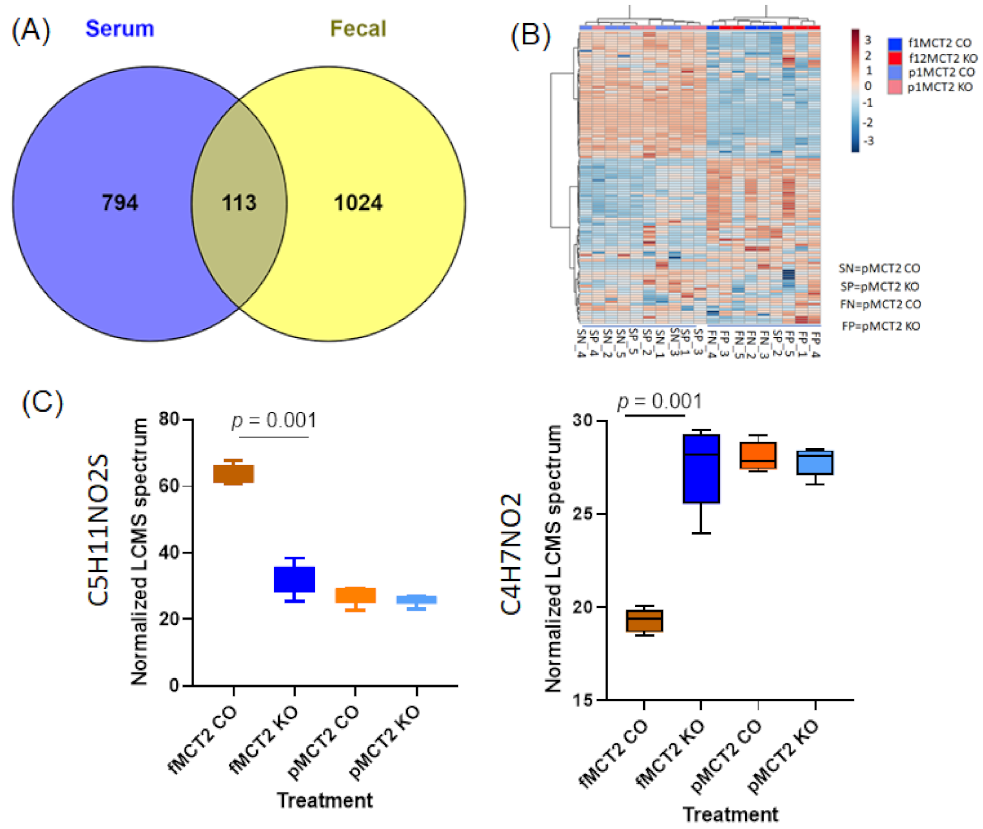
2.6. Fecal and Plasma Metabolomes
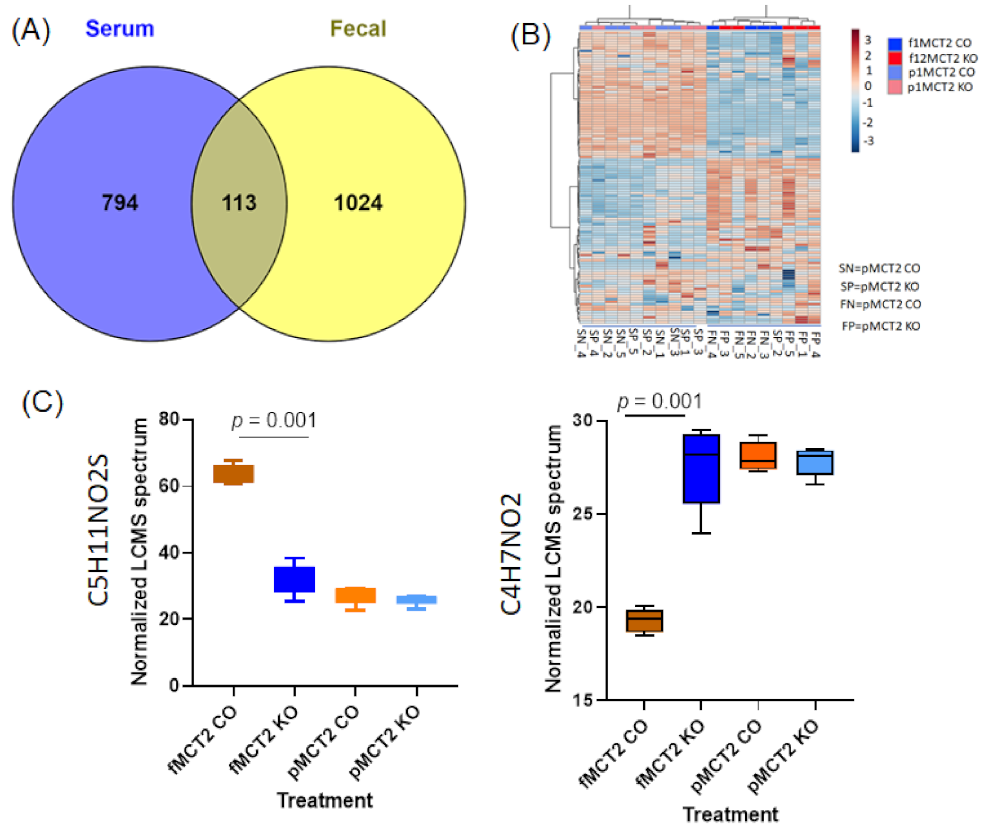
2.7. Comparison between Fecal and Plasma Metabolomes
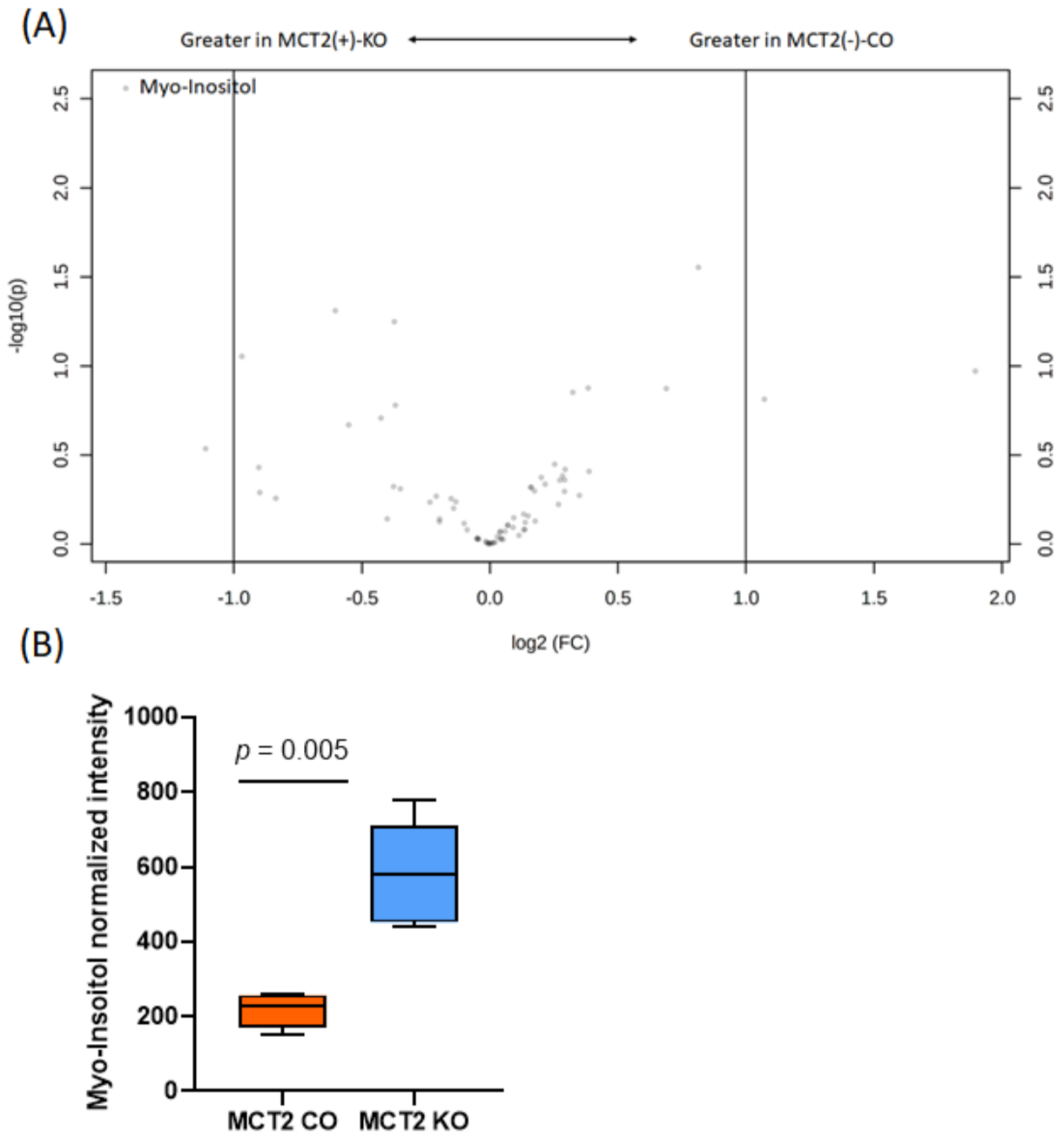
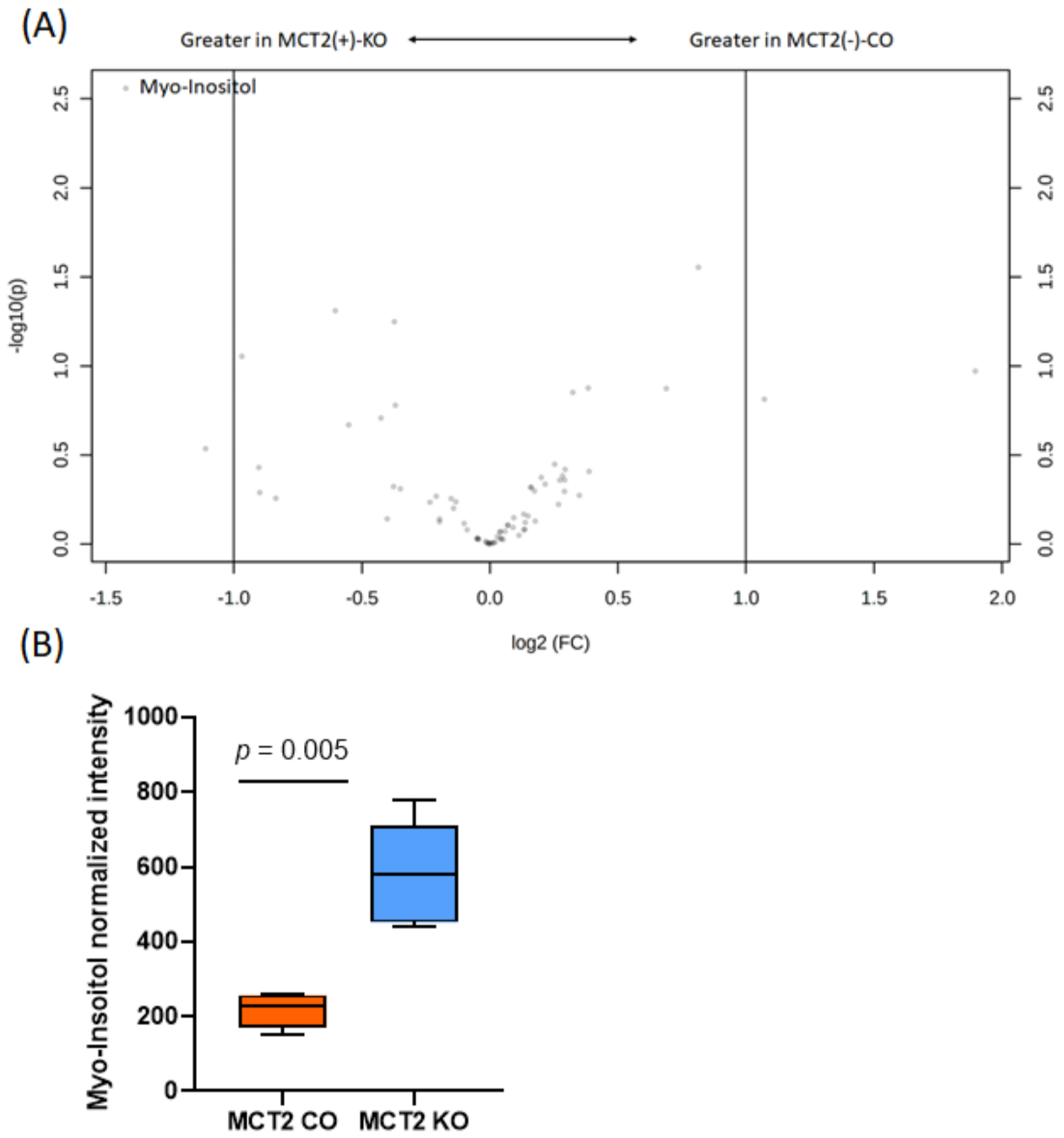
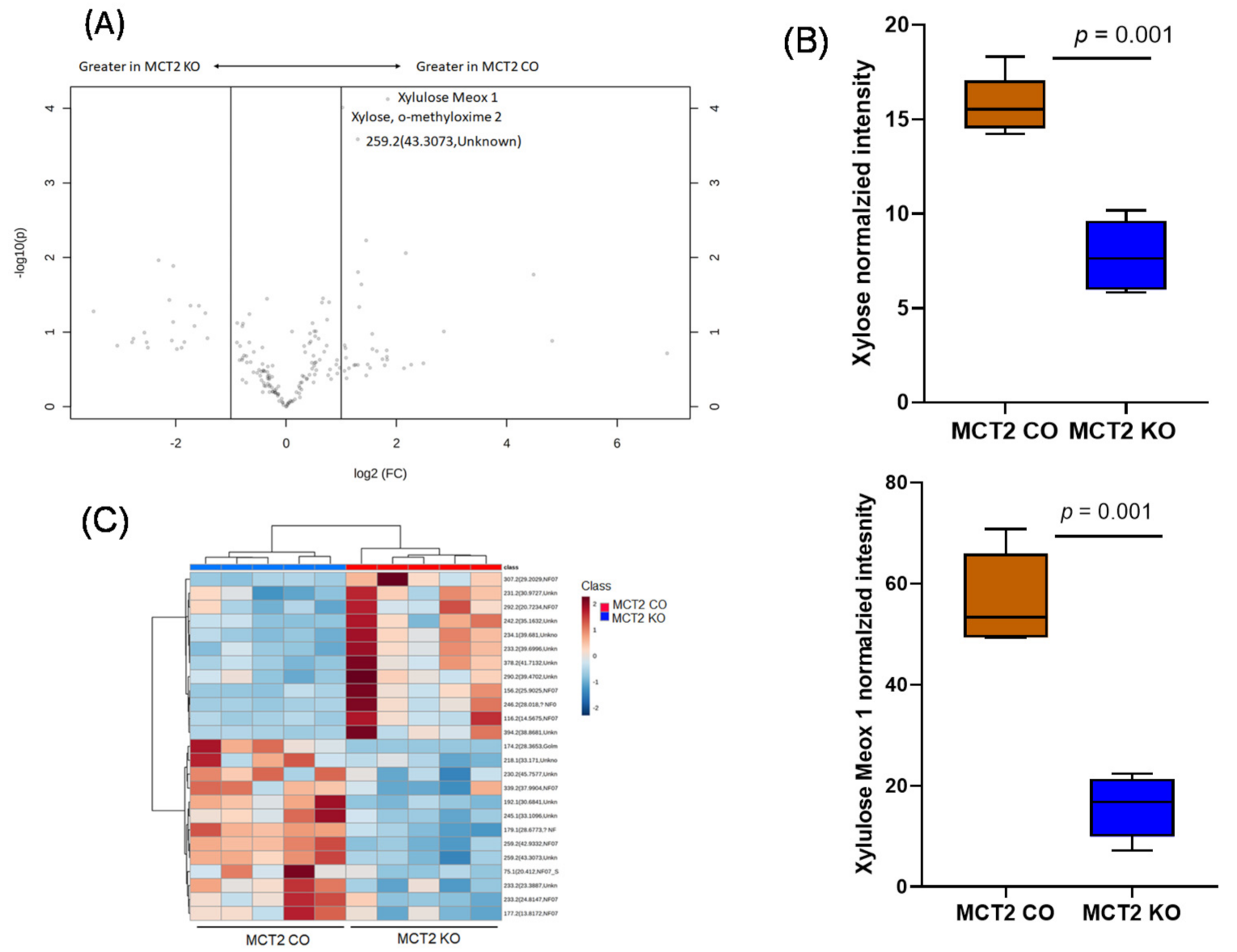
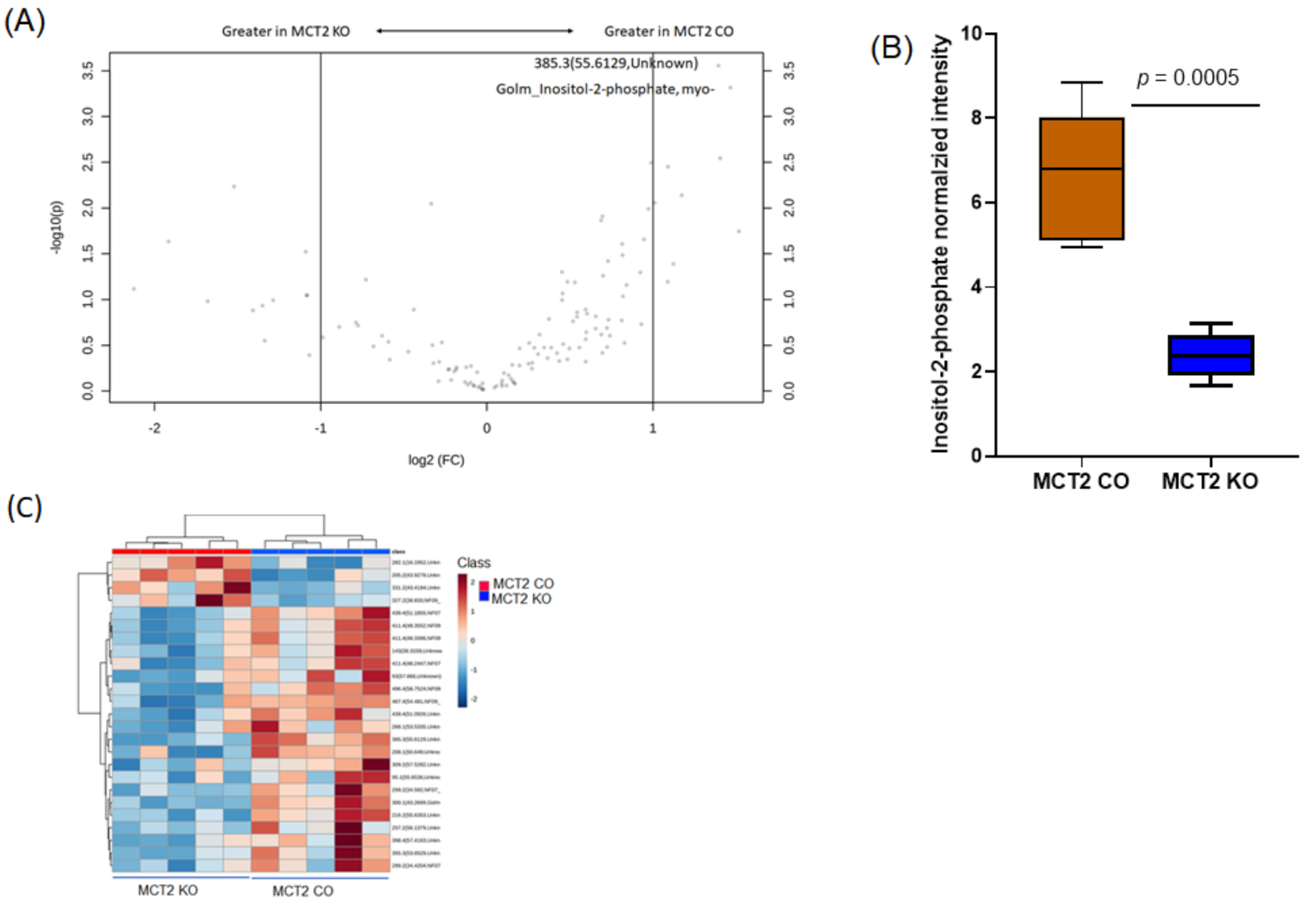
2.8. GC−MS for Plasma and Fecal Metabolomes
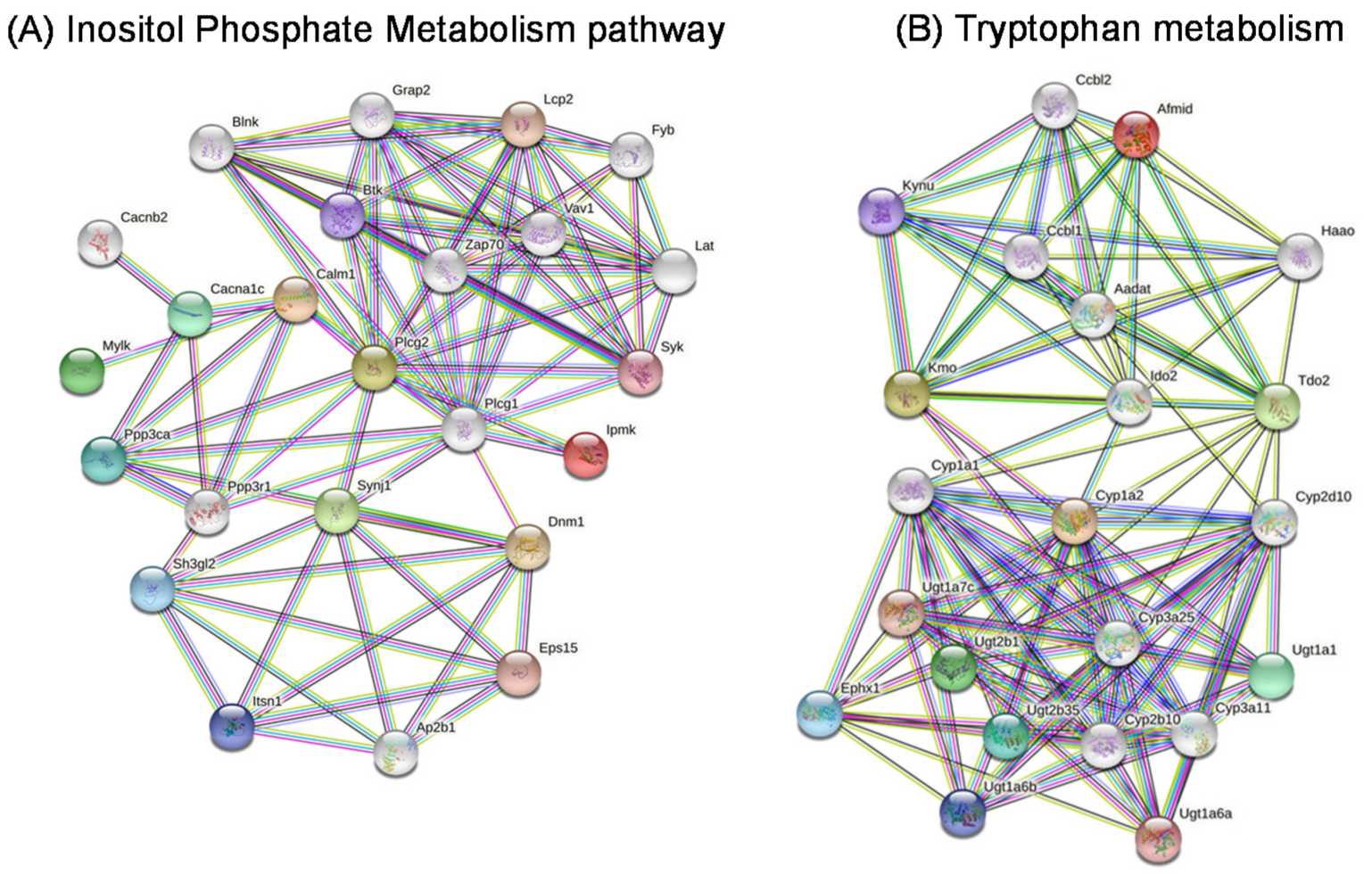
2.9. Metabolic Pathways
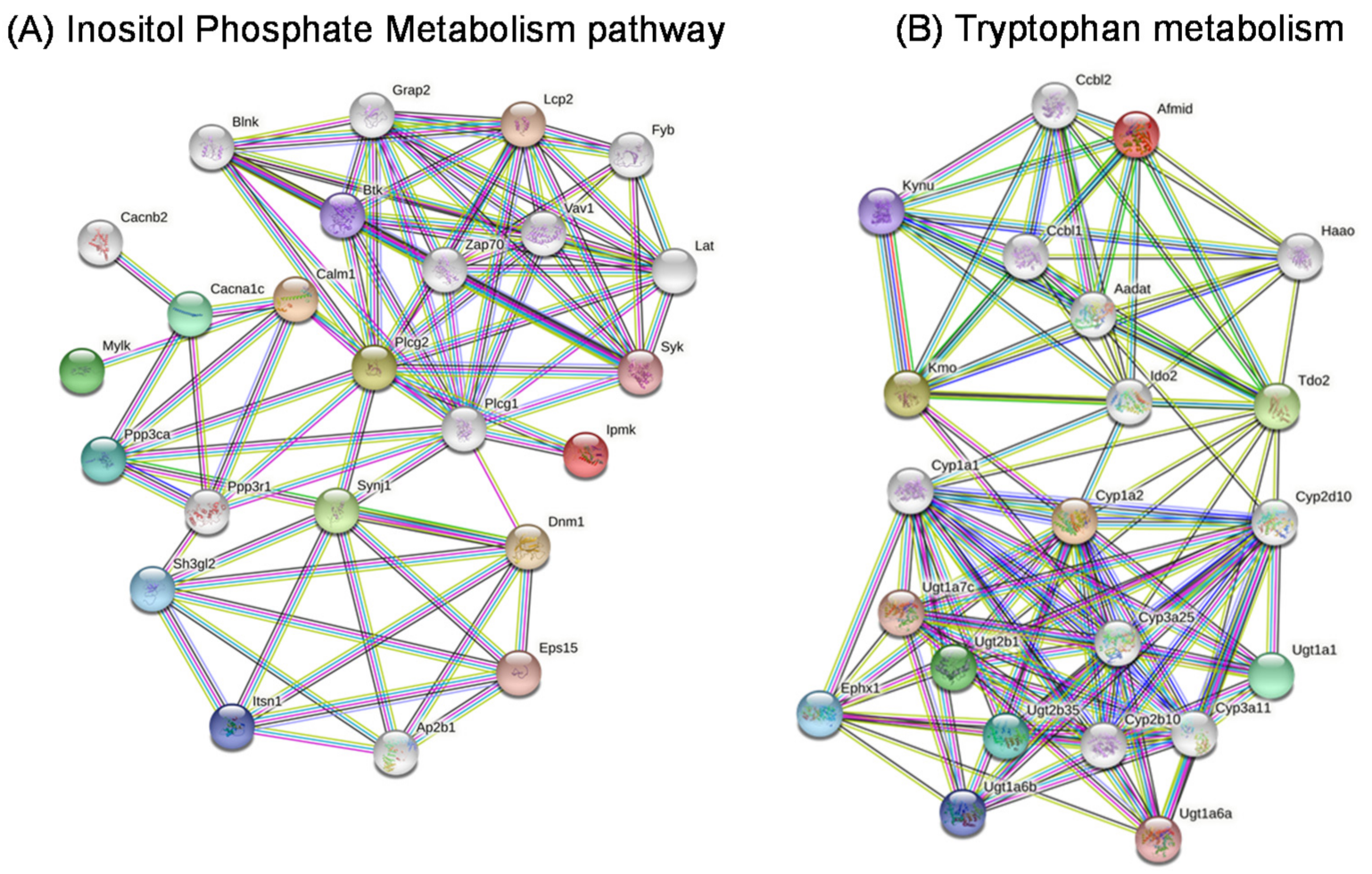
2.10. Gene Networks
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. MCT2 Mouse Genotyping
4.3. Tumor Cell Line
4.4. Tamoxifen and Subcutaneous Flank Tumor Model
4.5. Western Blots
4.6. Transmission Electron Microscopy and Mitochondrial Dysfunction
4.7. Lactate Measurement
4.8. Isolation Tumor Macrophage
4.9. RNA-seq Analysis of Tumor Macrophages
4.10. Functional Enrichment Analysis of DEGs
4.11. DNA Extraction
4.12. 16S rRNA Library Preparation and Sequencing
4.13. Informatics Analysis
4.14. Metabolomic Profiling
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family-Role and regulation. IUBMB Life 2011, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Meredith, D.; Fox, J.E.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: The ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Kouroupi, M.; Balaska, K.; Koukourakis, M.I. A Novel Lipofuscin-detecting Marker of Senescence Relates With Hypoxia, Dysregulated Autophagy and With Poor Prognosis in Non-small-cell-lung Cancer. Vivo 2020, 34, 3187–3193. [Google Scholar] [CrossRef]
- Bonatelli, M.; Fornari, I.F.; Bernécule, P.N.; Pinheiro, L.E.; Costa, R.F.A.; Longatto-Filho, A.; Junior, J.N.A.; Silva, E.C.A.; Cárcano, F.M.; Pinheiro, C. Expression of Glycolysis-Related Proteins in Cancer of Unknown Primary Origin. Front. Oncol. 2021, 11, 682665. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, I.M.; Balaska, K.; Mitrakas, A.G.; Harris, A.L.; Koukourakis, M.I. Programmed death-1 receptor (PD-1) and PD-ligand-1 (PD-L1) expression in non-small cell lung cancer and the immune-suppressive effect of anaerobic glycolysis. Med Oncol. 2019, 36, 76. [Google Scholar] [CrossRef]
- Fets, L.; Driscoll, P.C.; Grimm, F.; Jain, A.; Nunes, P.M.; Gounis, M.; Doglioni, G.; Papageorgiou, G.; Ragan, T.J.; Campos, S.; et al. MCT2 mediates concentration-dependent inhibition of glutamine metabolism by MOG. Nat. Chem. Biol. 2018, 14, 1032–1042. [Google Scholar] [CrossRef]
- Valença, I.; Ferreira, A.; Correia, M.; Kühl, S.; Van Roermund, C.; Waterham, H.; Máximo, V.; Islinger, M.; Ribeiro, D. Prostate Cancer Proliferation Is Affected by the Subcellular Localization of MCT2 and Accompanied by Significant Peroxisomal Alterations. Cancers 2020, 12, 3152. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.; Sivridis, E. Comparison of Metabolic Pathways between Cancer Cells and Stromal Cells in Colorectal Carcinomas: A Metabolic Survival Role for Tumor-Associated Stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathupala, S.P.; Parajuli, P.; Sloan, A.E. Silencing of Monocarboxylate Transporters via Small Interfering Ribonucleic Acid Inhibits Glycolysis and Induces Cell Death in Malignant Glioma: An in Vitro Study. Neurosurgery 2004, 55, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef]
- Li, W.; Deng, Y.; Chu, Q.; Zhang, P. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019, 447, 41–47. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [Green Version]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The Long-Term Stability of the Human Gut Microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [Green Version]
- Leystra, A.A.; Clapper, M.L. Gut Microbiota Influences Experimental Outcomes in Mouse Models of Colorectal Cancer. Genes 2019, 10, 900. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Tang, Q.; Yu, S.; Xie, M.; Xie, Y.; Chen, G.; Chen, L. Role of the oral microbiota in cancer evolution and progression. Cancer Med. 2020, 9, 6306–6321. [Google Scholar] [CrossRef]
- Chinen, T.; Rudensky, A.Y. The effects of commensal microbiota on immune cell subsets and inflammatory responses. Immunol. Rev. 2011, 245, 45–55. [Google Scholar] [CrossRef]
- Sethi, V.; Kurtom, S.; Tarique, M.; Lavania, S.; Malchiodi, Z.; Hellmund, L.; Zhang, L.; Sharma, U.; Giri, B.; Garg, B.; et al. Gut Microbiota Promotes Tumor Growth in Mice by Modulating Immune Response. Gastroenterology 2018, 155, 33–37.e6. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gharaibeh, R.Z.; Newsome, R.C.; Jobin, C. Amending microbiota by targeting intestinal inflammation with TNF blockade attenuates development of colorectal cancer. Nat. Rev. Cancer 2020, 1, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Li, B.; Guo, Y.; Wu, L.; Kou, F.; Yang, L. Signatures of Multi-Omics Reveal Distinct Tumor Immune Microenvironment Contributing to Immunotherapy in Lung Adenocarcinoma. Front Immunol. 2021, 12, 723172. [Google Scholar] [CrossRef]
- Poole, R.C.; Halestrap, A. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am. J. Physiol. Physiol. 1993, 264, C761–C782. [Google Scholar] [CrossRef]
- Zhang, B.; Jin, Q.; Xu, L.; Li, N.; Meng, Y.; Chang, S.; Zheng, X.; Wang, J.; Chen, Y.; Neculai, D.; et al. Cooperative transport mechanism of human monocarboxylate transporter 2. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Zubricky, R.; Coughlin, L.; Jassim, A.H.; Inman, D.M. MCT2 overexpression rescues metabolic vulnerability and protects retinal ganglion cells in two models of glaucoma. Neurobiol. Dis. 2020, 141, 104944. [Google Scholar] [CrossRef]
- Lund, J.; Aas, V.; Tingstad, R.H.; Van Hees, A.; Nikolic, N. Utilization of lactic acid in human myotubes and interplay with glucose and fatty acid metabolism. Sci. Rep. 2018, 8, 9814. [Google Scholar] [CrossRef] [PubMed]
- Schwickert, G.; Walenta, S.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in human cervical cancer with incidence of metastasis. Cancer Res. 1995, 55, 4757–4759. [Google Scholar] [PubMed]
- Walenta, S.; Salameh, A.; Lyng, H.; Evensen, J.F.; Mitze, M.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am. J. Pathol. 1997, 150, 409–415. [Google Scholar]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2020, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Shan, T.; Chen, S.; Chen, X.; Wu, T.; Yang, Y.; Li, S.; Ma, J.; Zhao, J.; Lin, W.; Li, W.; et al. M2TAM subsets altered by lactic acid promote Tcell apoptosis through the PDL1/PD1 pathway. Oncol. Rep. 2020, 44, 1885–1894. [Google Scholar] [CrossRef]
- Pyaskovskaya, O.N.; Kolesnik, D.L.; Prokhorova, I.V.; Burlaka capital A., C.; Gorbach, O.I.; Solyanik, G.I. Tumor microenvironment changes tumor cell sensitivity to action of energy metabolism modifiers. Exp. Oncol. 2020, 42, 192–196. [Google Scholar] [CrossRef]
- Li, K.; Lin, C.; He, Y.; Lu, L.; Xu, K.; Tao, B.; Xia, Z.; Zeng, R.; Mao, Y.; Luo, Z.; et al. Engineering of Cascade-Responsive Nanoplatform to Inhibit Lactate Efflux for Enhanced Tumor Chemo-Immunotherapy. ACS Nano 2020, 14, 14164–14180. [Google Scholar] [CrossRef]
- Lee, I.; Lee, S.J.; Kang, W.K.; Park, C. Inhibition of monocarboxylate transporter 2 induces senescence-associated mitochondrial dysfunction and suppresses progression of colorectal malignancies in vivo. Mol. Cancer Ther. 2012, 11, 2342–2351. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM functions-it takes two to tango. FEBS J. 2017, 285, 700–716. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martin-Perez, R.; Serneels, J.; Prenen, H.; Ghesquiere, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Chu, Z.; Zhu, L.; Yang, T.; Wang, P.; Liu, F.; Huang, Y.; Zhang, F.; Zhang, X.; Ding, W.; et al. 2-Deoxy-d-Glucose Treatment Decreases Anti-inflammatory M2 Macrophage Polarization in Mice with Tumor and Allergic Airway Inflammation. Front. Immunol. 2017, 8, 637. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in human cancer. Int. J. Biochem. Cell Biol. 2009, 41, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in gliomas and their vascular microenvironment. Biochim. Biophys. Acta Bioenerg. 2010, 1807, 602–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, F.; Corbelli, A.; Affatato, R.; Chilà, R.; Chiappa, M.; Brunelli, L.; Fruscio, R.; Pastorelli, R.; Fiordaliso, F.; Damia, G. Mitochondrial structural alterations in ovarian cancer patient-derived xenografts resistant to cisplatin. Am. J. Cancer Res. 2021, 11, 2303–2311. [Google Scholar]
- Signorile, A.; De Rasmo, D.; Cormio, A.; Musicco, C.; Rossi, R.; Fortarezza, F.; Palese, L.L.; Loizzi, V.; Resta, L.; Scillitani, G.; et al. Human Ovarian Cancer Tissue Exhibits Increase of Mitochondrial Biogenesis and Cristae Remodeling. Cancers 2019, 11, 1350. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Wang, X.; Guo, Y.; Yan, J.; Abuduwaili, A.; Aximujiang, K.; Yan, J.; Wu, M. Gut microbiota influence tumor development and Alter interactions with the human immune system. J. Exp. Clin. Cancer Res. 2021, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Tsuei, J.; Chau, T.; Mills, D.; Wan, Y.J. Bile acid dysregulation, gut dysbiosis, and gastrointestinal cancer. Exp. Biol. Med. 2014, 239, 1489–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The Human Microbiome and Cancer. Cancer Prev. Res. 2017, 10, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, T.; Mikó, E.; Vida, A.; Sebő, S.; Toth, J.; Csonka, T.; Boratkó, A.; Ujlaki, G.; Lente, G.; Kovács, P.; et al. Cadaverine, a metabolite of the microbiome, reduces breast cancer aggressiveness through trace amino acid receptors. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Wang, Z.; Ammar, E.; Zhang, A.; Wang, L.; Lin, M.; Yang, S.-T. Engineering Propionibacterium freudenreichii subsp. shermanii for enhanced propionic acid fermentation: Effects of overexpressing propionyl-CoA:Succinate CoA transferase. Metab. Eng. 2015, 27, 46–56. [Google Scholar] [CrossRef]
- Tan, J.; Yu, C.-Y.; Wang, Z.-H.; Chen, H.-Y.; Guan, J.; Chen, Y.-X.; Fang, J.-Y. Genetic variants in the inositol phosphate metabolism pathway and risk of different types of cancer. Sci. Rep. 2015, 5, 8473. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chen, S.; Zhong, J.; Teng, K.; Yin, Y. Crosstalk between Tryptophan Metabolism and Cardiovascular Disease, Mechanisms, and Therapeutic Implications. Oxidative Med. Cell. Longev. 2017, 2017, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 1–30. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Campillo, N.; Torres, M.; Vilaseca, A.; Nonaka, P.N.; Gozal, D.; Roca-Ferrer, J.; Picado, C.; Montserrat, J.M.; Farre, R.; Navajas, D.; et al. Role of Cyclooxygenase-2 on Intermittent Hypoxia-Induced Lung Tumor Malignancy in a Mouse Model of Sleep Apnea. Sci. Rep. 2017, 7, 44693. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Almendros, I.; Gileles-Hillel, A.; Akbarpour, M.; Trzepizur, W.; Mokhlesi, B.; Huang, L.; Andrade, J.; Farre, R.; Gozal, D. Circulating exosomes potentiate tumor malignant properties in a mouse model of chronic sleep fragmentation. Oncotarget 2016, 7, 54676–54690. [Google Scholar] [CrossRef] [Green Version]
- Almendros, I.; Khalyfa, A.; Trzepizur, W.; Gileles-Hillel, A.; Huang, L.; Akbarpour, M.; Andrade, J.; Farre, R.; Gozal, D. Tumor Cell Malignant Properties Are Enhanced by Circulating Exosomes in Sleep Apnea. Chest 2016, 150, 1030–1041. [Google Scholar] [CrossRef]
- Khalyfa, A.; Mutskov, V.; Carreras, A.; Hakim, F.; Gozal, D. Sleep Fragmentation During Late Gestation Induces Metabolic Perturbations and Epigenetic Changes in Adiponectin Gene Expression in Male Adult Offspring Mice. Diabetes 2014, 63, 3230–3241. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Chong, X.; Peng, R.; Sun, Y.; Zhang, L.; Zhang, Z. Identification of Key Genes in Gastric Cancer by Bioinformatics Analysis. BioMed Res. Int. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.A.; Caporaso, J.G.; Lauber, C.L.; Berg-Lyons, D.; Fierer, N.; Knight, R. PrimerProspector: De novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 2011, 27, 1159–1161. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2010, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Loy, A.; Maixner, F.; Wagner, M.; Horn, M. probeBase—An online resource for rRNA-targeted oligonucleotide probes: New features 2007. Nucleic Acids Res. 2007, 35, D800-4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Sumner, B.W.; Bhatia, A.; Sarma, S.J.; Sumner, L.W. UHPLC-MS Analyses of Plant Flavonoids. Curr. Protoc. Plant Biol. 2019, 4, e20085. [Google Scholar] [CrossRef] [Green Version]
- Vu, D.C.; Lei, Z.; Sumner, L.W.; Coggeshall, M.V.; Lin, C.-H. Identification and quantification of phytosterols in black walnut kernels. J. Food Compos. Anal. 2018, 75, 61–69. [Google Scholar] [CrossRef]
- Vieira-Potter, V.J.; Cross, T.-W.L.; Swanson, K.; Sarma, S.J.; Lei, Z.; Sumner, L.W.; Rosenfeld, C.S. Soy-Induced Fecal Metabolome Changes in Ovariectomized and Intact Female Rats: Relationship with Cardiometabolic Health. Sci. Rep. 2018, 8, 16896. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Enrichment Analysis | #Term ID | Term Description | False Discovery Rate |
|---|---|---|---|
| Molecular function | GO:0031994 | insulin-like growth factor I binding | 1.5 × 10−10 |
| GO:0031995 | insulin-like growth factor II binding | 2.9 × 10−9 | |
| GO:0005158 | insulin receptor binding | 5.1 × 10−4 | |
| GO:0005159 | insulin-like growth factor receptor binding | 3.1 × 10−3 | |
| GO:0008083 | growth factor activity | 2.5 × 10−2 | |
| GO:0004859 | phospholipase inhibitor activity | 2.9 × 10−2 | |
| GO:0005515 | protein binding | 2.9 × 10−2 | |
| GO:0003677 | DNA binding | 4.2 × 10−2 | |
| GO:0098772 | molecular function regulator | 4.2 × 10−2 | |
| GO:0005160 | transforming growth factor beta receptor binding | 4.9 × 10−2 | |
| GO:0005178 | integrin binding | 4.9 × 10−2 | |
| Cellular Component | GO:0005615 | extracellular space | 6.5 × 10−7 |
| GO:0000786 | nucleosome | 3.8 × 10−6 | |
| GO:0000788 | nuclear nucleosome | 7.6 × 10−5 | |
| GO:0005576 | extracellular region | 1.5 × 10−4 | |
| GO:0000785 | chromatin | 2.1 × 10−5 | |
| GO:0000790 | nuclear chromatin | 2.3 × 10−5 | |
| GO:0000228 | nuclear chromosome | 1.6 × 10−3 | |
| GO:0042568 | insulin-like growth factor binary complex | 2.1 × 10−3 | |
| GO:0035867 | alphav-beta3 integrin-IGF-1-IGF1R complex | 4.9 × 10−3 | |
| GO:0005694 | chromosome | 1.2 × 10−2 | |
| GO:0001518 | voltage-gated sodium channel complex | 2.5 × 10−2 | |
| GO:0005751 | mitochondrial respiratory chain complex IV | 4.4 × 10−2 | |
| Biological process | GO:0043567 | regulation of insulin-like growth factor receptor | 1.4 × 10−7 |
| GO:0043568 | positive regulation of insulin-like growth factor receptor | 4.6 × 10−4 | |
| GO:0034728 | nucleosome organization | 5.1 × 10−4 | |
| GO:0001649 | osteoblast differentiation | 1.4 × 10−3 | |
| GO:0006323 | DNA packaging | 1.4 × 10−3 | |
| GO:0014910 | regulation of smooth muscle cell migration | 1.4 × 10−3 | |
| GO:0019556 | histidine catabolic process to glutamate and formamide | 1.4 × 10−3 | |
| GO:0019557 | histidine catabolic process to glutamate and formate | 1.4 × 10−3 | |
| GO:0042246 | tissue regeneration | 1.4 × 10−3 | |
| GO:0006325 | chromatin organization | 1.6 × 10−3 | |
| GO:0006333 | chromatin assembly or disassembly | 1.8 × 10−3 | |
| GO:0010906 | regulation of glucose metabolic process | 1.8 × 10−3 | |
| GO:0090031 | positive regulation of steroid hormone biosynthetic process | 1.8 × 10−3 | |
| GO:0048009 | insulin-like growth factor receptor signaling pathway | 2.7 × 10−3 | |
| GO:0045725 | positive regulation of glycogen biosynthetic process | 4.3 × 10−3 |
| KEGG ID | Term Description | False Discovery Rate |
|---|---|---|
| mmu00340 | Histidine metabolism | 0.0066 |
| mmu04115 | p53 signaling pathway | 0.0066 |
| mmu04610 | Complement and coagulation cascades | 0.0066 |
| mmu05202 | Transcriptional misregulation in cancer | 0.0066 |
| mmu05215 | Prostate cancer | 0.0066 |
| mmu04350 | TGF-beta signaling pathway | 0.0091 |
| mmu01522 | Endocrine resistance | 0.0104 |
| mmu05322 | Systemic lupus erythematosus | 0.0104 |
| mmu04066 | HIF-1 signaling pathway | 0.0122 |
| mmu05205 | Proteoglycans in cancer | 0.0187 |
| mmu04068 | FoxO signaling pathway | 0.0244 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalyfa, A.; Qiao, Z.; Raju, M.; Shyu, C.-R.; Coghill, L.; Ericsson, A.; Gozal, D. Monocarboxylate Transporter-2 Expression Restricts Tumor Growth in a Murine Model of Lung Cancer: A Multi-Omic Analysis. Int. J. Mol. Sci. 2021, 22, 10616. https://doi.org/10.3390/ijms221910616
Khalyfa A, Qiao Z, Raju M, Shyu C-R, Coghill L, Ericsson A, Gozal D. Monocarboxylate Transporter-2 Expression Restricts Tumor Growth in a Murine Model of Lung Cancer: A Multi-Omic Analysis. International Journal of Molecular Sciences. 2021; 22(19):10616. https://doi.org/10.3390/ijms221910616
Chicago/Turabian StyleKhalyfa, Abdelnaby, Zhuanhong Qiao, Murugesan Raju, Chi-Ren Shyu, Lyndon Coghill, Aaron Ericsson, and David Gozal. 2021. "Monocarboxylate Transporter-2 Expression Restricts Tumor Growth in a Murine Model of Lung Cancer: A Multi-Omic Analysis" International Journal of Molecular Sciences 22, no. 19: 10616. https://doi.org/10.3390/ijms221910616
APA StyleKhalyfa, A., Qiao, Z., Raju, M., Shyu, C.-R., Coghill, L., Ericsson, A., & Gozal, D. (2021). Monocarboxylate Transporter-2 Expression Restricts Tumor Growth in a Murine Model of Lung Cancer: A Multi-Omic Analysis. International Journal of Molecular Sciences, 22(19), 10616. https://doi.org/10.3390/ijms221910616